Natural Killer Cell Dysfunction and Its Role in COVID-19



Abstract
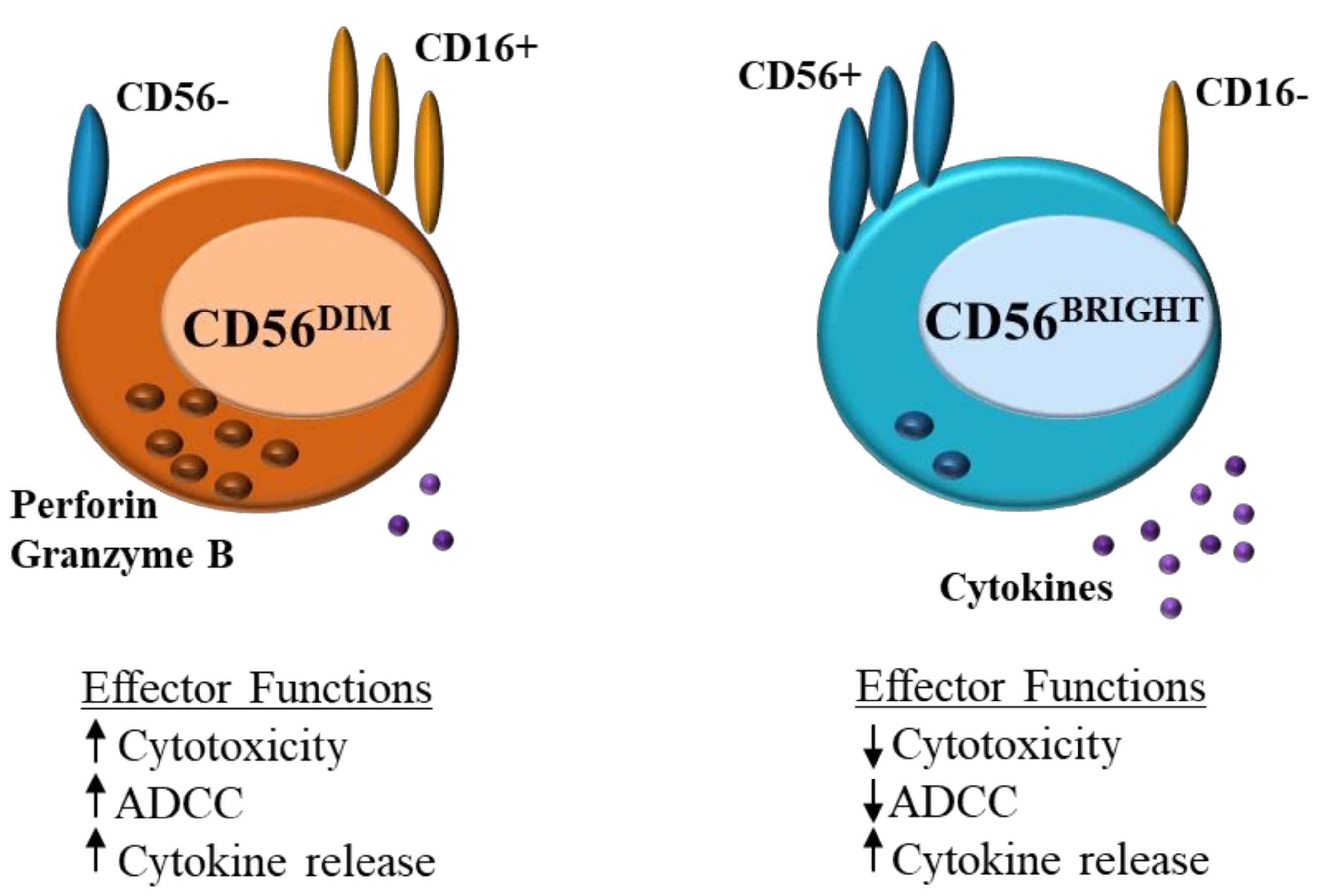
1. Natural Killer Cell Role in Immune Regulation
2. Unbalanced Immune Responses in Autoimmune and Systemic Conditions
2.1. Age and Sex Related Immunoscenescence
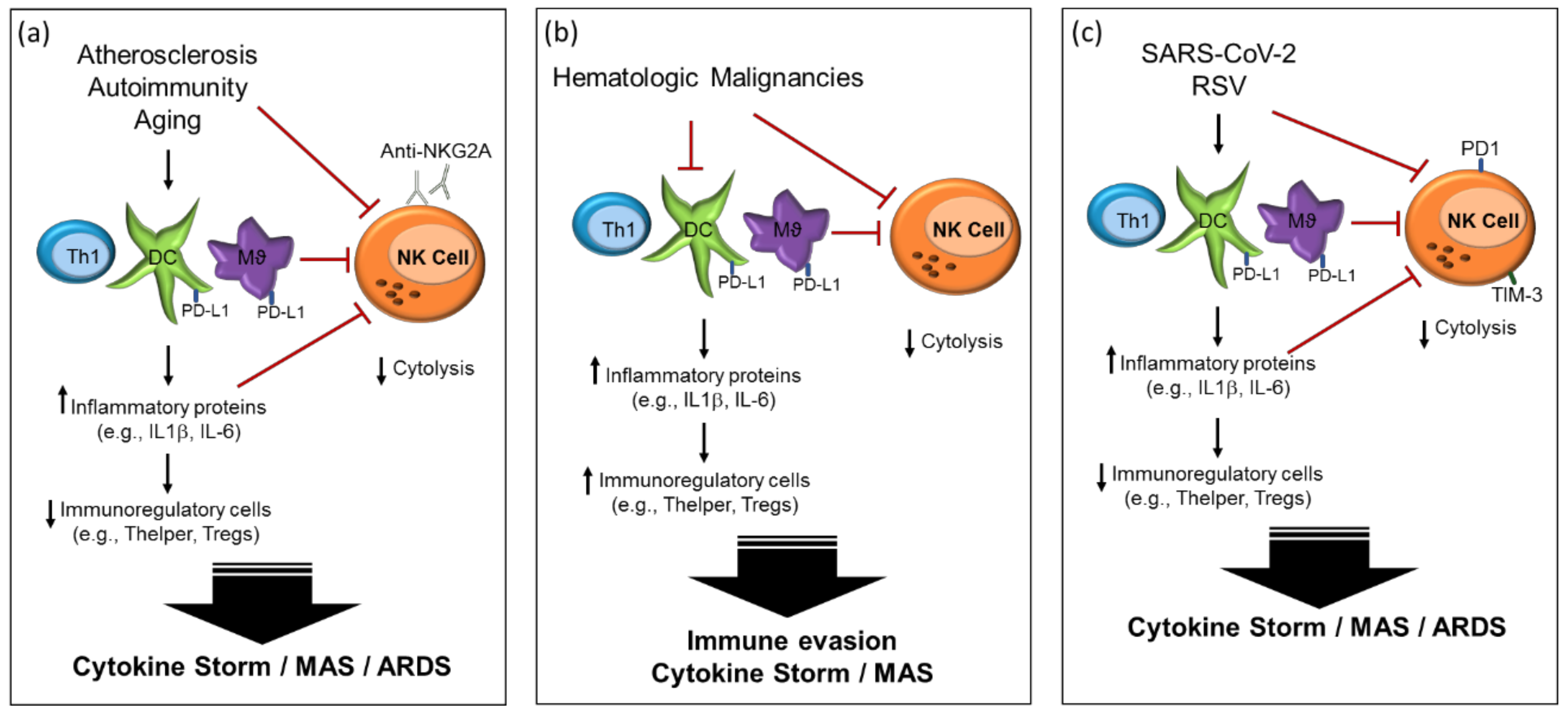
2.2. Atherosclerosis and Cardiovascular Disease
2.3. Autoimmunity
2.4. Hematologic Malignancies
2.5. Hemophagocytic Lymphohistiocytosis
3. Viral Infection Immune Dysregulation
3.1. Pandemic 2009 (H1N1) Influenza A (Pandemic A(H1N1) 2009)
3.2. Severe Acute Respiratory Syndrome (SARS)
3.3. Respiratory Syncytial Virus (RSV)
4. Immunological Features of COVID-19
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ARDS | Acute respiratory distress syndrome |
| HLH | Hemophagocytic lymphohistiocytosis |
| NK | Natural killer |
| MHC | Major histocompatibility complex |
| KIR | Killer cell immunoglobulin receptors |
| IFN | Interferon |
| TLR | Toll-like receptor |
| ADCC | Antibody-dependent cellular cytotoxicity |
| TNF | Tumor necrosis factor |
| MIP | Macrophage inflammatory protein |
| DC | Dendritic cells |
| NCR | Natural cytotoxicity receptors |
| LDL | Low density lipoprotein |
| Th1 | T helper cell 1 |
| SLE | Systemic lupus erythematosus |
| HLA | Human leukocyte antigen |
| IL | Interleukin |
| PD-1 | Programmed cell death protein |
| MAS | Macrophage activation syndrome |
| RSV | Respiratory syncytial virus |
| SARS | Severe acquired respiratory syndrome |
| ACE2 | Angiotensin-converting enzyme 2 |
| ANKL | Aggressive natural killer leukemia |
| JIA | Juvenile idiopathic arthritis |
| MS | Multiple sclerosis |
References
- Schuster, I.S.; Coudert, J.D.; Andoniou, C.E.; Degli-Esposti, M.A. “Natural Regulators”: NK Cells as Modulators of T Cell Immunity. Front. Immunol. 2016, 7, 235. [Google Scholar]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar]
- Karre, K. NK cells, MHC class I molecules and the missing self. Scand. J. Immunol. 2002, 55, 221–228. [Google Scholar]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar]
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar]
- Chan, C.J.; Smyth, M.J.; Martinet, L. Molecular mechanisms of natural killer cell activation in response to cellular stress. Cell Death Differ. 2014, 21, 5–14. [Google Scholar]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar]
- Zhang, X.; Ing, S.; Fraser, A.; Chen, M.; Khan, O.; Zakem, J.; Davis, W.; Quinet, R. Follicular helper T cells: New insights into mechanisms of autoimmune diseases. Ochsner J. 2013, 13, 131–139. [Google Scholar]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar]
- Gubbels Bupp, M.R.; Potluri, T.; Fink, A.L.; Klein, S.L. The Confluence of Sex Hormones and Aging on Immunity. Front. Immunol. 2018, 9, 1269. [Google Scholar]
- Ponnappan, S.; Ponnappan, U. Aging and immune function: Molecular mechanisms to interventions. Antioxid. Redox Signal. 2011, 14, 1551–1585. [Google Scholar]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar]
- Zhang, Y.; Wallace, D.L.; de Lara, C.M.; Ghattas, H.; Asquith, B.; Worth, A.; Griffin, G.E.; Taylor, G.P.; Tough, D.F.; Beverley, P.C.; et al. In vivo kinetics of human natural killer cells: The effects of ageing and acute and chronic viral infection. Immunology 2007, 121, 258–265. [Google Scholar]
- Hazeldine, J.; Lord, J.M. The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Res. Rev. 2013, 12, 1069–1078. [Google Scholar]
