Abstract
Cerebral small vessel disease (CSVD) is a leading cause of cognitive decline in elderly people and development of Alzheimer’s disease (AD). Blood–brain barrier (BBB) leakage is a key pathophysiological mechanism of amyloidal CSVD. Sleep plays a crucial role in keeping health of the central nervous system and in resistance to CSVD. The deficit of sleep contributes to accumulation of metabolites and toxins such as beta-amyloid in the brain and can lead to BBB disruption. Currently, sleep is considered as an important informative platform for diagnosis and therapy of AD. However, there are no effective methods for extracting of diagnostic information from sleep characteristics. In this review, we show strong evidence that slow wave activity (SWA) (0–0.5 Hz) during deep sleep reflects glymphatic pathology, the BBB leakage and memory deficit in AD. We also discuss that diagnostic and therapeutic targeting of SWA in AD might lead to be a novel era in effective therapy of AD. Moreover, we demonstrate that SWA can be pioneering non-invasive and bed–side technology for express diagnosis of the BBB permeability. Finally, we review the novel data about the methods of detection and enhancement of SWA that can be biomarker and a promising therapy of amyloidal CSVD and CSVD associated with the BBB disorders.
1. Sleep as a Potential Biomarker of Alzheimer’S Disease
Why do we need to sleep and how long should we sleep? Such very highly active people as Margaret Thatcher resent the idea of spending one third of their lives asleep and train themselves to get by with significantly less sleep than others. The surrealist painter Salvador Dali claimed to sleep for only three or four hours every night and to compensate for this by taking short naps during the day. On the other hand, Einstein liked to sleep about 14 h a day. However, regardless of the regime of sleep, without it, we become tired, our brain functions less well and prolonged sleep deprivation can be fatal. Indeed, the brain eats itself after short and chronic sleep loss via microglial activation and astrocytic phagocytosis of synaptic elements [1]. Insufficient sleep leads to sterile inflammation in the absence of infection [2,3,4] and to enhanced permeability of the blood–brain barrier (BBB) [3,5]. The total sleep deprivation of rats produced their death [6]. In humans the longest time of awakeness of 11 days is accompanied by hallucination and various cognitive deficiencies [7]. Thus, it seems obvious that sleep plays an important role in restoration of brain functions. However, what exactly is being restored by sleep remains unanswered. The functions of sleep have been speculated in the ancient works such as “Aristotle’s Theory of ‘Sleep and Dreams’” [8]. Aristotle proposed that sleep helps the body cleans its blood at the end of the day. More than 2000 years later, researchers confirmed Aristotle’s idea that sleep has a crucial function of clearance of metabolites and neurotoxic wastes from the brain accumulated in the awake central nervous system (CNS). So, Xie et al. demonstrated that the CSF tracer influx into the mouse brain is largely reduced by 95% in the awake state [9]. However, during deep sleep the brain’s interstitial fluid (ISF) volume expands (compared with wakefulness) by 60% via astrocytic aquaporin-4 (AQP4) water channels, resulting in faster waste removal, including toxins such as beta-amyloid (A). The Fultz et al. in human studies discovered the close correlation between oscillation of CSF, which clears metabolic waste products from the brain, and EEG delta band during deep sleep [10].
Measuring how people sleep can be a promising approach to screen for Alzheimer’s disease (AD) [11,12,13]. People with AD have poor sleep, they often wake up and their nights become less refreshing as memory loss and other symptoms worsen [14,15,16]. The poor sleep quality and short sleep duration are associated with increased A deposition Clinical studies have shown that A content in CSF is lower in sleep than wakefulness There is evidence that A clearance is increased during sleep due to increased ISF bulk flow [9]. Furthermore, excessive daytime sleepiness in older adults is associated with increased longitudinal A accumulation [17]. Thus, there are growing body of evidence that disturbance of cleaning processes during sleep is a putative marker of AD pathology, at least in part, via an A mechanism [18]. However, not all sleep can be an effective marker of AD.
Sleep consists of rapid eye movement (REM) associated with dreams and non-rapid eye movement (NREM) or deep sleep. REM sleep is characterized by desynchronization of EEG dynamics with faster oscillations and low voltage waveforms [19]. Human NREM sleep is subdivided into four stages and is defined as synchronous of EEG activity including sleep spindles (12–14 Hz) or K-complex (stage 2) and slow wave activity (0–4 Hz) in delta band (stage 3) [20]. The slow wave activity (SWA) is a major rhythm of deep sleep. The SWA is strongly controlled and a deficit of sleep induces a compensatory increasing of the SWA time [21,22]. Conversely, preceding daytime nap is accompanied by a reduction of the SWA time during subsequent nocturnal sleep [22]. The nature of SWA is not recognized yet, but there are growing body of evidence that SWA plays an important role in regulation of quality of sleep and in its restorative and clearing functions [10,23,24,25,26,27,28,29]. The sleep efficiency and depth are directly related to SWA [25,26,30,31,32,33]. NREM SWA considered as a promising intervention target for AD [23]. SWA sleep plays crucial role in memory consolidation and the SWA disturbances are associated with AD in patients and animal models [23]. Studies in animal models have found decreased SWA in P301S tau transgenic mice [34]. Both young and adult mice with a model of amyloidosis (APPswePS1dE9) demonstrate decrease in the cortical SWA power but not frequency with significantly reducing the time of NREM sleep [35,36]. Tg2576 and 3xTg-AD mouse models are characterized by the low time of SWA [37,38].
In humans, atrophy and A accumulation in the medial prefrontal cortex is correlated with both decreased NREM-SWA and impaired overnight hippocampus-dependent memory consolidation in cognitively normal older adults [39,40]. Lucey et al. reported relationship between NREM-SWA and AD pathology, particularly tauopathy, and that this association was most evident at the lowest frequencies of NREM-SWA [11]. The changes in NREM-SWA, especially at 1 to 2Hz, might be able to discriminate tau pathology and cognitive impairment either before or at the earliest stages of symptomatic AD. The SWA disruption was also reported in patients with mild cognitive impairments [41]. The impairment of quality of sleep in cognitive normal old people could predict A and tau accumulation in the brain. [42]. Taking into account all of the above, we suppose that slow wave sleep may be potential biomarker of AD that also have been discussed in other reviews [23,42].
Why SWA reflects AD pathology remains unknown but there is strong evidence that it can be related to recently discovered correlation between SAW and activation of glymphatic clearance during deep sleep [24]. Neither alpha nor gamma wave dynamics correlated with glymphatic funtions. The SWA contributes to the efficiency of fluid influx into the brain and clearance of waste and A from the brain [9,43]. The reduced glymphatic perivascular flow with aging may facilitate the development of AD [44,45] due to the slower transit time that will cause greater cellular binding/update of A and apolipoprotein E (apoE) [46]. The decreased glymphatic fluid transport after insufficient sleep may be related to an increase interstitial noradrenaline (NE) level [9,47,48,49] leading to NE-mediated decrease of astrocytic volume [50] and vasoconstriction of pial arteries [51]. Thus, the impaired CSF and ISF flow during sleep deficit can contribute to the reduced glymphatic fluid transport.
There is intrigue idea that the body posture during sleep can be used as an additional sign in diagnosis of impairment of glymphatic functions [52]. There is hypothesis that the most popular sleep posture (lateral) has evolved to optimize waste removal, including A, during sleep and that posture must be considered in diagnostic imaging procedures developed in the future to assess CSF-ISF transport in humans [52].
