Established and Emerging Mechanisms in the Pathogenesis of Arrhythmogenic Cardiomyopathy: A Multifaceted Disease

Abstract
1. Introduction
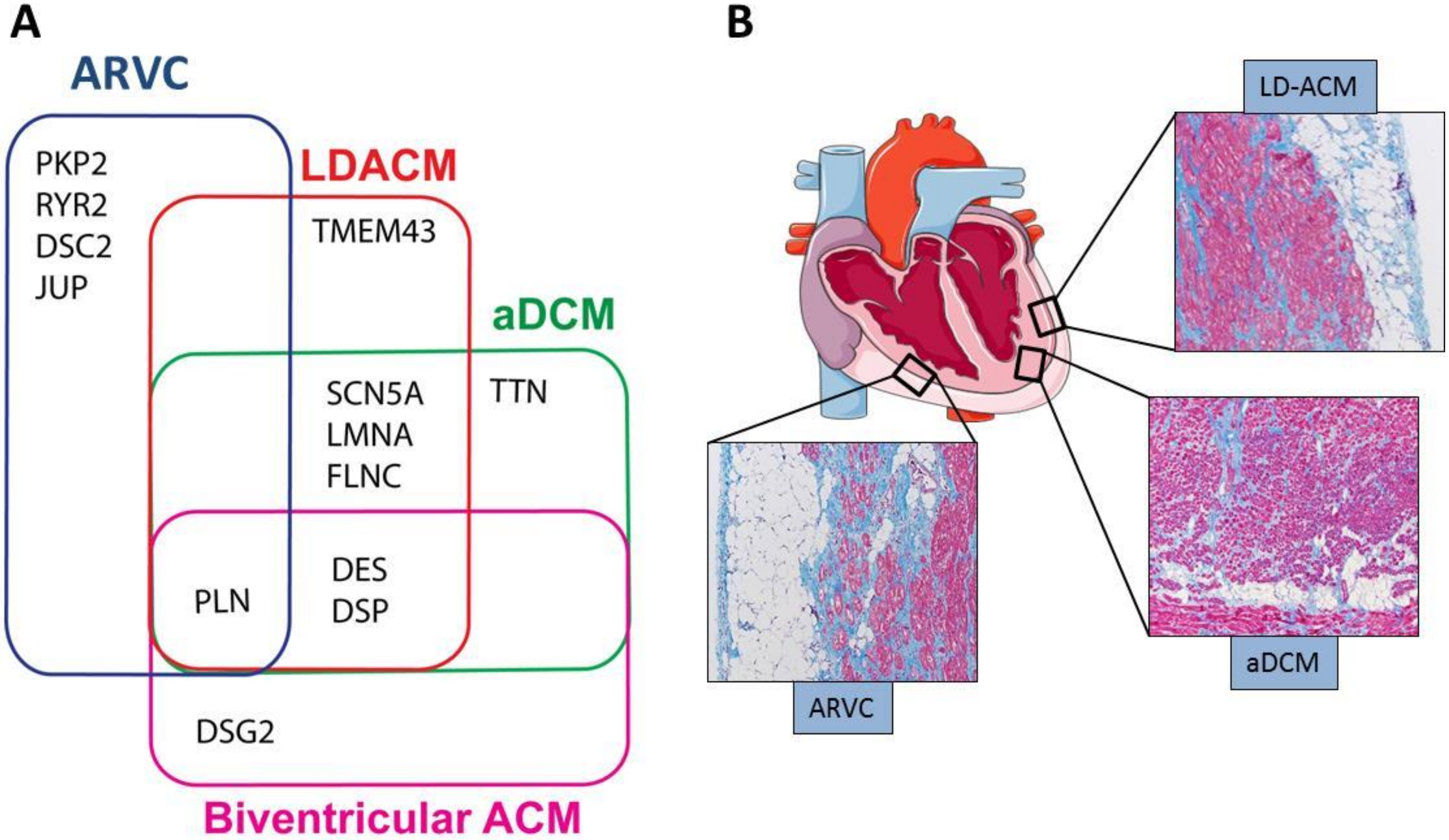
2. Genetics, Clinical and Histological Hallmarks
2.1. Genetics
2.2. Clinical Phenotypic Expressions of ACM
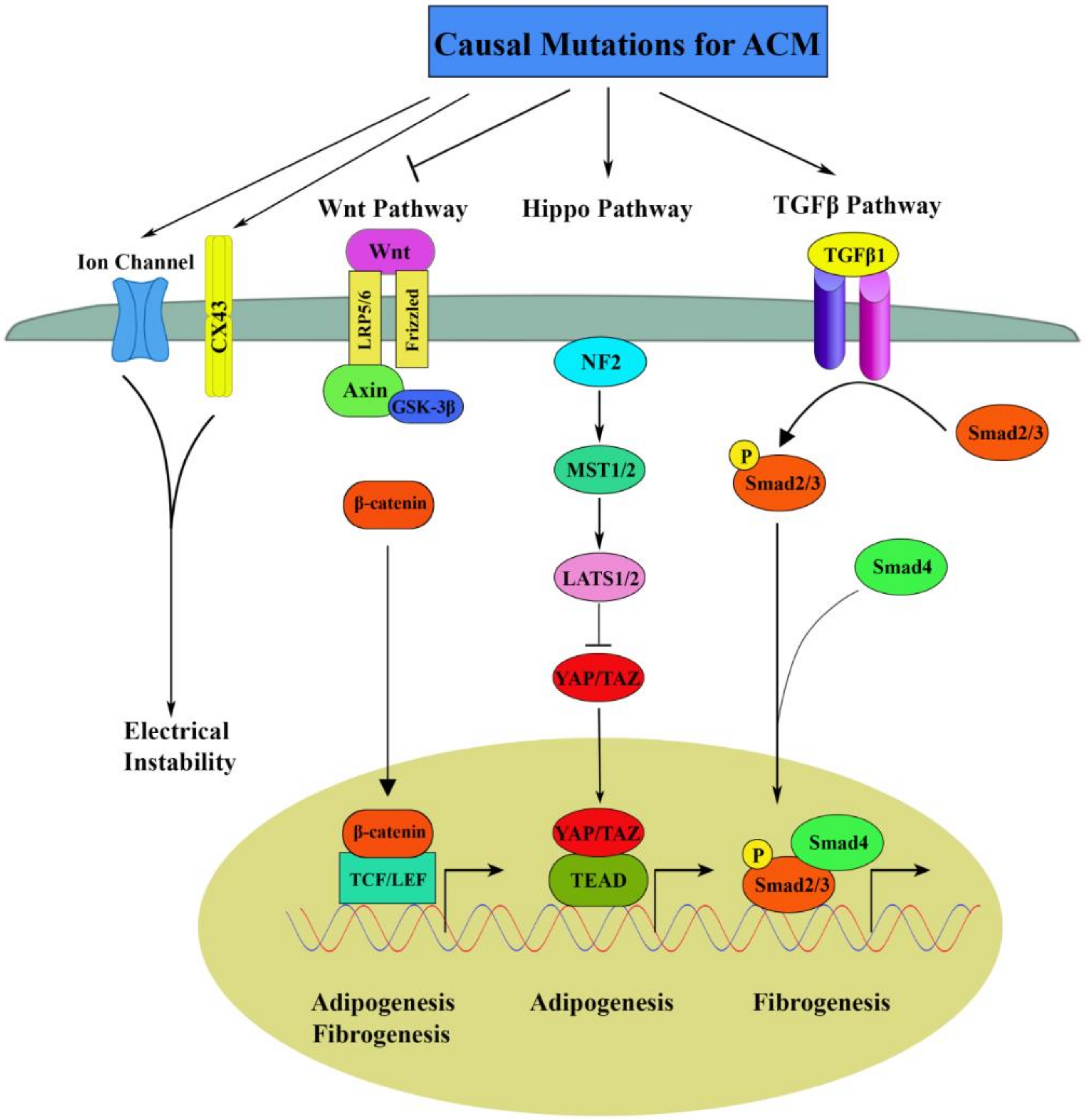
3. Molecular Pathogenic Pathways
3.1. Canonical Wnt Signaling in ACM
3.2. Hippo Signaling in ACM
3.3. TGFβ Signaling in ACM
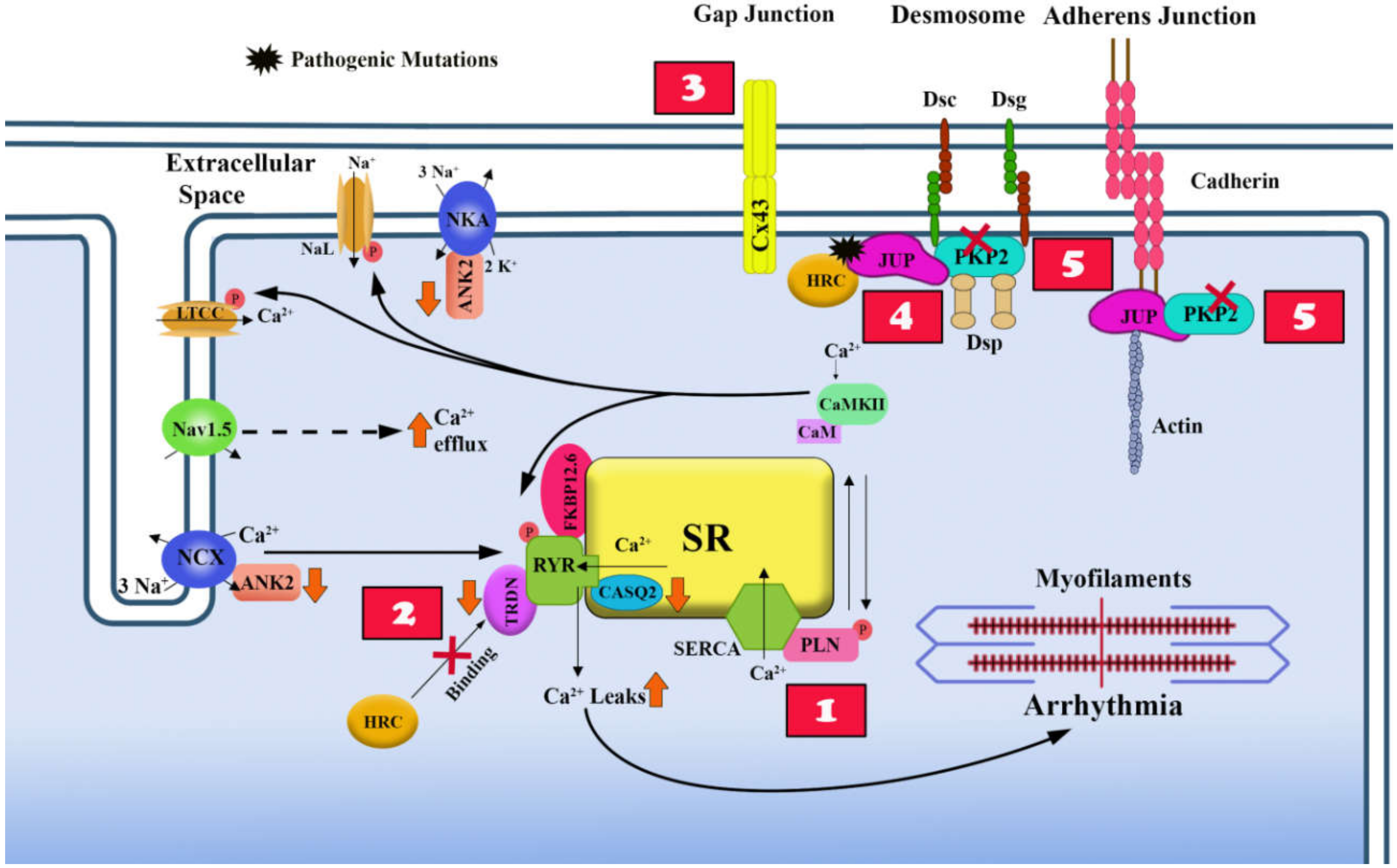
4. Electrical Instability in ACM
4.1. Overview of Excitation-Contraction Coupling in Heart and its Dysfunction in ACM
4.2. Effects of Desmosome Mutations on the Electrical Properties of the Cell Membrane
4.3. Ca2+ Homeostasis and Non-Desmosomal ACM Causal Genes
4.4. Ca2+ Homeostasis and Desmosomal ACM Causal Genes
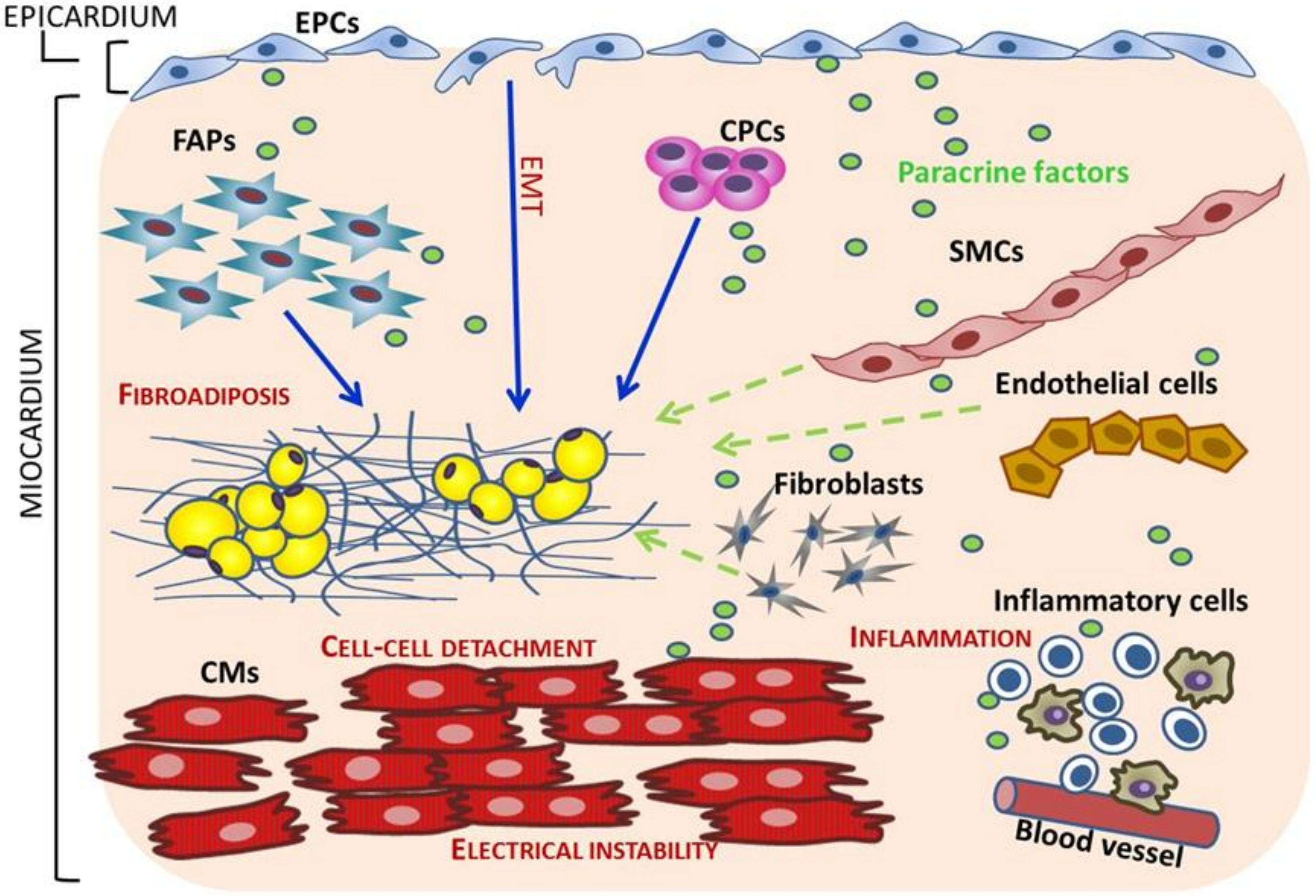
5. Cell Sources of Fibro-Adipocytes in ACM
5.1. Trans-Differentiation of Adult Cardiomyocytes
5.2. Cardiac Progenitor Cells (CPCs) switch from Myogenesis to Adipogenesis
5.3. Mesenchymal Cells and Cardiac Fibro-Adipocyte Progenitors Differentiation to Adipocytes
5.4. Epicardial Progenitor Cells (EPCs)
6. Novel Mechanisms in the Pathogenesis of ACM
6.1. Inflammation and Autoimmunity in ACM
6.2. Paracrine Factors as Regulators of Fibroadiposis in ACM
6.3. Effects of ACM Causal Mutations on Different Cell Types
6.4. Cell Types Cross-Talk in ACM
7. Novel Therapeutic Options in ACM
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lombardi, R.; Marian, A.J. Arrhythmogenic right ventricular cardiomyopathy is a disease of cardiac stem cells. Curr. Opin. Cardiol. 2010, 25, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Bauce, B.; Corrado, D.; Thiene, G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2011, 9, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.P.J.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Den Haan, A.D.; Tan, B.Y.; Zikusoka, M.N.; Ibañez Lladó, L.; Jain, R.; Daly, A.; Tichnell, C.; James, C.; Amat-Alarcon, N.; Abraham, T.; et al. Comprehensive desmosome mutation analysis in north americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Fressart, V.; Duthoit, G.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Dubourg, O.; Delacretaz, E.; Cosnay, P.; et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Spectrum of mutations and clinical impact in practice. Europace 2010, 12, 861–888. [Google Scholar] [CrossRef]
- Xu, Z.; Zhu, W.; Wang, C.; Huang, L.; Zhou, Q.; Hu, J.; Cheng, X.; Hong, K. Genotype-phenotype relationship in patients with arrhythmogenic right ventricular cardiomyopathy caused by desmosomal gene mutations: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 41387. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef]
- Rübsam, M.; Broussard, J.A.; Wickström, S.A.; Nekrasova, O.; Green, K.J.; Niessen, C.M. Adherens Junctions and Desmosomes Coordinate Mechanics and Signaling to Orchestrate Tissue Morphogenesis and Function: An Evolutionary Perspective. Cold Spring Harb. Perspect. Biol. 2018, 10, a029207. [Google Scholar] [CrossRef]
- Sheikh, F.; Ross, R.S.; Chen, J. Cell-cell connection to cardiac disease. Trends Cardiovasc. Med. 2009, 19, 182–190. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm. 2019, 16, e373–e407. [Google Scholar] [CrossRef] [PubMed]