- Thoren, F.B.; Riise, R.E.; Ousback, J.; Della Chiesa, M.; Alsterholm, M.; Marcenaro, E.; Pesce, S.; Prato, C.; Cantoni, C.; Bylund, J.; et al. Human NK Cells induce neutrophil apoptosis via an NKp46- and Fas-dependent mechanism. J. Immunol. 2012, 188, 1668–1674. [Google Scholar]
- Hazeldine, J.; Hampson, P.; Lord, J.M. Reduced release and binding of perforin at the immunological synapse underlies the age-related decline in natural killer cell cytotoxicity. Aging Cell 2012, 11, 751–759. [Google Scholar]
- Andrews, D.M.; Estcourt, M.J.; Andoniou, C.E.; Wikstrom, M.E.; Khong, A.; Voigt, V.; Fleming, P.; Tabarias, H.; Hill, G.R.; van der Most, R.G.; et al. Innate immunity defines the capacity of antiviral T cells to limit persistent infection. J. Exp. Med. 2010, 207, 1333–1343. [Google Scholar]
- Yovel, G.; Shakhar, K.; Ben-Eliyahu, S. The effects of sex, menstrual cycle, and oral contraceptives on the number and activity of natural killer cells. Gynecol. Oncol. 2001, 81, 254–262. [Google Scholar]
- Bonaccorsi, I.; Spinelli, D.; Cantoni, C.; Barilla, C.; Pipito, N.; De Pasquale, C.; Oliveri, D.; Cavaliere, R.; Carrega, P.; Benedetto, F.; et al. Symptomatic Carotid Atherosclerotic Plaques Are Associated With Increased Infiltration of Natural Killer (NK) Cells and Higher Serum Levels of NK Activating Receptor Ligands. Front. Immunol. 2019, 10, 1503. [Google Scholar]
- Xia, M.; Guerra, N.; Sukhova, G.K.; Yang, K.; Miller, C.K.; Shi, G.P.; Raulet, D.H.; Xiong, N. Immune activation resulting from NKG2D/ligand interaction promotes atherosclerosis. Circulation 2011, 124, 2933–2943. [Google Scholar]
- Hagberg, N.; Theorell, J.; Hjorton, K.; Spee, P.; Eloranta, M.L.; Bryceson, Y.T.; Ronnblom, L. Functional anti-CD94/NKG2A and anti-CD94/NKG2C autoantibodies in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2015, 67, 1000–1011. [Google Scholar]
- Kastrukoff, L.F.; Lau, A.; Wee, R.; Zecchini, D.; White, R.; Paty, D.W. Clinical relapses of multiple sclerosis are associated with ‘novel’ valleys in natural killer cell functional activity. J. Neuroimmunol. 2003, 145, 103–114. [Google Scholar]
- Rodriguez-Martin, E.; Picon, C.; Costa-Frossard, L.; Alenda, R.; Sainz de la Maza, S.; Roldan, E.; Espino, M.; Villar, L.M.; Alvarez-Cermeno, J.C. Natural killer cell subsets in cerebrospinal fluid of patients with multiple sclerosis. Clin. Exp. Immunol. 2015, 180, 243–249. [Google Scholar]
- Segerberg, F.; Lundtoft, C.; Reid, S.; Hjorton, K.; Leonard, D.; Nordmark, G.; Carlsten, M.; Hagberg, N. Autoantibodies to Killer Cell Immunoglobulin-Like Receptors in Patients With Systemic Lupus Erythematosus Induce Natural Killer Cell Hyporesponsiveness. Front. Immunol. 2019, 10, 2164. [Google Scholar]
- Villanueva, J.; Lee, S.; Giannini, E.H.; Graham, T.B.; Passo, M.H.; Filipovich, A.; Grom, A.A. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res. Ther. 2005, 7, R30–R37. [Google Scholar]
- De Charette, M.; Houot, R. Hide or defend, the two strategies of lymphoma immune evasion: Potential implications for immunotherapy. Haematologica 2018, 103, 1256–1268. [Google Scholar]
- Kwong, Y.L. Natural killer-cell malignancies: Diagnosis and treatment. Leukemia 2005, 19, 2186–2194. [Google Scholar]
- Menter, T.; Tzankov, A. Mechanisms of Immune Evasion and Immune Modulation by Lymphoma Cells. Front. Oncol. 2018, 8, 54. [Google Scholar]
- Rosado, F.G.; Kim, A.S. Hemophagocytic lymphohistiocytosis: An update on diagnosis and pathogenesis. Am. J. Clin. Pathol 2013, 139, 713–727. [Google Scholar]