A plagues target synapses, contributing abnormalities in excitatory and inhibitory neurotransmission leading neural network disruption that can be responsible for reducing the time of SWA [35,53,54,55,56]. Indeed, more than 20% of cortical neurons exhibit hyperactivity surrounding A [35] and blocking of neuron depolarization by the gamma-aminobutyric acid A (GABAA) improves SWA deficit in mice with AD [35,54]. The AAP mice demonstrate the deficit of the excitatory neurotransmitter glutamate and SWA, while the glutamate receptor antagonist alleviated hyperactivity and restored SWA [35,54]. Optogenetically increasing neural activity in hippocampal causes elevation of level of A in ISF and A depositions in the brain in mice with AD as well as augmentation of neural calcium content and decrease in the synaptic spine density [35,36,57]. Optogenetic-induced neural hyperactivity is accompanied by elevated release and propagation of tau in htau mice [58]. Taken together, A-mediated synaptic inhibition, which leads to neural hyperactivity, can be another possible mechanism underling the SWA deficit in AD.
The aberrant astrocytic activity can also contribute to the SWA disruption in AD. Astrocytes maintain glutamate and GABA recycling and form the tripartite synapses to regulate synaptic transmission via calcium signaling [59,60]. A depositions disrupt astrocytic morphology, astrocytic calcium signaling and glutamate/GABA recycling [61,62,63]. The blocking of astrocytic calcium transients resulted in decrease the number of astrocytes and neurons participating in regulation of SWA oscillations [64]. The astrocytic-mediated modulation of slow oscillations via intracalcium transient and extracellular glutamate triggers SWA [65,66]. Thus, the astrocytic abnormalities induced by A accumulation in the brain may contribute to aberrant neural firing and lead to the hyperactivity of neurons, thus perturbation SWA. The further animal studies of the role of astrocytes in the SWA deficit in AD can shed light on mechanisms of SWA disturbances in AD and also might be point to a novel therapeutic methods for AD. Thus, sleep is natural factor, which activates clearance of accumulated metabolites from the brain, including A, via increasing interstitial space and connective flow. Sleep disorders, especially SWA, are closely associated with reducing of glymphatic clearance of metabolites and toxins such as A that can be an important informative platform for development of new promising strategies in early diagnosis of AD (Figure 1).
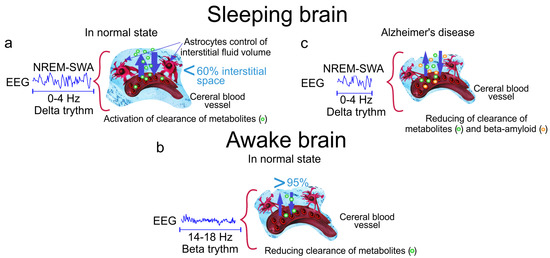
Figure 1.
The cleaning power of a slow wave activity (SWA) during deep sleep. (a) The slow wave sleep is accompanied by increasing the interstitial fluid (ISF) volume by 60% via astrocytic-aquaporin (AQP)-channels that contributes augmentation of metabolic clearance; (b) Wakefulness reduces diffusion of metabolites by 95% via decreasing the ISF volume; (c) Alzheimer’s disease is associated with accumulation of A in the brain tissues due to reducing of the time of SWA and suppression of clearance of toxic protein from the brain.
2. Slow Wave Activity as a Biomarker of Disruption of Blood-Brain Barrier
Sleep and the blood–brain barrier (BBB) are two important gamers in scenarios of the homeostasis of the central nervous system (CNS). Sleep is essential for maintenance of the health of the CNS via clearance of metabolites and neurotoxic wastes from the brain [9,10].
It is generally accepted that the BBB acts as the blood–brain interface protecting the CNS from the penetration of microorganisms and toxins from the blood. Therefore, the methods for opening the BBB are usually used for brain drug delivery and therapy of CNS diseases [67]. However, the latest findings changed our understanding of the role of BBB in the keeping of the CNS health. The BBB opening is accompanied by activation of clearance of macromolecules from the brain [68,69]. It can explain why the BBB opening without pharmacological therapy contributes for the clearance of A in patients with AD and in mouse models of amyloidosis [70,71,72,73]. The interrelation between sleep and the BBB opening is not known but both conditions are interlinked with activation of clearance of macromolecules and toxins from the brain [9,68,69]. Thus, neurological activity during sleep is expected to be similar those during the BBB opening. Indeed, both sleep [9] and the BBB opening [70,71,72,73] are accompanied by clearance of A from the brain. The sleep is characterized by the coupled oscillations of SWA and CSF, which cleans metabolic waste products from the brain [9,10], that becomes stronger with more low frequency EEG oscillations [24]. The SWA in EEG dynamics is also associated with BBB opening in humans and animals [74,75,76] and the BBB disruption causes activation of clearance of macromolecules from the brain [68,69].
The mechanisms of BBB-mediated changes of neural activity are not fully understood. The BBB opening can affect EEG activity by direct and indirect ways. Direct influence of an increased BBB permeability on the EED dynamics is generation signals of the BBB via electrophysiological properties of brain endothelial cells forming the BBB. The signals generated by the BBB originate from a trans-endothelial voltage between blood and brain tissue This voltage is a consequence of unequal endothelial cell apical and basolateral membrane potentials [77]. The ion influx/efflux changes the BBB permeability via brain endothelial cells membrane depolarization affecting the cell stiffness via molecular mechanisms underlying cortical actin cytoskeleton [77,78]. These changes of cell potential cause up to mV-level shifts in human scalp EEG [78,79]. Kiviniemi et al. observed that the intact BBB maintains a positive voltage, while the BBB leakage is characterized by a negative shift in this parameter [74].
In the 1970s, it was discovered that large-amplitude brain-potential shifts originate from a potential difference, which can occur during the BBB opening induced by respiratory acidosis in different animals species, including cats, money, and rats [80,81,82]. On the one hand, there is evidence suggesting that the BBB acts as a non-neuronal signal generator of mV-level slow shifts measured at scalp [79,82]. On the other hand, the BBB-signals can also be coupled to neuronal function, since low level frequency oscillations in the human brain are synchronized with faster cortical EEG oscillations and they are associated with the slow fluctuations in brain excitability [79,83,84].
Indirect influences of the BBB on the EEG behavior are astrocytes which are essential for the formation and maintenance of the BBB. Reduction in astrocyte number in the mPFC was associated with impaired cognitive flexibility and reduced power across delta (1–4 Hz), alpha (12–20 Hz), and gamma (30–80 Hz) frequency ranges [85,86]. The astrocytic mechanism of EEG modulation can be mediated via astrocyte-related regulation of the synaptic conductance [87,88,89], which are involved in electrically induced EEG-activated states in cortical neurons
We consider that the clearance of different compounds from the brain can be possible bridge between the similar changes in EEG dynamics co-occur during sleep and the BBB opening (Figure 2). It is believed that an increase in the volume of the interstitial fluid (ISF) contributes by drainage of water-soluble metabolites from ISF to CSF compartments [90].The sleep is associated with an increase in the ISF volume that is accompanied activation of macromolecular diffusion in the brain tissues [9,91]. The astrocytes rapidly and significantly change their volume, making a decisive contribution to the change in the total proportion of volume of ISF [92,93,94]. These dynamic astrocyte volume changes may represent a previously unappreciated yet fundamental mechanism by which astrocytes regulate brain rhythm during sleep [95,96] and the BBB integrity [97,98].
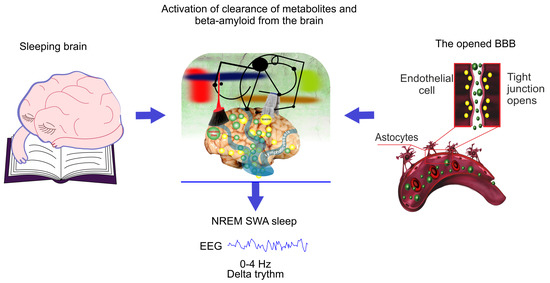
Figure 2.