- Bauce, B.; Rampazzo, A.; Basso, C.; Mazzotti, E.; Rigato, I.; Steriotis, A.; Beffagna, G.; Lorenzon, A.; De Bortoli, M.; Pilichou, K.; et al. Clinical phenotype and diagnosis of arrhythmogenic right ventricular cardiomyopathy in pediatric patients carrying desmosomal gene mutations. Heart Rhythm. 2011, 8, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Zorzi, A.; Vanoli, E.; Gronda, E. Current challenges in sudden cardiac death prevention. Heart Fail Rev. 2020, 25, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Goff, Z.D.; Calkins, H. Sudden death related cardiomyopathies—Arrhythmogenic right ventricular cardiomyopathy, arrhythmogenic cardiomyopathy, and exercise-induced cardiomyopathy. Prog. Cardiovasc. Dis. 2019, 62, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Jhund, P.S.; Petrie, M.C.; Claggett, B.L.; Barlera, S.; Cleland, J.G.F.; Dargie, H.J.; Granger, C.B.; Kjekshus, J.; Køber, L.; et al. Declining Risk of Sudden Death in Heart Failure. N. Engl. J. Med. 2017, 377, 41–51. [Google Scholar] [PubMed]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef]
- Lombardi, R.; da Graca Cabreira-Hansen, M.; Bell, A.; Fromm, R.R.; Willerson, J.T.; Marian, A.J. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2011, 109, 1342–1353. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Invest. 2006, 116, 2012–2021. [Google Scholar] [CrossRef]
- Calore, M.; Lorenzon, A.; Vitiello, L.; Poloni, G.; Khan, M.A.F.; Beffagna, G.; Dazzo, E.; Sacchetto, C.; Polishchuk, R.; Sabatelli, P.; et al. A novel murine model for arrhythmogenic cardiomyopathy points to a pathogenic role of Wnt signalling and miRNA dysregulation. Cardiovasc. Res. 2019, 115, 739–751. [Google Scholar] [CrossRef]
- Giuliodori, A.; Beffagna, G.; Marchetto, G.; Fornetto, C.; Vanzi, F.; Toppo, S.; Facchinello, N.; Santimaria, M.; Vettori, A.; Rizzo, S.; et al. Loss of cardiac Wnt/beta-catenin signalling in desmoplakin-deficient AC8 zebrafish models is rescuable by genetic and pharmacological intervention. Cardiovasc. Res. 2018, 114, 1082–1097. [Google Scholar] [CrossRef]
- Li, J.; Swope, D.; Raess, N.; Cheng, L.; Muller, E.J.; Radice, G.L. Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Mol. Cell Biol. 2011, 31, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Padrón-Barthe, L.; Villalba-Orero, M.; Gómez-Salinero, J.M.; Domínguez, F.; Román, M.; Larrasa-Alonso, J.; Ortiz-Sánchez, P.; Martínez, F.; López-Olañeta, M.; Bonzón-Kulichenko, E.; et al. Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3beta. Circulation 2019, 140, 1188–1204. [Google Scholar] [CrossRef] [PubMed]
- Noorman, M.; Hakim, S.; Kessler, E.; Groeneweg, J.A.; Cox, M.G.; Asimaki, A.; van Rijen, H.V.; van Stuijvenberg, L.; Chkourko, H.; van der Heyden, M.A.; et al. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013, 10, 412–4129. [Google Scholar] [CrossRef]
- Akdis, D.; Brunckhorst, C.; Duru, F.; Saguner, A.M. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythm. Electrophysiol. Rev. 2016, 5, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, S.R.; Gard, J.J.; Protonotarios, N.; Tsatsopoulou, A.; Spiliopoulou, C.; Anastasakis, A.; Squarcioni, C.P.; McKenna, W.J.; Thiene, G.; Basso, C.; et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 2004, 1, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Sato, P.Y.; Musa, H.; Coombs, W.; Guerrero-Serna, G.; Patiño, G.A.; Taffet, S.M.; Isom, L.L.; Delmar, M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 2009, 105, 523–526. [Google Scholar] [CrossRef]
- Rizzo, S.; Lodder, E.M.; Verkerk, A.O.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.R. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 2012, 95, 409–418. [Google Scholar] [CrossRef]