- Van Erp, E.A.; Feyaerts, D.; Duijst, M.; Mulder, H.L.; Wicht, O.; Luytjes, W.; Ferwerda, G.; van Kasteren, P.B. Respiratory Syncytial Virus Infects Primary Neonatal and Adult Natural Killer Cells and Affects Their Antiviral Effector Function. J. Infect. Dis. 2019, 219, 723–733. [Google Scholar]
- Van Erp, E.A.; Lakerveld, A.J.; de Graaf, E.; Larsen, M.D.; Schepp, R.M.; Hipgrave Ederveen, A.L.; Ahout, I.M.; de Haan, C.A.; Wuhrer, M.; Luytjes, W.; et al. Natural killer cell activation by respiratory syncytial virus-specific antibodies is decreased in infants with severe respiratory infections and correlates with Fc-glycosylation. Clin. Transl. Immunology 2020, 9, e1112. [Google Scholar]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 533–535. [Google Scholar]
- Ilhan, F.; Kalkanli, S.T. Atherosclerosis and the role of immune cells. World J. Clin. Cases 2015, 3, 345–352. [Google Scholar]
- Blankenberg, S.; Rupprecht, H.J.; Bickel, C.; Espinola-Klein, C.; Rippin, G.; Hafner, G.; Ossendorf, M.; Steinhagen, K.; Meyer, J. Cytomegalovirus infection with interleukin-6 response predicts cardiac mortality in patients with coronary artery disease. Circulation 2001, 103, 2915–2921. [Google Scholar]
- Martinez-Rodriguez, J.E.; Munne-Collado, J.; Rasal, R.; Cuadrado, E.; Roig, L.; Ois, A.; Muntasell, A.; Baro, T.; Alameda, F.; Roquer, J.; et al. Expansion of the NKG2C+ natural killer-cell subset is associated with high-risk carotid atherosclerotic plaques in seropositive patients for human cytomegalovirus. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2653–2659. [Google Scholar]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. NK cells in autoimmune diseases: Linking innate and adaptive immune responses. Autoimmun. Rev. 2018, 17, 142–154. [Google Scholar]
- Pan, L.; Lu, M.P.; Wang, J.H.; Xu, M.; Yang, S.R. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J. Pediatr. 2020, 16, 19–30. [Google Scholar]
- Martini, A. Systemic juvenile idiopathic arthritis. Autoimmun. Rev. 2012, 12, 56–59. [Google Scholar]
- Sawhney, S.; Woo, P.; Murray, K.J. Macrophage activation syndrome: A potentially fatal complication of rheumatic disorders. Arch. Dis. Child. 2001, 85, 421–426. [Google Scholar]
- Fraussen, J.; de Bock, L.; Somers, V. B cells and antibodies in progressive multiple sclerosis: Contribution to neurodegeneration and progression. Autoimmun. Rev. 2016, 15, 896–899. [Google Scholar]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Runzi, A.; Kuhlmann, T.; Posevitz-Fejfar, A.; Schwab, N.; Schneider-Hohendorf, T.; Herich, S.; Held, K.; Konjević, M. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2973–E2982. [Google Scholar]
- Upadhyay, R.; Hammerich, L.; Peng, P.; Brown, B.; Merad, M.; Brody, J.D. Lymphoma: Immune evasion strategies. Cancers (Basel) 2015, 7, 736–762. [Google Scholar]
- Steidl, C.; Shah, S.P.; Woolcock, B.W.; Rui, L.; Kawahara, M.; Farinha, P.; Johnson, N.A.; Zhao, Y.; Telenius, A.; Neriah, S.B.; et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377–381. [Google Scholar]
- Roemer, M.G.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced beta2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar]
- Chambers, A.M.; Wang, J.; Lupo, K.B.; Yu, H.; Atallah Lanman, N.M.; Matosevic, S. Adenosinergic Signaling Alters Natural Killer Cell Functional Responses. Front. Immunol. 2018, 9, 2533. [Google Scholar]
- Ishida, F. Aggressive NK-Cell Leukemia. Front. Pediatr. 2018, 6, 292. [Google Scholar]
- Mace, E.M.; Orange, J.S. Genetic Causes of Human NK Cell Deficiency and Their Effect on NK Cell Subsets. Front. Immunol. 2016, 7, 545. [Google Scholar]