Schematic illustration of hypothesis that the EEG characteristics of non-rapid eye movement (NREM) SWA sleep is similar for natural sleep and the blood–brain barrier (BBB) disruption due to the same mechanism of activation of clearance of metabolites and toxins such as A from sleeping brain and from the areas surrounding the opened BBB.
The SWA would be the cleaning power of sleep [24]. Indeed, during awake stage and REM sleep, the brain’s rhythms are desynchronized due to cacophony of neuron activity connecting different parts of brain [19,20]. During SWA sleep, the neuron dynamics demonstrates synchronous activity and inactivity changing each other over brief periods. We think that these oscillations of synchronous activity and inactivity of delta rhythm may be helping in “brain rinsing” and to move brain fluids and waste products through brain tissues like sea waves move salt and water. The reducing of slow wave sleep might be one of important diagnostic symptom of altered clearance of the brain that can contribute development of neurodegenerative diseases via accumulation of toxins in the brain [11,12].
3. Slow Sleep Wave Enhancement Is Promising Therapy of Alzheimer’S Disease
Current, there are no therapy of AD [99,100,101]. The majority of clinical approaches are focus on using monoclonal antibodies as passive immunotherapy [102]. However, pharmaceutical companies such as Biogen, Johnson & Johnson, Pfizer announced the cancellation of funding for the synthesis of antibodies for the treatment of AD due to the failure of clinical trials (Biogen/Eisai Halt Phase 3 Aducanumab Trials. https://www.alzforum.org/news/research-news/biogeneisai-haltphase-3-aducanumab-trial).
Obviously, in the next couple of decades, the main strategies for a treatment of AD will be non-invasive methods of stimulation of clearance of the toxic A from the brain. The enhancement of SWA sleep is discussed as a promising tool in therapy of AD and rescue sleep-dependent memory consolidation [23].
Auditory stimulation of SWA appeared to be more effective in increasing SWA and improvement of memory consolidation as well as cognitive functions [103,104,105,106,107]. This method is based on uses of “pink noise” (50-millisecond bursts) that is synchronized with neural cortical activity of delta band and increases the time of SWA [105,106,107]. The morphology, topography and propagation pattern of auditory-stimulated SWA are very similar to those of SWA observed during natural sleep [105,106].
Auditory stimulation is used in overnight and nap studies [107,108,109,110,111]. Overnight studies began 5 min after falling into NREM for the first time and ended 210 min later [109,110,111]. In afternoon nap studies, auditory stimulation is used intermittently regime with 90-min nap session [107,112,113]. The auditory-mediated SWA enhancement is hypothesized to be the results of “bottom up” activation of large populations of cortical neurons as the same process that is underlying arousing the organism The intensity of sensory stimulation has to be strong enough to trigger SWA enhancement, but no so strong to cause awakening. Thus, the intensity of stimulation has to be strong enough to trigger the activation of the reticular ascending system (ARAS) playing a crucial role in arousal, but not so strong as to cause a full-blown awakening. The idea that the arousal systems can be functionally parceled according to the magnitude of stimulation was first by Moruzzi in 1950s. He considered that for mild sensory stimulation only some portions of the activating ARAS might be activated, while the entire system could be recruited only by more intense stimuli [114,115].
Therefore, the optimization of acoustic stimulation of SWA such as intensity, frequency, and timing is in the trend of development of breakthrough technologies in the enhancement of SWA [103,104,105,106,107].
Transcranial electrical (tDCS) and magnetic stimulation have been successfully applied to enhance SWA with the aim to improve quality of sleep and behavior/cognitive functions [116,117,118,119,120,121,122]. tDCS (1–20 Hz) triggers SWA that is indistinguishable from those during natural sleep [118,121]. tDCS induces a widespread electrical potential field with a focus on fronto-cortical areas. In majority of studies, tDCS is delivered at 0.75 Hz for 5 min intervals separated by 1 min off periods after SWA onset [116,117]. However, the long-term effects of repeated exposure of both these methods remain unknown [105]. The complex of response of activated/deactivated cortical fields following two these methods and difficulties of characterization of precise mechanism of tDCS and magnetic stimulation make unpredictable effects of these approaches. Since these methods are currently impractical and their safety is questionable, especially for chronic long-term exposure, natural physiological sensory stimulation of SWA is more preferable.
Pharmacological methods can also lead increase the time of SWA and might be alternative strategies relying on electric, magnetic, or auditory stimulation of SWA for improving of quality of sleep and brain functions. The administration of tiagabine [123,124], gaboxadol [125], sodium oxybate [126,127,128], baclofen [128], olanzapine [129,130], interleukin-6 [131] has been demonstrated as pharmacological method for SWA enhancing. Tiagabine, gaboxadol, sodium oxybate and baclofen increases SWA via the inhibition of neurotransmitter GABA [132]. Olanzapine is an antagonist of the serotonin2C (5-HT2C) receptor, which is involved in SWA regulation [133]. Interleukin-6 as proinflammatory cytokinine stimulates neuromedulatory mechanisms of regulation of SWA nature [134]. The pharmacological SWA enhancement is not new idea and were discussed in these reviews [134,135,136,137,138]. Anesthesia such as ketamine/xylazine or dexmedetomidine increase in EEG delta power [139,140] and significantly enhance glymphatic influx [24].
Optogenetic modulation of SWA is a leading-edge research method that can be used for restoring brain oscillatory brain activity, including SWA. This method is based on light cell-targeting manipulation of proteins expression in cells leading to modulation of the neural activity within neural circuits interest. Optogenetic-mediated activation of neural nitric oxide synthase (nNOS) or somatostatine neurons is useful for restoring SWA [141]. The optogenetically evoked responses in nNOS-positive cells of the cerebral cortex improve memory, sleep quality and prolong the time of SWA [142]. Thus, SWA restoration provides a promising novel therapeutic target for AD. Development of breakthrough strategies targeting SWA during NREM sleep might be a promising therapeutic tool to slow memory decline in the elderly or in healthy individuals at risk for developing AD as well as to delay progression of the disease in patients with AD (Figure 3).
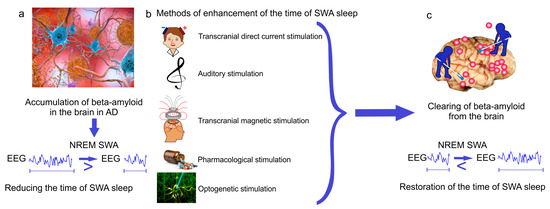
Figure 3.
New breakthrough strategies targeting SWA sleep for therapy of Alzheimer’s disease (AD). (a) Alzheimer’s disease is characterized by A-mediated reducing the time of SWA sleep; (b,c) methods of enhancement of SWA oscillations restore the time of SWA sleep and memory via improvement of glymphatic clearance of A from the brain.
4. Conclusions
Cerebral small vessel disease (CSVD) is an important course of cognitive decline and AD. An key pathophysiological mechanism of CSVD is BBB leakage leading to progression of CSVD. Currently, there are no specific preventive or therapeutic measures to improve CSVD. Sleep can be a novel biomarker and a promising therapeutic target for amyloid CSVD and for CSVD associated with BBB disruption. Amyloid CSVD such as AD is correlated with SWA deficit at early stages of disease and was found in asymptomatic cognitively normal adults. Since SWA disruption is an early event, it can be an early biomarker for AD. The level of A in the brain depends on quality of SWA and progression of AD is associated with reducing the time of SWA. Thus, the SWA pattern has the potential to be used as prognostic tool of severity of AD. The latest findings clearly suggest that SWA can be pioneering non-invasive and bed–side technology for express diagnosis of BBB leakage. Thus, SWA restoration provides a promising novel therapeutic target for amyloid CSVD and for CSVD associated with the BBB disruption. A better understanding of the link between SWA disruptions and reducing of clearance A from the brain associated with memory decline in AD may shed light on the mechanisms of AD. The methods of detection and enhancement of SWA during NREM sleep can open a new era in novel strategies of early therapy of CSVD, including improving memory in patients with AD or in healthy individuals at risk for AD.