- Oxford, E.M.; Musa, H.; Maass, K.; Coombs, W.; Taffet, S.M.; Delmar, M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ. Res. 2007, 101, 703–711. [Google Scholar] [CrossRef]
- Lyon, R.C.; Mezzano, V.; Wright, A.T.; Pfeiffer, E.; Chuang, J.; Banares, K.; Castaneda, A.; Ouyang, K.; Cui, L.; Contu, R.; et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum. Mol. Genet. 2014, 23, 1134–1150. [Google Scholar] [CrossRef]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Pérez-Hernández, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca(2+)i Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.Y.; et al. Evidence from family studies for autoimmunity in arrhythmogenic right ventricular cardiomyopathy: Associations of circulating anti-heart and anti-intercalated disk autoantibodies with disease severity and family history. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
- D’amati, G.; Di Gioia, C.R.; Giordano, C.; Gallo, P. Myocyte transdifferentiation: A possible pathogenetic mechanism for arrhythmogenic right ventricular cardiomyopathy. Arch. Pathol. Lab. Med. 2000, 124, 287–290. [Google Scholar]
- Fujita, S.; Terasaki, F.; Otsuka, K.; Katashima, T.; Kanzaki, Y.; Kawamura, K.; Tanaka, T.; Kitaura, Y. Markedly increased intracellular lipid droplets and disruption of intercellular junctions in biopsied myocardium from a patient with arrhythmogenic right ventricular cardiomyopathy. Heart Vessels 2008, 23, 440–444. [Google Scholar] [CrossRef]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef]
- Gurha, P.; Chen, X.; Lombardi, R.; Willerson, J.T.; Marian, A.J. Knockdown of Plakophilin 2 downregulates miR-184 through CpG hypermethylation and suppression of the E2F1 pathway and leads to enhanced adipogenesis in vitro. Circ. Res. 2016, 119, 731–750. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Chen, S.N.; Ruggiero, A.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Cardiac Fibro-Adipocyte Progenitors Express Desmosome Proteins and Preferentially Differentiate to Adipocytes Upon Deletion of the Desmoplakin Gene. Circ. Res. 2016, 119, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Dong, J.; Rodriguez, G.; Bell, A.; Leung, T.K.; Schwartz, R.J.; Willerson, J.T.; Brugada, R.; Marian, A.J. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2009, 104, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Van Tintelen, J.P.; Entius, M.M.; Bhuiyan, Z.A.; Jongbloed, R.; Wiesfeld, A.C.; Wilde, A.A.; van der Smagt, J.; Boven, L.G.; Mannens, M.M.; van Langen, I.M.; et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2006, 113, 1650–1658. [Google Scholar] [CrossRef]
- Li Mura, I.E.; Bauce, B.; Nava, A.; Fanciulli, M.; Vazza, G.; Mazzotti, E.; Rigato, I.; De Bortoli, M.; Beffagna, G.; Lorenzon, A.; et al. Identification of a PKP2 gene deletion in a family with arrhythmogenic right ventricular cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 1226–1231. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef]
- Gehmlich, K.; Asimaki, A.; Cahill, T.J.; Ehler, E.; Syrris, P.; Zachara, E.; Re, F.; Avella, A.; Monserrat, L.; Saffitz, J.E.; et al. Novel missense mutations in exon 15 of desmoglein-2: Role of the intracellular cadherin segment in arrhythmogenic right ventricular cardiomyopathy? Heart Rhythm. 2010, 7, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef]
- Al-Sabeq, B.; Krahn, A.D.; Conacher, S.; Klein, G.J.; Laksman, Z. Arrhythmogenic right ventricular cardiomyopathy with recessive inheritance related to a new homozygous desmocollin-2 mutation. Can. J. Cardiol. 2014, 30, 696.e1–696.e3. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef]