- Mace, E.M.; Orange, J.S. Emerging insights into human health and NK cell biology from the study of NK cell deficiencies. Immunol. Rev. 2019, 287, 202–225. [Google Scholar]
- Vandenhaute, J.; Wouters, C.H.; Matthys, P. Natural Killer Cells in Systemic Autoinflammatory Diseases: A Focus on Systemic Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome. Front. Immunol. 2019, 10, 3089. [Google Scholar]
- Crayne, C.B.; Albeituni, S.; Nichols, K.E.; Cron, R.Q. The Immunology of Macrophage Activation Syndrome. Front. Immunol. 2019, 10, 119. [Google Scholar]
- Terrell, C.E.; Jordan, M.B. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+) T cells and dendritic cells. Blood 2013, 121, 5184–5191. [Google Scholar]
- Gholam, C.; Grigoriadou, S.; Gilmour, K.C.; Gaspar, H.B. Familial haemophagocytic lymphohistiocytosis: Advances in the genetic basis, diagnosis and management. Clin. Exp. Immunol. 2011, 163, 271–283. [Google Scholar]
- Carter, S.J.; Tattersall, R.S.; Ramanan, A.V. Macrophage activation syndrome in adults: Recent advances in pathophysiology, diagnosis and treatment. Rheumatology (Oxford) 2019, 58, 5–17. [Google Scholar]
- Sandler, R.D.; Carter, S.; Kaur, H.; Francis, S.; Tattersall, R.S.; Snowden, J.A. Haemophagocytic lymphohistiocytosis (HLH) following allogeneic haematopoietic stem cell transplantation (HSCT)-time to reappraise with modern diagnostic and treatment strategies? Bone Marrow Transplant. 2020, 55, 307–316. [Google Scholar]
- Schulert, G.S.; Zhang, M.; Fall, N.; Husami, A.; Kissell, D.; Hanosh, A.; Zhang, K.; Davis, K.; Jentzen, J.M.; Napolitano, L.; et al. Whole-Exome Sequencing Reveals Mutations in Genes Linked to Hemophagocytic Lymphohistiocytosis and Macrophage Activation Syndrome in Fatal Cases of H1N1 Influenza. J. Infect. Dis. 2016, 213, 1180–1188. [Google Scholar]
- Vastert, S.J.; van Wijk, R.; D’Urbano, L.E.; de Vooght, K.M.; de Jager, W.; Ravelli, A.; Magni-Manzoni, S.; Insalaco, A.; Cortis, E.; van Solinge, W.W.; et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford) 2010, 49, 441–449. [Google Scholar]
- Hot, A.; Toh, M.L.; Coppere, B.; Perard, L.; Madoux, M.H.; Mausservey, C.; Desmurs-Clavel, H.; Ffrench, M.; Ninet, J. Reactive hemophagocytic syndrome in adult-onset Still disease: Clinical features and long-term outcome: A case-control study of 8 patients. Medicine (Baltimore) 2010, 89, 37–46. [Google Scholar]
- Dawood, F.S.; Iuliano, A.D.; Reed, C.; Meltzer, M.I.; Shay, D.K.; Cheng, P.Y.; Bandaranayake, D.; Breiman, R.F.; Brooks, W.A.; Buchy, P.; et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: A modelling study. Lancet. Infect. Dis. 2012, 12, 687–695. [Google Scholar]
- Witczak, A.; Prystupa, A.; Kurys-Denis, E.; Borys, M.; Czuczwar, M.; Niemcewicz, M.; Kocik, J.; Michalak, A.; Pietrzak, A.; Chodorowska, G.; et al. Acute respiratory distress syndrome (ARDS) complicating influenza A/H1N1v infection—A clinical approach. Ann. Agric. Environ. Med. 2013, 20, 820–822. [Google Scholar]
- To, K.K.; Hung, I.F.; Li, I.W.; Lee, K.L.; Koo, C.K.; Yan, W.W.; Liu, R.; Ho, K.Y.; Chu, K.H.; Watt, C.L.; et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin. Infect. Dis. 2010, 50, 850–859. [Google Scholar]
- Guo, H.; Kumar, P.; Malarkannan, S. Evasion of natural killer cells by influenza virus. J. Leukoc. Biol. 2011, 89, 189–194. [Google Scholar]
- Achdout, H.; Manaster, I.; Mandelboim, O. Influenza virus infection augments NK cell inhibition through reorganization of major histocompatibility complex class I proteins. J. Virol. 2008, 82, 8030–8037. [Google Scholar]