Author Contributions
Conceptualization, O.S.-G.; writing—original draft preparation, O.S.-G. and D.P.; writing—review and editing, O.S.-G., D.P. and T.P.; project administration, J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Grants of Russian Science Foundation: #20-15-00090, #19-15-00201, RFBR grants #19-515-55016, #20-015-00308-a, and RF Government Grant #075-15-2019-1885.
Acknowledgments
We thank Saranceva Elena for preparation of figures for this review.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| A | Beta-amyliod |
| APQ4 | Aquaporin(s)-4 |
| ARAS | reticular ascending system |
| BBB | Blood–Brain Barrier |
| CSF | Cerebral spinal fluid |
| CSVD | Cerebral Small Vessel Disease |
| GABAA | Gamma-aminobutyric acid A |
| ISF | Interstitial fluid |
| NA | Noradrenaline |
| NREM | Non repaid eyes movement |
| nNOS | Neuronal nitric oxide synthase |
| REM | Rapid eye movement |
| SWA | Slow wave activity |
| tDCS | Transcranial electrical stimulation |
References
- Bellesi, M.; de Vivo, L.; Chini, M.; Gilli, F.; Tononi, G.; Cirelli, C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J. Neurosci. 2017, 37, 5263–5273. [Google Scholar] [CrossRef]
- Mullington, J.M.; Simpson, N.S.; Meier-Ewert, H.K.; Haack, M. Sleep loss and inflammation. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Alvarado, G.; Pavón, L.; Castillo-García, S.A.; Hernández, M.E.; Domínguez-Salazar, E.; Velázquez-Moctezuma, J.; Gómez-González, B. Sleep loss as a factor to induce cellular and molecular inflammatory variations. Clin. Dev. Immunol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Lahtinen, A.; Puttonen, S.; Vanttola, P.; Viitasalo, K.; Sulkava, S.; Pervjakova, N.; Joensuu, A.; Salo, P.; Toivola, A.; Härmä, M.; et al. A distinctive DNA methylation pattern in insufficient sleep. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- He, J.; Hsuchou, H.; He, Y.; Kastin, A.J.; Wang, Y.; Pan, W. Sleep restriction impairs blood–brain barrier function. J. Neurosci. 2014, 34, 14697–14706. [Google Scholar] [CrossRef] [PubMed]
- Everson, C.A.; Bergmann, B.M.; Rechtschaffen, A. Sleep deprivation in the rat: III. Total sleep deprivation. Sleep 1989, 12, 13–21. [Google Scholar] [CrossRef]
- Ross, J.J. Neurological findings after prolonged sleep deprivation. Arch. Neurol. 1965, 12, 399–403. [Google Scholar] [CrossRef]
- Papachristou, C.S. Aristotle’s Theory of ‘Sleep and Dreams’ in the light of Modern and Contemporary Experimental Research. Electron. J. Philos. 2014, 17, 1–47. [Google Scholar] [CrossRef]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef]
- Fultz, N.E.; Bonmassar, G.; Setsompop, K.; Stickgold, R.A.; Rosen, B.R.; Polimeni, J.R.; Lewis, L.D. Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science 2019, 366, 628–631. [Google Scholar] [CrossRef]
- Lucey, B.P.; McCullough, A.; Landsness, E.C.; Toedebusch, C.D.; McLeland, J.S.; Zaza, A.M.; Fagan, A.M.; McCue, L.; Xiong, C.; Morris, J.C.; et al. Reduced non–rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Lutsey, P.L.; Misialek, J.R.; Mosley, T.H.; Gottesman, R.F.; Punjabi, N.M.; Shahar, E.; MacLehose, R.; Ogilvie, R.P.; Knopman, D.; Alonso, A. Sleep characteristics and risk of dementia and Alzheimer’s disease: The atherosclerosis risk in communities study. Alzheimer Dement 2018, 14, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, A.R.; Larrick, J.W. Sleep facilitates clearance of metabolites from the brain: Glymphatic function in aging and neurodegenerative diseases. Rejuvenation Res. 2013, 16, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Weldemichael, D.A.; Grossberg, G.T. Circadian rhythm disturbances in patients with Alzheimer’s disease: A review. Int. J. Alzheimer Dis. 2010, 2010. [Google Scholar] [CrossRef]
- McCurry, S.M.; Logsdon, R.G.; Teri, L.; Gibbons, L.E.; Kukull, W.A.; Bowen, J.D.; McCormick, W.C.; Larson, E.B. Characteristics of sleep disturbance in community-dwelling Alzheimer’s disease patients. J. Geriatr. Psychiatry Neurol. 1999, 12, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Tworoger, S.S.; Lee, S.; Schernhammer, E.S.; Grodstein, F. The association of self-reported sleep duration, difficulty sleeping, and snoring with cognitive function in older women. Alzheimer Dis. Assoc. Disord. 2006, 20, 41–48. [Google Scholar] [CrossRef]
- Carvalho, D.Z.; Knopman, D.S.; Boeve, B.F.; Lowe, V.J.; Roberts, R.O.; Mielke, M.M.; Przybelski, S.A.; Machulda, M.M.; Petersen, R.C.; Jack, C.R.; et al. Association of excessive daytime sleepiness with longitudinal β-amyloid accumulation in elderly persons without dementia. JAMA Neurol. 2018, 75, 672–680. [Google Scholar] [CrossRef]
- Lucey, B.P.; Bateman, R.J. Amyloid-β diurnal pattern: Possible role of sleep in Alzheimer’s disease pathogenesis. Neurobiol. Aging 2014, 35, S29–S34. [Google Scholar] [CrossRef]
- Carskadon, M.A.; Dement, W.C. Normal human sleep: An overview. Princ. Pract. Sleep Med. 2005, 4, 13–23. [Google Scholar]
- Iber, C. The AASM manual for the scoring of sleep and associated events: Rules. Terminol. Tech. Specif. 2007, 176, 2012. [Google Scholar]
- Borbély, A.A. A two process model of sleep regulation. Hum Neurobiol. 1982, 1, 195–204. [Google Scholar] [PubMed]
- Borb, A.A.; Achermann, P. Sleep homeostasis and models of sleep regulation. J. Biol. Rhythm. 1999, 14, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.F.; Gerashchenko, D.; Timofeev, I.; Bacskai, B.J.; Kastanenka, K.V. Slow Wave Sleep Is a Promising Intervention Target for Alzheimer’s Disease. Front. Neurosci. 2020, 14, 705. [Google Scholar] [CrossRef] [PubMed]
- Hablitz, L.M.; Vinitsky, H.S.; Sun, Q.; Stæger, F.F.; Sigurdsson, B.; Mortensen, K.N.; Lilius, T.O.; Nedergaard, M. Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci. Adv. 2019, 5, eaav5447. [Google Scholar] [CrossRef]
- Hauner, K.K.; Howard, J.D.; Zelano, C.; Gottfried, J.A. Stimulus-specific enhancement of fear extinction during slow-wave sleep. Nat. Neurosci. 2013, 16, 1553. [Google Scholar] [CrossRef]
- Keklund, G.; ÅKerstedt, T. Objective components of individual differences in subjective sleep quality. J. Sleep Res. 1997, 6, 217–220. [Google Scholar] [CrossRef]
- Diekelmann, S.; Born, J. The memory function of sleep. Nat. Rev. Neurosci. 2010, 11, 114–126. [Google Scholar] [CrossRef]
- Rasch, B.; Born, J. About sleep’s role in memory. Physiol. Rev. 2013, 93, 681–766. [Google Scholar] [CrossRef]