- Jona, I.; Nanasi, P. Cardiomyopathies and sudden cardiac death caused by RyR2 mutations: Are the channels the beginning and the end? Cardiovasc. Res. 2006, 71, 416–418. [Google Scholar] [CrossRef]
- Van der Heijden, J.F.; Hassink, R.J. The phospholamban p.Arg14del founder mutation in Dutch patients with arrhythmogenic cardiomyopathy. Neth Heart J. 2013, 21, 284–285. [Google Scholar] [CrossRef]
- Van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Van Tintelen, J.P.; Van Gelder, I.C.; Asimaki, A.; Suurmeijer, A.J.; Wiesfeld, A.C.; Jongbloed, J.D.; van den Wijngaard, A.; Kuks, J.B.; van Spaendonck-Zwarts, K.Y.; Notermans, N.; et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009, 6, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Brun, F.; Barnes, C.V.; Sinagra, G.; Slavov, D.; Barbati, G.; Zhu, X.; Graw, S.L.; Spezzacatene, A.; Pinamonti, B.; Merlo, M.; et al. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2014, 51, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Hall, C.L.; Akhtar, M.M.; Sabater-Molina, M.; Futema, M.; Asimaki, A.; Protonotarios, A.; Dalageorgou, C.; Pittman, A.M.; Suarez, M.P.; Aguilera, B.; et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int. J. Cardiol. 2020, 307, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwaag, P.A.; van Rijsingen, I.A.; de Ruiter, R.; Nannenberg, E.A.; Groeneweg, J.A.; Post, J.G.; Hauer, R.N.; van Gelder, I.C.; van den Berg, M.P.; van der Harst, P.; et al. Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth Heart J. 2013, 21, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Pinamonti, B.; Dragos, A.M.; Pyxaras, S.A.; Merlo, M.; Pivetta, A.; Barbati, G.; Di Lenarda, A.; Morgera, T.; Mestroni, L.; Sinagra, G. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: Results from a 10-year registry. Eur. Heart J. 2011, 32, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.J.; McKenna, W.J. Left-dominant arrhythmogenic cardiomyopathy: An under-recognized clinical entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef]
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149. [Google Scholar] [CrossRef]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic Genotypes in Familial Dilated Cardiomyopathy: Implications for Genetic Testing and Clinical Management. Heart Lung. Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef]
- Zegkos, T.; Panagiotidis, T.; Parcharidou, D.; Efthimiadis, G. Emerging concepts in arrhythmogenic dilated cardiomyopathy. Heart Fail Rev. 2020. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020. [Google Scholar] [CrossRef]
- Roux-Buisson, N.; Gandjbakhch, E.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; Cosnay, P.; et al. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: Results of a systematic screening. Heart Rhythm. 2014, 11, 1999–2009. [Google Scholar] [CrossRef]
- Van Opbergen, C.J.M.; Noorman, M.; Pfenniger, A.; Copier, J.S.; Vermij, S.H.; Li, Z.; van der Nagel, R.; Zhang, M.; de Bakker, J.M.T.; Glass, A.M.; et al. Plakophilin-2 Haploinsufficiency Causes Calcium Handling Deficits and Modulates the Cardiac Response Towards Stress. Int. J. Mol. Sci. 2019, 20, 4076. [Google Scholar] [CrossRef] [PubMed]
- Delmar, M.; McKenna, W.J. The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circ. Res. 2010, 107, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Bass-Zubek, A.E.; Godsel, L.M.; Delmar, M.; Green, K.J. Plakophilins: Multifunctional scaffolds for adhesion and signaling. Curr. Opin. Cell Biol. 2009, 21, 708–716. [Google Scholar] [CrossRef]