- Giamarellos-Bourboulis, E.J.; Raftogiannis, M.; Antonopoulou, A.; Baziaka, F.; Koutoukas, P.; Savva, A.; Kanni, T.; Georgitsi, M.; Pistiki, A.; Tsaganos, T.; et al. Effect of the novel influenza A (H1N1) virus in the human immune system. PLoS ONE 2009, 4, e8393. [Google Scholar]
- Huang, Y.; Zhu, W.; Zeng, X.; Li, S.; Li, X.; Lu, C. Innate and adaptive immune responses in patients with pandemic influenza A(H1N1)pdm09. Arch. Virol. 2013, 158, 2267–2272. [Google Scholar]
- Ozdemir, H.; Ciftci, E.; Ince, E.U.; Ertem, M.; Ince, E.; Dogru, U. Hemophagocytic lymphohistiocytosis associated with 2009 pandemic influenza A (H1N1) virus infection. J. Pediatr. Hematol. Oncol. 2011, 33, 135–137. [Google Scholar]
- Gu, J.; Korteweg, C. Pathology and pathogenesis of severe acute respiratory syndrome. Am. J. Pathol. 2007, 170, 1136–1147. [Google Scholar]
- Gu, J.; Taylor, C.R. Acute immunodeficiency, multiple organ injury, and the pathogenesis of SARS. Appl. Immunohistochem. Mol. Morphol. 2003, 11, 281–282. [Google Scholar]
- Nagata, N.; Iwata-Yoshikawa, N.; Taguchi, F. Studies of severe acute respiratory syndrome coronavirus pathology in human cases and animal models. Vet. Pathol. 2010, 47, 881–892. [Google Scholar]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar]
- National Research Project for SARS, Beijing Group. The involvement of natural killer cells in the pathogenesis of severe acute respiratory syndrome. Am. J. Clin. Pathol. 2004, 121, 507–511. [Google Scholar]
- Zhan, J.; Deng, R.; Tang, J.; Zhang, B.; Tang, Y.; Wang, J.K.; Li, F.; Anderson, V.M.; McNutt, M.A.; Gu, J. The spleen as a target in severe acute respiratory syndrome. FASEB J. 2006, 20, 2321–2328. [Google Scholar]
- Hsueh, P.R.; Chen, P.J.; Hsiao, C.H.; Yeh, S.H.; Cheng, W.C.; Wang, J.L.; Chiang, B.L.; Chang, S.C.; Chang, F.Y.; Wong, W.W.; et al. Patient data, early SARS epidemic, Taiwan. Emerg. Infect. Dis. 2004, 10, 489–493. [Google Scholar]
- Wong, C.K.; Lam, C.W.; Wu, A.K.; Ip, W.K.; Lee, N.L.; Chan, I.H.; Lit, L.C.; Hui, D.S.; Chan, M.H.; Chung, S.S.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar]
- Bhat, R.; Farrag, M.A.; Almajhdi, F.N. Double-edged role of natural killer cells during RSV infection. Int. Rev. Immunol. 2020, 1–12. [Google Scholar] [CrossRef]
- Taleb, S.A.; Al Thani, A.A.; Al Ansari, K.; Yassine, H.M. Human respiratory syncytial virus: Pathogenesis, immune responses, and current vaccine approaches. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1817–1827. [Google Scholar]
- Drajac, C.; Laubreton, D.; Riffault, S.; Descamps, D. Pulmonary Susceptibility of Neonates to Respiratory Syncytial Virus Infection: A Problem of Innate Immunity? J. Immunol. Res. 2017, 2017, 8734504. [Google Scholar]
- Malinczak, C.A.; Lukacs, N.W.; Fonseca, W. Early-Life Respiratory Syncytial Virus Infection, Trained Immunity and Subsequent Pulmonary Diseases. Viruses 2020, 12, 505. [Google Scholar]
- Swedan, S.; Andrews, J.; Majumdar, T.; Musiyenko, A.; Barik, S. Multiple functional domains and complexes of the two nonstructural proteins of human respiratory syncytial virus contribute to interferon suppression and cellular location. J. Virol. 2011, 85, 10090–100100. [Google Scholar]
- World Health Organization. Coronavirus disease (COVID-19) Pandemic. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 24 August 2020).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). medRxiv 2020, 11, 827. [Google Scholar]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet. Respir. Med. 2020, 8, 420–422. [Google Scholar]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 1–11. [Google Scholar]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar]