- Tononi, G.; Cirelli, C. Sleep and the price of plasticity: From synaptic and cellular homeostasis to memory consolidation and integration. Neuron 2014, 81, 12–34. [Google Scholar] [CrossRef]
- Huber, R.; Ghilardi, M.F.; Massimini, M.; Tononi, G. Local sleep and learning. Nature 2004, 430, 78–81. [Google Scholar] [CrossRef]
- ÅKerstedt, T.; Hume, K.; Minors, D.; Waterhouse, J. Good sleep—Its timing and physiological sleep characteristics. J. Sleep Res. 1997, 6, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Hoch, C.C.; Reynolds, C.F., III; Kupfer, D.J.; Berman, S.R.; Houck, P.R.; Stack, J.A. Empirical note: Self-report versus recorded sleep in healthy seniors. Psychophysiology 1987, 24, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Kryger, M.; Steljes, D.; Pouliot, Z.; Neufeld, H.; Odynski, T. Subjective versus objective evaluation of hypnotic efficacy: Experience with zolpidem. Sleep 1991, 14, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Holth, J.K.; Mahan, T.E.; Robinson, G.O.; Rocha, A.; Holtzman, D.M. Altered sleep and EEG power in the P301S Tau transgenic mouse model. Ann. Clin. Transl. Neurol. 2017, 4, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Kastanenka, K.V.; Hou, S.S.; Shakerdge, N.; Logan, R.; Feng, D.; Wegmann, S.; Chopra, V.; Hawkes, J.M.; Chen, X.; Bacskai, B.J. Optogenetic restoration of disrupted slow oscillations halts amyloid deposition and restores calcium homeostasis in an animal model of Alzheimer’s disease. PLoS ONE 2017, 12, e0170275. [Google Scholar] [CrossRef]
- Kastanenka, K.V.; Calvo-Rodriguez, M.; Hou, S.S.; Zhou, H.; Takeda, S.; Arbel-Ornath, M.; Lariviere, A.; Lee, Y.F.; Kim, A.; Hawkes, J.M.; et al. Frequency-dependent exacerbation of Alzheimer’s disease neuropathophysiology. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Kent, B.A.; Strittmatter, S.M.; Nygaard, H.B. Sleep and EEG power spectral analysis in three transgenic mouse models of Alzheimer’s disease: APP/PS1, 3xTgAD, and Tg2576. J. Alzheimer Dis. 2018, 64, 1325–1336. [Google Scholar] [CrossRef]
- Castano-Prat, P.; Perez-Mendez, L.; Perez-Zabalza, M.; Sanfeliu, C.; Giménez-Llort, L.; Sanchez-Vives, M.V. Altered slow (<1 Hz) and fast (beta and gamma) neocortical oscillations in the 3xTg-AD mouse model of Alzheimer’s disease under anesthesia. Neurobiol. Aging 2019, 79, 142–151. [Google Scholar]
- Mander, B.A.; Rao, V.; Lu, B.; Saletin, J.M.; Lindquist, J.R.; Ancoli-Israel, S.; Jagust, W.; Walker, M.P. Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nat. Neurosci. 2013, 16, 357. [Google Scholar] [CrossRef]
- Mander, B.A.; Marks, S.M.; Vogel, J.W.; Rao, V.; Lu, B.; Saletin, J.M.; Ancoli-Israel, S.; Jagust, W.J.; Walker, M.P. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat. Neurosci. 2015, 18, 1051–1057. [Google Scholar] [CrossRef]
- Westerberg, C.E.; Mander, B.A.; Florczak, S.M.; Weintraub, S.; Mesulam, M.M.; Zee, P.C.; Paller, K.A. Concurrent impairments in sleep and memory in amnestic mild cognitive impairment. J. Int. Neuropsychol. Soc. JINS 2012, 18, 490. [Google Scholar] [CrossRef] [PubMed]
- Winer, J.R.; Mander, B.A.; Helfrich, R.F.; Maass, A.; Harrison, T.M.; Baker, S.L.; Knight, R.T.; Jagust, W.J.; Walker, M.P. Sleep as a potential biomarker of tau and β-amyloid burden in the human brain. J. Neurosci. 2019, 39, 6315–6324. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Kostrikov, S.; Mehta, R.I.; Nedergaard, M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin. Sci. 2017, 131, 2257–2274. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef]
- Achariyar, T.M.; Li, B.; Peng, W.; Verghese, P.B.; Shi, Y.; McConnell, E.; Benraiss, A.; Kasper, T.; Song, W.; Takano, T.; et al. Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol. Neurodegener. 2016, 11, 1–20. [Google Scholar] [CrossRef]
- Hipólide, D.C.; Moreira, K.M.; Barlow, K.B.; Wilson, A.A.; Nobrega, J.N.; Tufik, S. Distinct effects of sleep deprivation on binding to norepinephrine and serotonin transporters in rat brain. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 297–303. [Google Scholar] [CrossRef]
- Irwin, M.; Thompson, J.; Miller, C.; Gillin, J.C.; Ziegler, M. Effects of sleep and sleep deprivation on catecholamine and interleukin-2 levels in humans: Clinical implications. J. Clin. Endocrinol. Metab. 1999, 84, 1979–1985. [Google Scholar] [CrossRef]
- Kato, M.; Phillips, B.G.; Sigurdsson, G.; Narkiewicz, K.; Pesek, C.A.; Somers, V.K. Effects of sleep deprivation on neural circulatory control. Hypertension 2000, 35, 1173–1175. [Google Scholar] [CrossRef]
- O’Donnell, J.; Zeppenfeld, D.; McConnell, E.; Pena, S.; Nedergaard, M. Norepinephrine: A neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem. Res. 2012, 37, 2496–2512. [Google Scholar] [CrossRef]
- Cipolla, M.J.; Li, R.; Vitullo, L. Perivascular innervation of penetrating brain parenchymal arterioles. J. Cardiovasc. Pharmacol. 2004, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Xie, L.; Yu, M.; Kang, H.; Feng, T.; Deane, R.; Logan, J.; Nedergaard, M.; Benveniste, H. The effect of body posture on brain glymphatic transport. J. Neurosci. 2015, 35, 11034–11044. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Early network dysfunction in Alzheimer’s disease. Science 2019, 365, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Yamada, K.A.; Finn, M.B.; Sloviter, R.S.; Bales, K.R.; May, P.C.; Schoepp, D.D.; Paul, S.M.; Mennerick, S.; Holtzman, D.M. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 2005, 48, 913–922. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Kang, J.E.; Lee, J.; Stewart, F.R.; Verges, D.K.; Silverio, L.M.; Bu, G.; Mennerick, S.; Holtzman, D.M. Endocytosis is required for synaptic activity-dependent release of amyloid-β in vivo. Neuron 2008, 58, 42–51. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tanei, Z.i.; Hashimoto, T.; Wakabayashi, T.; Okuno, H.; Naka, Y.; Yizhar, O.; Fenno, L.E.; Fukayama, M.; Bito, H.; et al. Chronic optogenetic activation augments Aβ pathology in a mouse model of Alzheimer disease. Cell Rep. 2015, 11, 859–865. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Newman, E.A. New roles for astrocytes: Regulation of synaptic transmission. Trends Neurosci. 2003, 26, 536–542. [Google Scholar] [CrossRef]
- Galea, E.; Morrison, W.; Hudry, E.; Arbel-Ornath, M.; Bacskai, B.J.; Gómez-Isla, T.; Stanley, H.E.; Hyman, B.T. Topological analyses in APP/PS1 mice reveal that astrocytes do not migrate to amyloid-β plaques. Proc. Natl. Acad. Sci. USA 2015, 112, 15556–15561. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef]