- Puzzi, L.; Borin, D.; Gurha, P.; Lombardi, R.; Martinelli, V.; Weiss, M.; Andolfi, L.; Lazzarino, M.; Mestroni, L.; Marian, A.J.; et al. Knock Down of plakophillin 2 dysregulates adhesion pathway through upregulation of miR200b and alters the mechanical properties in cardiac cells. Cells 2019, 8, 1639. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb. Perspect Biol. 2012, 4, a007880. [Google Scholar] [CrossRef]
- Willert, K.; Shibamoto, S.; Nusse, R. Wnt-induced dephosphorylation of axin releases beta-catenin from the axin complex. Genes Dev. 1999, 13, 1768–1773. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Guttridge, D.C.; You, Z.; Zhang, Z.; Fribley, A.; Mayo, M.W.; Kitajewski, J.; Wang, C.Y. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell Biol. 2001, 152, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Longo, K.A.; Kennell, J.A.; Ochocinska, M.J.; Ross, S.E.; Wright, W.S. MacDougald, O.A. Wnt signaling protects 3T3-L1 preadipocytes from apoptosis through induction of insulin-like growth factors. J. Biol. Chem. 2002, 277, 38239–38244. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 2016, 64, 993–1008. [Google Scholar] [CrossRef]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef]
- Liu, G.; Ma, C.; Yang, H.; Zhang, P.Y. Transforming growth factor beta and its role in heart disease. Exp. Ther. Med. 2017, 13, 2123–2128. [Google Scholar] [CrossRef]
- Wrana, J.L. Signaling by the TGFbeta superfamily. Cold Spring Harb. Perspect Biol. 2013, 5, a011197. [Google Scholar] [CrossRef]
- Chaudhury, A.; Howe, P.H. The tale of transforming growth factor-beta (TGFbeta) signaling: A soigne enigma. IUBMB Life 2009, 61, 929–939. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.F.; Haneline, L.S.; Field, L.J. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef]
- Szekely, Y.; Arbel, Y. A Review of Interleukin-1 in Heart Disease: Where Do We Stand Today? Cardiol. Ther. 2018, 7, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Verma, G.; Datta, M. IL-1beta induces ER stress in a JNK dependent manner that determines cell death in human pancreatic epithelial MIA PaCa-2 cells. Apoptosis 2010, 15, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fu, S.; Wang, Y.; Yu, P.; Hu, J.; Gu, W.; Xu, X.M.; Lu, P. Interleukin-1beta mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. Mol. Cell Neurosci. 2007, 36, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M.; Balser, J.R.; George, A.L., Jr.; Anderson, M.E. Cardiac ion channels. Annu. Rev. Physiol. 2002, 64, 431–475. [Google Scholar] [CrossRef]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Traaseth, N.J.; Ha, K.N.; Verardi, R.; Shi, L.; Buffy, J.J.; Masterson, L.R.; Veglia, G. Structural and dynamic basis of phospholamban and sarcolipin inhibition of Ca(2+)-ATPase. Biochemistry 2008, 47, 3–13. [Google Scholar] [CrossRef]
- Assous, N.; Touzé, E.; Meune, C.; Kahan, A.; Allanore, Y. Cardiovascular disease in rheumatoid arthritis: Single-center hospital-based cohort study in France. Joint Bone Spine 2007, 74, 66–72. [Google Scholar] [CrossRef]
- Hof, I.E.; van der Heijden, J.F.; Kranias, E.G.; Sanoudou, D.; de Boer, R.A.; van Tintelen, J.P.; van der Zwaag, P.A.; Doevendans, P.A. Prevalence and cardiac phenotype of patients with a phospholamban mutation. Neth Heart J. 2019, 27, 64–69. [Google Scholar] [CrossRef]