- Uri, K.; Fagyas, M.; Kertesz, A.; Borbely, A.; Jenei, C.; Bene, O.; Csanadi, Z.; Paulus, W.J.; Edes, I.; Papp, Z.; et al. Circulating ACE2 activity correlates with cardiovascular disease development. J. Renin Angiotensin Aldosterone Syst. 2016, 17. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Penninger, J.M. Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 2006, 6, 271–276. [Google Scholar]
- Li, D.; Chen, Y.; Liu, H.; Jia, Y.; Li, F.; Wang, W.; Wu, J.; Wan, Z.; Cao, Y.; Zeng, R. Immune dysfunction leads to mortality and organ injury in patients with COVID-19 in China: Insights from ERS-COVID-19 study. Signal. Transduct. Target. Ther. 2020, 5, 62. [Google Scholar]
- Wang, F.; Nie, J.; Wang, H.; Zhao, Q.; Xiong, Y.; Deng, L.; Song, S.; Ma, Z.; Mo, P.; Zhang, Y. Characteristics of Peripheral Lymphocyte Subset Alteration in COVID-19 Pneumonia. J. Infect. Dis. 2020, 221, 1762–1769. [Google Scholar]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar]
- Ndhlovu, L.C.; Lopez-Verges, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martinez-Colon, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Rogers, A.J.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar]
- Crinier, A.; Milpied, P.; Escaliere, B.; Piperoglou, C.; Galluso, J.; Balsamo, A.; Spinelli, L.; Cervera-Marzal, I.; Ebbo, M.; Girard-Madoux, M.; et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 2018, 49, 971–986. [Google Scholar]
- Pedersen, S.F.; Ho, Y. SARS-CoV-2: A storm is raging. J. Clin. Investig. 2020, 130, 2202–2205. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G.; Toniato, E. IL-1 induces throboxane-A2 (TxA2) in COVID-19 causing inflammation and micro-thrombi: Inhibitory effect of the IL-1 receptor antagonist (IL-1Ra). J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.J.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.J.; et al. Platelet Gene Expression and Function in COVID-19 Patients. Blood 2020. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodriguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020. [Google Scholar] [CrossRef]
- Wang, R.; Jaw, J.J.; Stutzman, N.C.; Zou, Z.; Sun, P.D. Natural killer cell-produced IFN-gamma and TNF-alpha induce target cell cytolysis through up-regulation of ICAM-1. J. Leukoc. Biol. 2012, 91, 299–309. [Google Scholar]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar]
- Cifaldi, L.; Prencipe, G.; Caiello, I.; Bracaglia, C.; Locatelli, F.; De Benedetti, F.; Strippoli, R. Inhibition of natural killer cell cytotoxicity by interleukin-6: Implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol. 2015, 67, 3037–3046. [Google Scholar]
- Lassen, M.G.; Lukens, J.R.; Dolina, J.S.; Brown, M.G.; Hahn, Y.S. Intrahepatic IL-10 maintains NKG2A+Ly49- liver NK cells in a functionally hyporesponsive state. J. Immunol. 2010, 184, 2693–26701. [Google Scholar]
- Szkaradkiewicz, A.; Karpinski, T.M.; Drews, M.; Borejsza-Wysocki, M.; Majewski, P.; Andrzejewska, E. Natural killer cell cytotoxicity and immunosuppressive cytokines (IL-10, TGF-beta1) in patients with gastric cancer. J. Biomed. Biotechnol. 2010, 2010, 901564. [Google Scholar]
- Osman, M.S.; van Eeden, C.; Tervaert, J.W.C. Fatal COVID-19 infections: Is NK cell dysfunction a link with autoimmune HLH? Autoimmun. Rev. 2020, 19, 102561. [Google Scholar] [CrossRef]
- Leng, Z.; Zhu, R.; Hou, W.; Feng, Y.; Yang, Y.; Han, Q.; Shan, G.; Meng, F.; Du, D.; Wang, S.; et al. Transplantation of ACE2(-) Mesenchymal Stem Cells Improves the Outcome of Patients with COVID-19 Pneumonia. Aging Dis. 2020, 11, 216–228. [Google Scholar]