- Robinson, S.R. Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 2000, 36, 471–482. [Google Scholar] [CrossRef]
- Szabó, Z.; Héja, L.; Szalay, G.; Kékesi, O.; Füredi, A.; Szebényi, K.; Dobolyi, Á.; Orbán, T.I.; Kolacsek, O.; Tompa, T.; et al. Extensive astrocyte synchronization advances neuronal coupling in slow wave activity in vivo. Sci. Rep. 2017, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Poskanzer, K.E.; Yuste, R. Astrocytic regulation of cortical UP states. Proc. Natl. Acad. Sci. USA 2011, 108, 18453–18458. [Google Scholar] [CrossRef]
- Poskanzer, K.E.; Yuste, R. Astrocytes regulate cortical state switching in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, E2675–E2684. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.K.; Sharma, A.K.; Gupta, U. Blood brain barrier: An overview on strategies in drug delivery, realistic in vitro modeling and in vivo live tracking. Tissue Barriers 2016, 4, e1129476. [Google Scholar] [CrossRef] [PubMed]
- Semyachkina-Glushkovskaya, O.; Abdurashitov, A.; Dubrovsky, A.; Bragin, D.; Bragina, O.; Shushunova, N.; Maslyakova, G.; Navolokin, N.; Bucharskaya, A.; Tuchind, V.; et al. Application of optical coherence tomography for in vivo monitoring of the meningeal lymphatic vessels during opening of blood–brain barrier: Mechanisms of brain clearing. J. Biomed. Opt. 2017, 22, 121719. [Google Scholar] [CrossRef] [PubMed]
- Semyachkina-Glushkovskaya, O.; Chehonin, V.; Borisova, E.; Fedosov, I.; Namykin, A.; Abdurashitov, A.; Shirokov, A.; Khlebtsov, B.; Lyubun, Y.; Navolokin, N.; et al. Photodynamic opening of the blood-brain barrier and pathways of brain clearing. J. Biophotonics 2018, 11, e201700287. [Google Scholar] [CrossRef] [PubMed]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood–brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Jordão, J.F.; Thévenot, E.; Markham-Coultes, K.; Scarcelli, T.; Weng, Y.Q.; Xhima, K.; O’Reilly, M.; Huang, Y.; McLaurin, J.; Hynynen, K.; et al. Amyloid-β plaque reduction, endogenous antibody delivery and glial activation by brain-targeted, transcranial focused ultrasound. Exp. Neurol. 2013, 248, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Leinenga, G.; Götz, J. Scanning ultrasound removes amyloid-β and restores memory in an Alzheimer’s disease mouse model. Sci. Transl. Med. 2015, 7, 278ra33. [Google Scholar] [CrossRef]
- Burgess, A.; Dubey, S.; Yeung, S.; Hough, O.; Eterman, N.; Aubert, I.; Hynynen, K. Alzheimer disease in a mouse model: MR imaging–guided focused ultrasound targeted to the hippocampus opens the blood-brain barrier and improves pathologic abnormalities and behavior. Radiology 2014, 273, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Kiviniemi, V.; Korhonen, V.; Kortelainen, J.; Rytky, S.; Keinänen, T.; Tuovinen, T.; Isokangas, M.; Sonkajärvi, E.; Siniluoto, T.; Nikkinen, J.; et al. Real-time monitoring of human blood-brain barrier disruption. PLoS ONE 2017, 12, e0174072. [Google Scholar] [CrossRef]
- Pavlov, A.; Dubrovsky, A.; Koronovskii, A., Jr.; Pavlova, O.; Semyachkina-Glushkovskaya, O.; Kurths, J. Extended detrended fluctuation analysis of sound-induced changes in brain electrical activity. Chaos Solitons Fractals 2020, 139, 109989. [Google Scholar] [CrossRef]
- Pavlov, A.; Dubrovsky, A.; Koronovskii, A., Jr.; Pavlova, O.; Semyachkina-Glushkovskaya, O.; Kurths, J. Extended detrended fluctuation analysis of electroencephalograms signals during sleep and the opening of the blood–brain barrier. Chaos Interdiscip. J. Nonlinear Sci. 2020, 30, 073138. [Google Scholar] [CrossRef]
- Shuvaev, A.; Kuvacheva, N.; Morgun, A.; Khilazheva, E.; Salmina, A. The Role of Ion Channels Expressed in Cerebral Endothelial Cells in the Functional Integrity of the Blood-Brain Barrier (Review). Sovrem. Tehnol. V Med. 2016, 8, 241–250. [Google Scholar] [CrossRef]
- Callies, C.; Fels, J.; Liashkovich, I.; Kliche, K.; Jeggle, P.; Kusche-Vihrog, K.; Oberleithner, H. Membrane potential depolarization decreases the stiffness of vascular endothelial cells. J. Cell Sci. 2011, 124, 1936–1942. [Google Scholar] [CrossRef]
- Vanhatalo, S.; Voipio, J.; Kaila, K. Infraslow EEG activity. In Niedermeyer’s Electroencephalography: Basic Principles, Clinical Applications, and Related Fields; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011; pp. 741–747. [Google Scholar]
- Woody, C.; Marshall, W.; Besson, J.; Thompson, H.; Aleonard, P.; Albe-Fessard, D. Brain potential shift with respiratory acidosis in the cat and monkey. Am. J. Physiol. Leg. Content 1970, 218, 275–283. [Google Scholar] [CrossRef]
- Revest, P.A.; Jones, H.C.; Abbott, N.J. The transendothelial DC potential of rat blood-brain barrier vessels in situ. In Frontiers in Cerebral Vascular Biology; Springer: Berlin/Heidelberg, Germany, 1993; pp. 71–74. [Google Scholar]
- Revest, P.A.; Jones, H.C.; Abbott, N.J. Transendothelial electrical potential across pial vessels in anaesthetised rats: A study of ion permeability and transport at the blood-brain barrier. Brain Res. 1994, 652, 76–82. [Google Scholar] [CrossRef]
- Monto, S.; Palva, S.; Voipio, J.; Palva, J.M. Very slow EEG fluctuations predict the dynamics of stimulus detection and oscillation amplitudes in humans. J. Neurosci. 2008, 28, 8268–8272. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, T.; Kantola, J.; Abou Elseoud, A.; Lepola, P.; Suominen, K.; Starck, T.; Nikkinen, J.; Remes, J.; Tervonen, O.; Palva, S.; et al. Infra-slow EEG fluctuations are correlated with resting-state network dynamics in fMRI. J. Neurosci. 2014, 34, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Brockett, A.T.; Kane, G.A.; Monari, P.K.; Briones, B.A.; Vigneron, P.A.; Barber, G.A.; Bermudez, A.; Dieffenbach, U.; Kloth, A.D.; Buschman, T.J.; et al. Evidence supporting a role for astrocytes in the regulation of cognitive flexibility and neuronal oscillations through the Ca2+ binding protein S100β. PLoS ONE 2018, 13, e0195726. [Google Scholar] [CrossRef] [PubMed]
- Bellot-Saez, A.; Cohen, G.; van Schaik, A.; Ooi, L.; Morley, J.W.; Buskila, Y. Astrocytic modulation of cortical oscillations. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Henneberger, C.; Papouin, T.; Oliet, S.H.; Rusakov, D.A. Long-term potentiation depends on release of D-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef]