- Eijgenraam, T.R.; Boukens, B.J.; Boogerd, C.J.; Schouten, E.M.; van de Kolk, C.W.A.; Stege, N.M.; Te Rijdt, W.P.; Hoorntje, E.T.; van der Zwaag, P.A.; van Rooij, E.; et al. The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unreponsive to standard heart failure therapy. Sci. Rep. 2020, 10, 9819. [Google Scholar] [CrossRef]
- Nava, A.; Canciani, B.; Daliento, L.; Miraglia, G.; Buja, G.; Fasoli, G.; Martini, B.; Scognamiglio, R.; Thiene, G. Juvenile sudden death and effort ventricular tachycardias in a family with right ventricular cardiomyopathy. Int. J. Cardiol. 1988, 21, 111–126. [Google Scholar] [CrossRef]
- Kannankeril, P.J.; Mitchell, B.M.; Goonasekera, S.A.; Chelu, M.G.; Zhang, W.; Sood, S.; Kearney, D.L.; Danila, C.I.; De Biasi, M.; Wehrens, X.H.; et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 12179–12184. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Shin, D.W.; Hong, C.S.; Jeong, D.; Kim, D.H.; Park, W.J. Increased Ca2+ storage capacity in the sarcoplasmic reticulum by overexpression of HRC (histidine-rich Ca2+ binding protein). Biochem. Biophys. Res. Commun. 2003, 300, 192–196. [Google Scholar] [CrossRef]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef]
- Zhang, Y.; Gago-Lopez, N.; Li, N.; Zhang, Z.; Alver, N.; Liu, Y.; Martinson, A.M.; Mehri, A.; MacLellan, W.R. Single-cell imaging and transcriptomic analyses of endogenous cardiomyocyte dedifferentiation and cycling. Cell Discov. 2019, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, A.; Tsukada, S.; Kamiyama, M.; Yanagimoto, T.; Nakajima, M.; Maeda, S. Wnt5b partially inhibits canonical Wnt/beta-catenin signaling pathway and promotes adipogenesis in 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun. 2005, 330, 505–510. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 2011, 124 (Pt 21), 3654–3664. [Google Scholar] [CrossRef]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef]
- Winter, E.M.; Gittenberger-de Groot, A.C. Epicardium-derived cells in cardiogenesis and cardiac regeneration. Cell Mol. Life Sci. 2007, 64, 692–703. [Google Scholar] [CrossRef]
- Bollini, S.; Vieira, J.M.; Howard, S.; Dubè, K.N.; Balmer, G.M.; Smart, N.; Riley, P.R. Re-activated adult epicardial progenitor cells are a heterogeneous population molecularly distinct from their embryonic counterparts. Stem Cells Dev. 2014, 23, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Bollini, S.; Dubé, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Zangi, L.; Oliveira, M.S.; Ye, L.Y.; Ma, Q.; Sultana, N.; Hadas, Y.; Chepurko, E.; Später, D.; Zhou, B.; Chew, W.L.; et al. Insulin-Like Growth Factor 1 Receptor-Dependent Pathway Drives Epicardial Adipose Tissue Formation After Myocardial Injury. Circulation 2017, 135, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.; Butterfield, C.; Lin, R.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Invest. 2011, 121, 1894–1904. [Google Scholar] [CrossRef]
- Singh, A.; Ramesh, S.; Cibi, D.M.; Yun, L.S.; Li, J.; Li, L.; Manderfield, L.J.; Olson, E.N.; Epstein, J.A.; Singh, M.K. Hippo Signaling Mediators Yap and Taz Are Required in the Epicardium for Coronary Vasculature Development. Cell Rep. 2016, 15, 1384–1393. [Google Scholar] [CrossRef]
- Xiao, Y.; Hill, M.C.; Zhang, M.; Martin, T.J.; Morikawa, Y.; Wang, S.; Moise, A.R.; Wythe, J.D.; Martin, J.F. Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 2018, 45, 153.e6.–169.e6. [Google Scholar] [CrossRef]
- Ramjee, V.; Li, D.; Manderfield, L.J.; Liu, F.; Engleka, K.A.; Aghajanian, H.; Rodell, C.B.; Lu, W.; Ho, V.; Wang, T.; et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J. Clin. Invest. 2017, 127, 899–911. [Google Scholar] [CrossRef]