- Mazzoni, A.; Salvati, L.; Maggi, L.; Capone, M.; Vanni, A.; Spinicci, M.; Mencarini, J.; Caporale, R.; Peruzzi, B.; Antonelli, A.; et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J. Clin. Investig. 2020, 130. [Google Scholar] [CrossRef]
- Bar-On, Y.; Seidel, E.; Tsukerman, P.; Mandelboim, M.; Mandelboim, O. Influenza virus uses its neuraminidase protein to evade the recognition of two activating NK cell receptors. J. Infect. Dis. 2014, 210, 410–418. [Google Scholar]
- Draghi, M.; Pashine, A.; Sanjanwala, B.; Gendzekhadze, K.; Cantoni, C.; Cosman, D.; Moretta, A.; Valiante, N.M.; Parham, P. NKp46 and NKG2D recognition of infected dendritic cells is necessary for NK cell activation in the human response to influenza infection. J. Immunol. 2007, 178, 2688–2698. [Google Scholar]
- Li, F.; Zhu, H.; Sun, R.; Wei, H.; Tian, Z. Natural killer cells are involved in acute lung immune injury caused by respiratory syncytial virus infection. J. Virol. 2012, 86, 2251–2258. [Google Scholar]
- Liu, H.; Osterburg, A.R.; Flury, J.; Huang, S.; McCormack, F.X.; Cormier, S.A.; Borchers, M.T. NKG2D Regulation of Lung Pathology and Dendritic Cell Function Following Respiratory Syncytial Virus Infection. J. Infect. Dis. 2018, 218, 1822–1832. [Google Scholar]
- Shemer-Avni, Y.; Kundu, K.; Shemesh, A.; Brusilovsky, M.; Yossef, R.; Meshesha, M.; Solomon-Alemayehu, S.; Levin, S.; Gershoni-Yahalom, O.; Campbell, K.S.; et al. Expression of NKp46 Splice Variants in Nasal Lavage Following Respiratory Viral Infection: Domain 1-Negative Isoforms Predominate and Manifest Higher Activity. Front. Immunol. 2017, 8, 161. [Google Scholar]
- Zdrenghea, M.T.; Telcian, A.G.; Laza-Stanca, V.; Bellettato, C.M.; Edwards, M.R.; Nikonova, A.; Khaitov, M.R.; Azimi, N.; Groh, V.; Mallia, P.; et al. RSV infection modulates IL-15 production and MICA levels in respiratory epithelial cells. Eur. Respir. J. 2012, 39, 712–720. [Google Scholar]
- Muntasell, A.; Magri, G.; Pende, D.; Angulo, A.; Lopez-Botet, M. Inhibition of NKG2D expression in NK cells by cytokines secreted in response to human cytomegalovirus infection. Blood 2010, 115, 5170–5179. [Google Scholar]
- Vietzen, H.; Zoufaly, A.; Traugott, M.; Aberle, J.; Aberle, S.; Puchhammer-Stockl, E. NK cell receptor NKG2C deletion and HA-E variants are risk factors for severe COVID-19. Res. Square 2020. [Google Scholar] [CrossRef]
- NIH. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 13 April 2020).
- CHICTR Chinese Clinical Trial Registry. Available online: http://www.chictr.org.cn/ (accessed on 13 April 2020).
- Hunter, C.A.; Timans, J.; Pisacane, P.; Menon, S.; Cai, G.; Walker, W.; Aste-Amezaga, M.; Chizzonite, R.; Bazan, J.F.; Kastelein, R.A. Comparison of the effects of interleukin-1 alpha, interleukin-1 beta and interferon-gamma-inducing factor on the production of interferon-gamma by natural killer. Eur. J. Immunol. 1997, 27, 2787–2792. [Google Scholar]
- Celularity, I. Celularity Announces FDA Clearance of IND Application for CYNK-001 in Coronavirus, First in Cellular Therapy. Available online: https://www.prnewswire.com/news-releases/celularity-announces-fda-clearance-of-ind-application-for-cynk-001-in-coronavirus-first-in-cellular-therapy-301034141.html (accessed on 13 April 2020).
- NIH. A Phase I/II Study of Universal Off-the-shelf NKG2D-ACE2 CAR-NK Cells for Therapy of COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04324996 (accessed on 13 April 2020).
