Modulated Electro-Hyperthermia-Induced Tumor Damage Mechanisms Revealed in Cancer Models

 ,
,  , ,
, ,
Abstract
1. Introduction
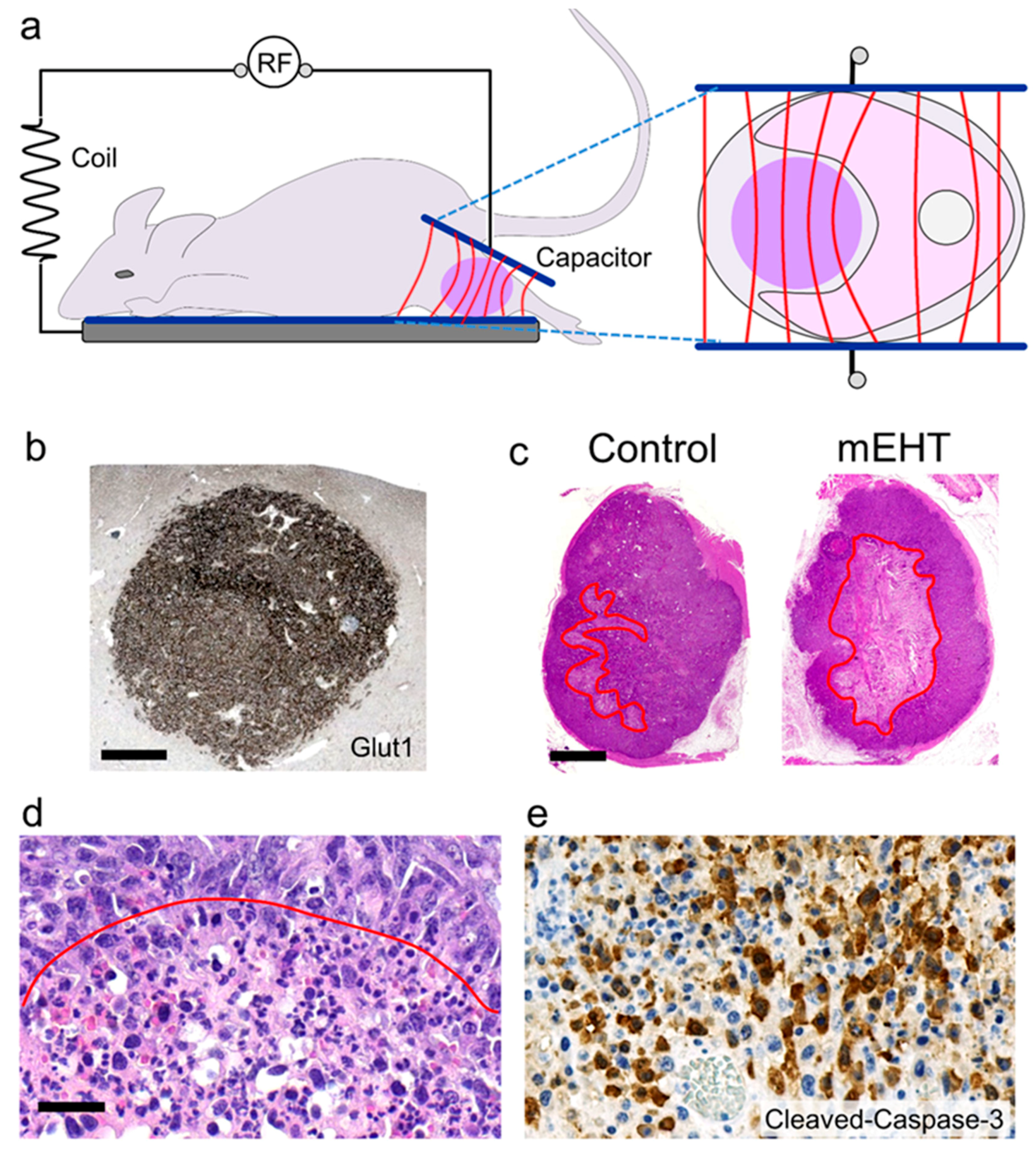
2. Standardized mEHT Treatment and Protocols