- Matthes, S.A.; Taffet, S.; Delmar, M. Plakophilin-2 and the migration, differentiation and transformation of cells derived from the epicardium of neonatal rat hearts. Cell Commun. Adhes 2011, 18, 73–84. [Google Scholar] [CrossRef]
- Lombardi, R.; Chen, S.N.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Desmoplakin Deficient Epicardial Progenitor Cells Mediate the Development of Fibroadiposis in Arrhythmogenic Cardiomyopathy. Circulation 2017, 136 (Suppl. 1). A20536. [Google Scholar]
- Liu, Q.; Huang, X.; Oh, J.; Lin, R.; Duan, S.; Yu, Y.; Yang, R.; Qiu, J.; Melero-Martin, J.M.; Pu, W.T.; et al. Epicardium-to-fat transition in injured heart. Cell Res. 2014, 24, 1367–1369. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Cavallero, S.; Patterson, M.; Shen, H.; Xu, J.; Kumar, S.R.; Sucov, H.M. Adipogenesis and epicardial adipose tissue: A novel fate of the epicardium induced by mesenchymal transformation and PPARgamma activation. Proc. Natl. Acad. Sci. USA 2015, 112, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.; de Bakker, J.M.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef] [PubMed]
- Campian, M.E.; Verberne, H.J.; Hardziyenka, M.; de Groot, E.A.; van Moerkerken, A.F.; van Eck-Smit, B.L.; Tan, H.L. Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.; Keeling, P.J.; McKenna, W.J.; Mann, J.M.; Bottazzo, G.F.; Zachara, E.; Mestroni, L.; Camerini, F. Evidence from family studies for autoimmunity in dilated cardiomyopathy. Lancet 1994, 344, 773–777. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef]
- Hodgkinson, C.P.; Bareja, A.; Gomez, J.A.; Dzau, V.J. Emerging Concepts in Paracrine Mechanisms in Regenerative Cardiovascular Medicine and Biology. Circ. Res. 2016, 118, 95–107. [Google Scholar] [CrossRef]
- Mancuso, T.; Barone, A.; Salatino, A.; Molinaro, C.; Marino, F.; Scalise, M.; Torella, M.; Angelis, A.D.; Urbanek, K.; Torella, D.; et al. Unravelling the Biology of Adult Cardiac Stem Cell-Derived Exosomes to Foster Endogenous Cardiac Regeneration and Repair. Int. J. Mol. Sci. 2020, 21, 3725. [Google Scholar] [CrossRef]
- Routledge, D.; Scholpp, S. Mechanisms of intercellular Wnt transport. Development 2019, 146. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef]
- Lazar, E.; Benedek, T.; Korodi, S.; Rat, N.; Lo, J.; Benedek, I. Stem cell-derived exosomes—An emerging tool for myocardial regeneration. World J. Stem Cells 2018, 10, 106–115. [Google Scholar] [CrossRef]
- Ermakov, S.; Gerstenfeld, E.P.; Svetlichnaya, Y.; Scheinman, M.M. Use of flecainide in combination antiarrhythmic therapy in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2017, 14, 564–569. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Estimated Frequency (%) | Features | Mode of Inheritance | Refs |
|---|---|---|---|---|---|
| Desmosome | |||||
| PKP2 | Plakophilin 2 | ~40 | Haploinsufficiency. ACM; Carvajal syndrome; LD-ACM; Arrhythmogenic DCM | AD | [46,47,48] [45,49] |
| DSP | Desmoplakin | ~16 | biventricular ACM | AD, AR | |
| DSG2 | Desmoglein 2 | ~10 | Overlap with DCM; biventricular ACM | AD | [50,51] |
| DSC2 | Desmocollin 2 | ~8 | ACM | AD, AR | [52,53] |
| JUP | Junction plakoglobin | Rare | Naxos disease; ACM | AD, AR | [44,54] |
| Adherens Junction | |||||
| CTNNA3 | Catenin-α3 | Rare | ACM with incomplete penetrance; | AD | [65] |
| CDH2 | Cadherin 2 | Rare | No specific genotype–phenotype relationship identified | AD | [67,68] |
| Cytoskeletal/Nuclear Structure | |||||