- Schrezenmeier, E.; Dorner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar]
- Chen, Z.; Hu, J.; Zhang, Z.; Jiang, S.; Han, S.; Yan, D.; Zhuang, R.; Hu, B.; Zhang, Z. Efficacy of hydroxychloroquine in patients with COVID-19: Results of a randomized clinical trial. medRxiv 2020. [Google Scholar] [CrossRef]
- Shoenfeld, Y. Corona (COVID-19) time musings: Our involvement in COVID-19 pathogenesis, diagnosis, treatment and vaccine planning. Autoimmun. Rev. 2020, 19, 102538. [Google Scholar]
- Van den Borne, B.E.; Dijkmans, B.A.; de Rooij, H.H.; le Cessie, S.; Verweij, C.L. Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J. Rheumatol. 1997, 24, 55–60. [Google Scholar]
- Tartari, A.P.S.; Moreira, F.F.; Pereira, M.; Carraro, E.; Cidral-Filho, F.J.; Salgado, A.I.; Kerppers, I.I. Anti-inflammatory Effect of Ozone Therapy in an Experimental Model of Rheumatoid Arthritis. Inflammation 2020, 43, 985–993. [Google Scholar]
- Marini, S.; Maggiorotti, M.; Dardes, N.; Bonetti, M.; Martinelli, M.; Re, L.; Carinci, F.; Tavera, C. Italian Oxygen-Ozone Federation. Oxygen-ozone therapy as adjuvant in the current emergency in SARS-COV-2 infection: A clinical study. J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar] [CrossRef]
- Market, M.; Angka, L.; Martel, A.B.; Bastin, D.; Olanubi, O.; Tennakoon, G.; Boucher, D.M.; Ng, J.; Ardolino, M.; Auer, R.C. Flattening the COVID-19 Curve With Natural Killer Cell Based Immunotherapies. Front. Immunol. 2020, 11, 1512. [Google Scholar]
- O’Neill, L.A.J.; Netea, M.G. BCG-induced trained immunity: Can it offer protection against COVID-19? Nat. Rev. Immunol. 2020, 20, 335–337. [Google Scholar]
- Kandere-Grzybowska, K.; Kempuraj, D.; Cao, J.; Cetrulo, C.L.; Theoharides, T.C. Regulation of IL-1-induced selective IL-6 release from human mast cells and inhibition by quercetin. Br. J. Pharmacol. 2006, 148, 208–215. [Google Scholar]
- Theoharides, T.C.; Conti, P. Dexamethasone for COVID-19? Not so fast. J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar] [CrossRef]
- Cavalli, G.; Koenders, M.; Kalabokis, V.; Kim, J.; Tan, A.C.; Garlanda, C.; Mantovani, A.; Dagna, L.; Joosten, L.A.; Dinarello, C.A. Treating experimental arthritis with the innate immune inhibitor interleukin-37 reduces joint and systemic inflammation. Rheumatology (Oxford) 2016, 55, 2220–2229. [Google Scholar]
- Qi, F.; Liu, M.; Li, F.; Lv, Q.; Wang, G.; Gong, S.; Wang, S.; Xu, Y.; Bao, L.; Qin, C. Interleukin-37 Ameliorates Influenza Pneumonia by Attenuating Macrophage Cytokine Production in a MAPK-Dependent Manner. Front. Microbiol. 2019, 10, 2482. [Google Scholar]
- Ronconi, G.; Tete, G.; Kritas, S.K.; Gallenga, C.E.; Caraffa, A.; Ross, R.; Conti, P. SARS-CoV-2, which induces COVID-19, causes kawasaki-like disease in children: Role of pro-inflammatory and anti-inflammatory cytokines. J. Biol. Regul. Homeost. Agents 2020, 34, 767–773. [Google Scholar]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 327–331. [Google Scholar]
- Jung, Y.S.; Park, J.H.; Park, D.I.; Sohn, C.I.; Lee, J.M.; Kim, T.I. Physical Inactivity and Unhealthy Metabolic Status Are Associated with Decreased Natural Killer Cell Activity. Yonsei Med. J. 2018, 59, 554–562. [Google Scholar]



{kind=link}
{kind=link}
| Condition | Findings | Reference |
|---|---|---|
| Ageing | NK cell production and proliferation is reduced. | [12,14,15] |
| Higher frequency of mature NK cells. | ||
| NK cell cytotoxicity is reduced and pro-inflammatory cytokines increased. | ||
| Tissue damage and necrosis are increased due to reduced expression of NKp46+ resulting in decreased neutrophil apoptosis by NK cells and enhancement of neutrophil necrosis. | ||
| Atherosclerosis | Increased frequency of CD56BRIGHT NK cells leads to tissue damage and inflammation due to an increased cytokine production. | [20,21] |
| Increased expression of MICA/B, results in elevated activation of natural cytotoxicity receptor NKG2D, increasing NK cell activity. | ||
| Autoimmunity | SLE; Reduced number of cytotoxic NK cells. Anti-NKG2A antibodies result is dysregulated self-nonself-recognition. | [22,23,24,25,26] |
| JIA; Reduction in both cytolytic and inflammatory NK cell activity. Patients can develop MAS | ||
| MS; Reduction in CD56DIM NK cells results in decreased cytolytic activity and impaired regulation of CD4* T cells, resulting in increased inflammatory responses. | ||
| Hematologic | Reduced CD58 expression leads to reduced NK cell activation. | [27,28,29,30] |
| Malignancy | Increased CD47 and IDO expression suppress NK cells activity. | |
| Increased CXCR1 expression leads to increased CD56DIM NK cell activity resulting in necrosis, apoptosis and organ failure. | ||
| RSV | Decreased expression of NKG2D and NKp44 reducing cytolytic activity. | [31,32] |
| Increased IFN-γ expression inducing pro-inflammatory response. | ||
| COVID-19 | NK cells display exhausted phenotype and are reduced in number. | [33,34,35] |
| Reduced cytotoxicity through increased NKG2A expression. | ||
| Increased production of inflammatory cytokines and chemokines. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Eeden, C.; Khan, L.; Osman, M.S.; Cohen Tervaert, J.W. Natural Killer Cell Dysfunction and Its Role in COVID-19. Int. J. Mol. Sci. 2020, 21, 6351. https://doi.org/10.3390/ijms21176351
van Eeden C, Khan L, Osman MS, Cohen Tervaert JW. Natural Killer Cell Dysfunction and Its Role in COVID-19. International Journal of Molecular Sciences. 2020; 21(17):6351. https://doi.org/10.3390/ijms21176351
Chicago/Turabian Stylevan Eeden, Charmaine, Lamia Khan, Mohammed S. Osman, and Jan Willem Cohen Tervaert. 2020. "Natural Killer Cell Dysfunction and Its Role in COVID-19" International Journal of Molecular Sciences 21, no. 17: 6351. https://doi.org/10.3390/ijms21176351
APA Stylevan Eeden, C., Khan, L., Osman, M. S., & Cohen Tervaert, J. W. (2020). Natural Killer Cell Dysfunction and Its Role in COVID-19. International Journal of Molecular Sciences, 21(17), 6351. https://doi.org/10.3390/ijms21176351