- Takata, N.; Mishima, T.; Hisatsune, C.; Nagai, T.; Ebisui, E.; Mikoshiba, K.; Hirase, H. Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. 2011, 31, 18155–18165. [Google Scholar] [CrossRef]
- Bellesi, M.; de Vivo, L.; Tononi, G.; Cirelli, C. Effects of sleep and wake on astrocytes: Clues from molecular and ultrastructural studies. BMC Biol. 2015, 13, 66. [Google Scholar] [CrossRef]
- Chang, J.; Wang, R.; Li, C.; Wang, Y.; Chu, X.P. Transcranial Low-Level Laser Therapy for Depression and Alzheimer’s Disease. Neuropsychiatry 2018, 8, 477–483. [Google Scholar] [CrossRef]
- Ding, F.; O’Donnell, J.; Xu, Q.; Kang, N.; Goldman, N.; Nedergaard, M. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science 2016, 352, 550–555. [Google Scholar] [CrossRef]
- Pasantes-Morales, H.; Tuz, K. Volume changes in neurons: Hyperexcitability and neuronal death. In Mechanisms and Significance of Cell Volume Regulation; Karger Publishers: Berlin, Germany, 2006; Volume 152, pp. 221–240. [Google Scholar]
- Hübel, N.; Ullah, G. Anions govern cell volume: A case study of relative astrocytic and neuronal swelling in spreading depolarization. PLoS ONE 2016, 11, e0147060. [Google Scholar] [CrossRef]
- Florence, C.M.; Baillie, L.D.; Mulligan, S.J. Dynamic volume changes in astrocytes are an intrinsic phenomenon mediated by bicarbonate ion flux. PLoS ONE 2012, 7, e51124. [Google Scholar] [CrossRef] [PubMed]
- Fellin, T.; Ellenbogen, J.M.; De Pittà, M.; Ben-Jacob, E.; Halassa, M.M. Astrocyte regulation of sleep circuits: Experimental and modeling perspectives. Front. Comput. Neurosci. 2012, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Florian, C.; Fellin, T.; Munoz, J.R.; Lee, S.Y.; Abel, T.; Haydon, P.G.; Frank, M.G. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 2009, 61, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Krueger, M.; Härtig, W.; Reichenbach, A.; Bechmann, I.; Michalski, D. Blood-brain barrier breakdown after embolic stroke in rats occurs without ultrastructural evidence for disrupting tight junctions. PLoS ONE 2013, 8, e56419. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, L.; An, C.; Wang, R.; Yang, L.; Yu, W.; Li, P.; Gao, Y. The blood brain barrier in cerebral ischemic injury–Disruption and repair. Brain Hemorrhages 2020, 1, 34–53. [Google Scholar] [CrossRef]
- Dunkel, P.; Chai, C.L.; Sperlagh, B.; Huleatt, P.B.; Matyus, P. Clinical utility of neuroprotective agents in neurodegenerative diseases: Current status of drug development for Alzheimer’s, Parkinson’s and Huntington’s diseases, and amyotrophic lateral sclerosis. Expert Opin. Investig. Drugs 2012, 21, 1267–1308. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment combinations for Alzheimer’s disease: Current and future pharmacotherapy options. J. Alzheimer Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef]
- Van Dyck, C.H. Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: Pitfalls and promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef]
- Zhang, Y.; Gruber, R. Focus: Attention Science: Can Slow-Wave Sleep Enhancement Improve Memory? A Review of Current Approaches and Cognitive Outcomes. Yale J. Biol. Med. 2019, 92, 63. [Google Scholar]
- Garcia-Molina, G.; Tsoneva, T.; Jasko, J.; Steele, B.; Aquino, A.; Baher, K.; Pastoor, S.; Pfundtner, S.; Ostrowski, L.; Miller, B.; et al. Closed-loop system to enhance slow-wave activity. J. Neural Eng. 2018, 15, 066018. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, M.; Riedner, B.A.; Garcia-Molina, G.N.; Cirelli, C.; Tononi, G. Enhancement of sleep slow waves: Underlying mechanisms and practical consequences. Front. Syst. Neurosci. 2014, 8, 208. [Google Scholar] [CrossRef] [PubMed]
- Tononi, G.; Riedner, B.; Hulse, B.; Ferrarelli, F.; Sarasso, S. Enhancing sleep slow waves with natural stimuli. Medicamundi 2010, 54, 73–79. [Google Scholar]
- Ngo, H.V.V.; Martinetz, T.; Born, J.; Mölle, M. Auditory closed-loop stimulation of the sleep slow oscillation enhances memory. Neuron 2013, 78, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Papalambros, N.A.; Santostasi, G.; Malkani, R.G.; Braun, R.; Weintraub, S.; Paller, K.A.; Zee, P.C. Acoustic enhancement of sleep slow oscillations and concomitant memory improvement in older adults. Front. Hum. Neurosci. 2017, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- Weigenand, A.; Mölle, M.; Werner, F.; Martinetz, T.; Marshall, L. Timing matters: Open-loop stimulation does not improve overnight consolidation of word pairs in humans. Eur. J. Neurosci. 2016, 44, 2357–2368. [Google Scholar] [CrossRef]
- Leminen, M.M.; Virkkala, J.; Saure, E.; Paajanen, T.; Zee, P.C.; Santostasi, G.; Hublin, C.; Müller, K.; Porkka-Heiskanen, T.; Huotilainen, M.; et al. Enhanced memory consolidation via automatic sound stimulation during non-REM sleep. Sleep 2017, 40, zsx003. [Google Scholar] [CrossRef]
- Santostasi, G.; Malkani, R.; Riedner, B.; Bellesi, M.; Tononi, G.; Paller, K.A.; Zee, P.C. Phase-locked loop for precisely timed acoustic stimulation during sleep. J. Neurosci. Methods 2016, 259, 101–114. [Google Scholar] [CrossRef]
- Ong, J.L.; Lo, J.C.; Chee, N.I.; Santostasi, G.; Paller, K.A.; Zee, P.C.; Chee, M.W. Effects of phase-locked acoustic stimulation during a nap on EEG spectra and declarative memory consolidation. Sleep Med. 2016, 20, 88–97. [Google Scholar] [CrossRef]
- Ong, J.L.; Patanaik, A.; Chee, N.I.; Lee, X.K.; Poh, J.H.; Chee, M.W. Auditory stimulation of sleep slow oscillations modulates subsequent memory encoding through altered hippocampal function. Sleep 2018, 41, zsy031. [Google Scholar] [CrossRef]
- Moruzzi, G. The Physiological Properties of the Brain Stem Reticular System; Blackwell: Oxford, UK, 1954. [Google Scholar]
- Berlucchi, G. One or many arousal systems? Reflections on some of Giuseppe Moruzzi’s foresights and insights about the intrinsic regulation of brain activity. Arch. Ital. Biol. 1997, 135, 5–14. [Google Scholar] [PubMed]
- Marshall, L.; Mölle, M.; Hallschmid, M.; Born, J. Transcranial direct current stimulation during sleep improves declarative memory. J. Neurosci. 2004, 24, 9985–9992. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.; Helgadóttir, H.; Mölle, M.; Born, J. Boosting slow oscillations during sleep potentiates memory. Nature 2006, 444, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Dorokhov, V.B.; Taranov, A.I.; Narbut, A.M.; Sakharov, D.S.; Gruzdeva, S.S.; Tkachenko, O.N.; Arsen’ev, G.N.; Blochin, I.S.; Putilov, A.A. Effects of Exposure to a Weak Extremely Low Frequency Electromagnetic Field on Daytime Sleep Architecture and Length. Sleep Med. Res. 2019, 10, 97–102. [Google Scholar] [CrossRef][Green Version]