| LMNA | Lamin A/C | Rare | DCM with arrhythmias and high risk of sudden cardiac death; LD-ACM | - | [61] |
| DES | Desmin | Rare | Fully penetrant; LV and RV-dominant ACM: DCM; skeletal myopathies | AD | [62,63] |
| FLNC | Filamin C | Rare | LD- ACM; high risk of arrhythmias and sudden death; Arrhythmogenic-DCM | - | [70,71] |
| TMEM43 | Transmembrane protein 43 | Rare | Fully penetrant; affected men more severely than women; LD-ACM | AD | [60] |
| TTN | Titin | Rare | Higher risk of supraventricular tachycardia and progression to heart failure; arrhythmogenic DCM | - | [64,65] |
| Ion Transport | |||||
| RYR2 | Ryanodine receptor 2 | Rare | Overlap with DCM | AD | [56,57] |
| SCN5A | Nav1.5 | Rare | Prolonged QRS interval; arrhythmhenic DCM and LD-ACM | - | [66] |
| PLN | Phospholamban | Rare | Low prevalence; DCM,ACM; LD-ACM, biventricular ACM, arrhythogenic DCM | - | [58,72] |
| Cytokine | |||||
| TGFB3 | Transforming growth factor-β3 | Rare | No specific genotype–phenotype relationship identified | - | [55] |
| Phenotype | Chamber Involved | Histology | CMRI-LEG Distribution | ECG | Cardiac Function by Echocardiography and Cine-MRI |
|---|---|---|---|---|---|
| ARVC | RV | Fibroadiposis | Subepicardial enhancement. Involving the area between the anterior part of the pulmonary infundibulum, the apex, and the infero-posterior wall (so called triangle of dysplasia). Interventricular septum is not involved | Inverted T waves in right precordial leads. Epsilon wave in the right precordial leads. Non-sustained or sustained VT of LBBB morphology with superior axis | RV dilatation and reduced global function; regional RV dys- akinesias and/or RV wall aneurisms |
| LD-ACM | LV | Fibroadiposis | Subepicardial enhancement involving the inferior/lateral walls of the LV | Low voltages at limb leads; left axis deviation; negative T waves at the inferolateral leads.Ventricular extrasystoles of RBBB morphology | LV dilatation and reduced systolic function with preservation of RV global and regional function |
| a-DCM | LV or LV and RV | Interstitial Fibrosis > adiposis | Diffuse midwall enhancement of the LV; interventricular septum usually involved | Conduction system disease including sinus and/or AV nodal dysfunction, frequent atrial or ventricular arrhythmias | Mild to severe LV global dysfunction in absence of regional abnormalities; RV may or may not be involved |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, S.; Puthenvedu, D.; Lombardi, R.; Chen, S.N. Established and Emerging Mechanisms in the Pathogenesis of Arrhythmogenic Cardiomyopathy: A Multifaceted Disease. Int. J. Mol. Sci. 2020, 21, 6320. https://doi.org/10.3390/ijms21176320
Gao S, Puthenvedu D, Lombardi R, Chen SN. Established and Emerging Mechanisms in the Pathogenesis of Arrhythmogenic Cardiomyopathy: A Multifaceted Disease. International Journal of Molecular Sciences. 2020; 21(17):6320. https://doi.org/10.3390/ijms21176320
Chicago/Turabian StyleGao, Shanshan, Deepa Puthenvedu, Raffaella Lombardi, and Suet Nee Chen. 2020. "Established and Emerging Mechanisms in the Pathogenesis of Arrhythmogenic Cardiomyopathy: A Multifaceted Disease" International Journal of Molecular Sciences 21, no. 17: 6320. https://doi.org/10.3390/ijms21176320
APA StyleGao, S., Puthenvedu, D., Lombardi, R., & Chen, S. N. (2020). Established and Emerging Mechanisms in the Pathogenesis of Arrhythmogenic Cardiomyopathy: A Multifaceted Disease. International Journal of Molecular Sciences, 21(17), 6320. https://doi.org/10.3390/ijms21176320