- Eggert, T.; Dorn, H.; Sauter, C.; Nitsche, M.A.; Bajbouj, M.; Danker-Hopfe, H. No effects of slow oscillatory transcranial direct current stimulation (tDCS) on sleep-dependent memory consolidation in healthy elderly subjects. Brain Stimul. 2013, 6, 938–945. [Google Scholar] [CrossRef]
- Reato, D.; Gasca, F.; Datta, A.; Bikson, M.; Marshall, L.; Parra, L.C. Transcranial electrical stimulation accelerates human sleep homeostasis. PLoS Comput. Biol. 2013, 9, e1002898. [Google Scholar] [CrossRef]
- Massimini, M.; Ferrarelli, F.; Esser, S.K.; Riedner, B.A.; Huber, R.; Murphy, M.; Peterson, M.J.; Tononi, G. Triggering sleep slow waves by transcranial magnetic stimulation. Proc. Natl. Acad. Sci. USA 2007, 104, 8496–8501. [Google Scholar] [CrossRef]
- Lang, N.; Siebner, H.R.; Ward, N.S.; Lee, L.; Nitsche, M.A.; Paulus, W.; Rothwell, J.C.; Lemon, R.N.; Frackowiak, R.S. How does transcranial DC stimulation of the primary motor cortex alter regional neuronal activity in the human brain? Eur. J. Neurosci. 2005, 22, 495–504. [Google Scholar] [CrossRef]
- Walsh, J.K.; Randazzo, A.C.; Stone, K.; Eisenstein, R.; Feren, S.D.; Kajy, S.; Dickey, P.; Roehrs, T.; Roth, T.; Schweitzer, P.K. Tiagabine is associated with sustained attention during sleep restriction: Evidence for the value of slow-wave sleep enhancement? Sleep 2006, 29, 433. [Google Scholar]
- Feld, G.B.; Wilhelm, I.; Ma, Y.; Groch, S.; Binkofski, F.; Mölle, M.; Born, J. Slow wave sleep induced by GABA agonist tiagabine fails to benefit memory consolidation. Sleep 2013, 36, 1317–1326. [Google Scholar] [CrossRef]
- Walsh, J.K.; Snyder, E.; Hall, J.; Randazzo, A.C.; Griffin, K.; Groeger, J.; Eisenstein, R.; Feren, S.D.; Dickey, P.; Schweitzer, P.K. Slow wave sleep enhancement with gaboxadol reduces daytime sleepiness during sleep restriction. Sleep 2008, 31, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.M. Memory Encoding and Sleep-Dependent Consolidation during Sleep Restriction with and without Pharmacologically Enhanced Slow Wave Sleep. Ph.D. Thesis, Saint Louis University, St. Louis, MO, USA, 2009. [Google Scholar]
- Walsh, J.K.; Hall-Porter, J.M.; Griffin, K.S.; Dodson, E.R.; Forst, E.H.; Curry, D.T.; Eisenstein, R.D.; Schweitzer, P.K. Enhancing slow wave sleep with sodium oxybate reduces the behavioral and physiological impact of sleep loss. Sleep 2010, 33, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Vienne, J.; Lecciso, G.; Constantinescu, I.; Schwartz, S.; Franken, P.; Heinzer, R.; Tafti, M. Differential effects of sodium oxybate and baclofen on EEG, sleep, neurobehavioral performance, and memory. Sleep 2012, 35, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Göder, R.; Fritzer, G.; Gottwald, B.; Lippmann, B.; Seeck-Hirschner, M.; Serafin, I.; Aldenhoff, J. Effects of olanzapine on slow wave sleep, sleep spindles and sleep-related memory consolidation in schizophrenia. Pharmacopsychiatry 2008, 41, 92–99. [Google Scholar] [CrossRef]
- Lazowski, L.K.; Townsend, B.; Hawken, E.R.; Jokic, R.; du Toit, R.; Milev, R. Sleep architecture and cognitive changes in olanzapine-treated patients with depression: A double blind randomized placebo controlled trial. BMC Psychiatry 2014, 14, 202. [Google Scholar] [CrossRef]
- Benedict, C.; Scheller, J.; Rose-John, S.; Born, J.; Marshall, L. Enhancing influence of intranasal interleukin-6 on slow-wave activity and memory consolidation during sleep. FASEB J. 2009, 23, 3629–3636. [Google Scholar] [CrossRef]
- Gottesmann, C. GABA mechanisms and sleep. Neuroscience 2002, 111, 231–239. [Google Scholar] [CrossRef]
- Sharpley, A.L.; Vassallo, C.M.; Cowen, P.J. Olanzapine increases slow-wave sleep: Evidence for blockade of central 5-HT2C receptors in vivo. Biol. Psychiatry 2000, 47, 468–470. [Google Scholar] [CrossRef]
- Harvey, N.L. The link between lymphatic function and adipose biology. Ann. N. Y. Acad. Sci. 2008, 1131, 82–88. [Google Scholar] [CrossRef]
- Mathias, S.; Wetter, T.C.; Steiger, A.; Lancel, M. The GABA uptake inhibitor tiagabine promotes slow wave sleep in normal elderly subjects. Neurobiol. Aging 2001, 22, 247–253. [Google Scholar] [CrossRef]
- Lancel, M.; Wetter, T.C.; Steiger, A.; Mathias, S. Effect of the GABAA agonist gaboxadol on nocturnal sleep and hormone secretion in healthy elderly subjects. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E130–E137. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, O.; Montpiaisir, J.; Lamarre, M.; Bedard, M. The effect of gamma-hydroxybutyrate on nocturnal and diurnal sleep of normal subjects: Further considerations on REM sleep-triggering mechanisms. Sleep 1990, 13, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Spath-Schwalbe, E.; Hansen, K.; Schmidt, F.; Schrezenmeier, H.; Marshall, L.; Burger, K.; Fehm, H.L.; Born, J. Acute effects of recombinant human interleukin-6 on endocrine and central nervous sleep functions in healthy men. J. Clin. Endocrinol. Metab. 1998, 83, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, H.; Lee, H.; Ding, F.; Sun, Q.; Al-Bizri, E.; Makaryus, R.; Probst, S.; Nedergaard, M.; Stein, E.A.; Lu, H. Anesthesia with dexmedetomidine and low-dose isoflurane increases solute transport via the glymphatic pathway in rat brain when compared with high-dose isoflurane. Anesthesiol. J. Am. Soc. Anesthesiol. 2017, 127, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Musizza, B.; Stefanovska, A.; McClintock, P.V.; Paluš, M.; Petrovčič, J.; Ribarič, S.; Bajrović, F.F. Interactions between cardiac, respiratory and EEG-δ oscillations in rats during anaesthesia. J. Physiol. 2007, 580, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Gerashchenko, D.; Wisor, J.P.; Burns, D.; Reh, R.K.; Shiromani, P.J.; Sakurai, T.; Horacio, O.; Kilduff, T.S. Identification of a population of sleep-active cerebral cortex neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 10227–10232. [Google Scholar] [CrossRef]
- Gerashchenko, D.; Schmidt, M.A.; Zielinski, M.R.; Moore, M.E.; Wisor, J.P. Sleep state dependence of optogenetically evoked responses in neuronal nitric oxide synthase-positive cells of the cerebral cortex. Neuroscience 2018, 379, 189–201. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).