Methods Used for Demonstration Images
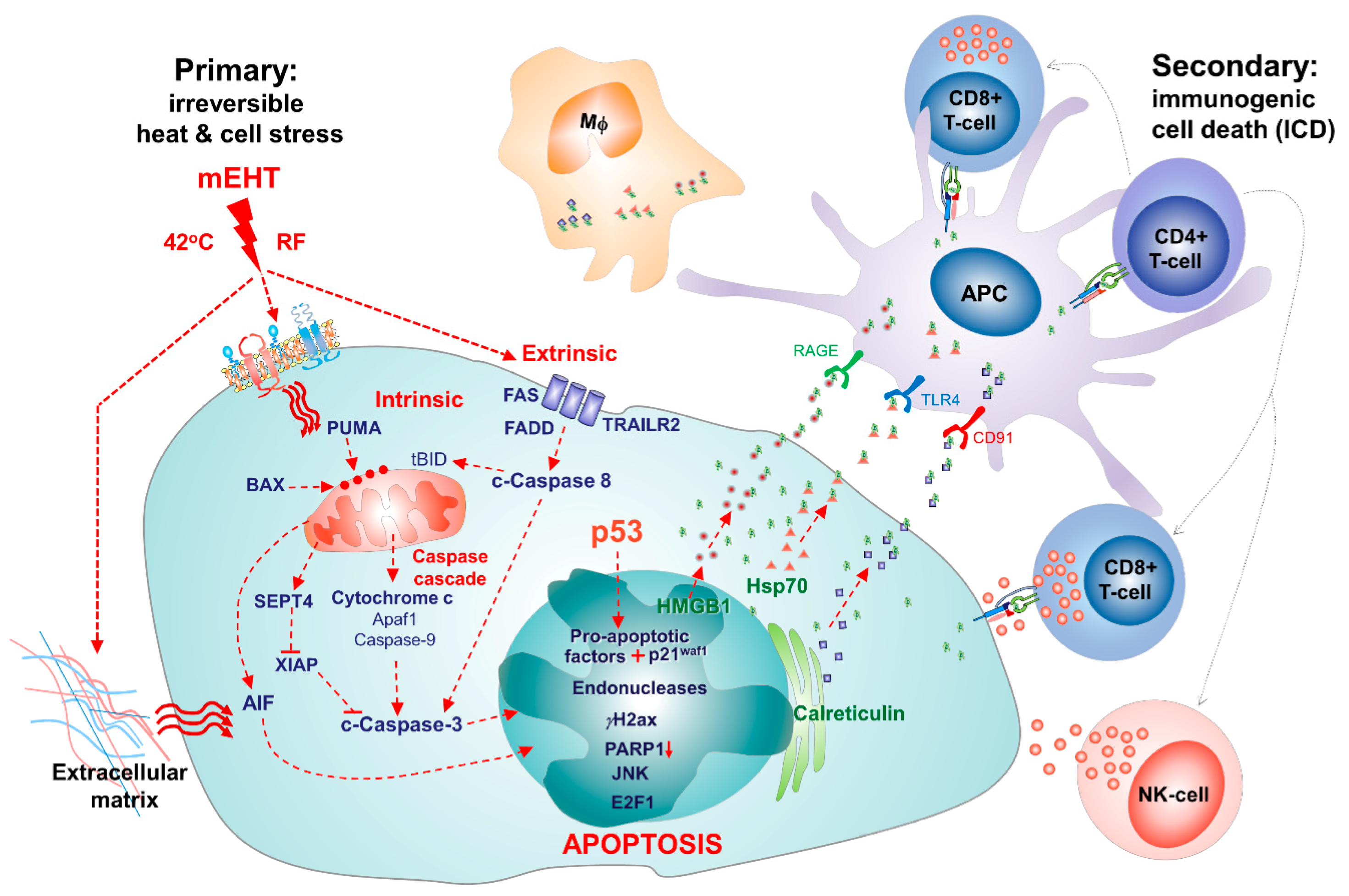
3. mEHT-Induced Apoptosis
3.1. Caspase-Dependent Apoptosis
3.2. Caspase-Independent Apoptosis
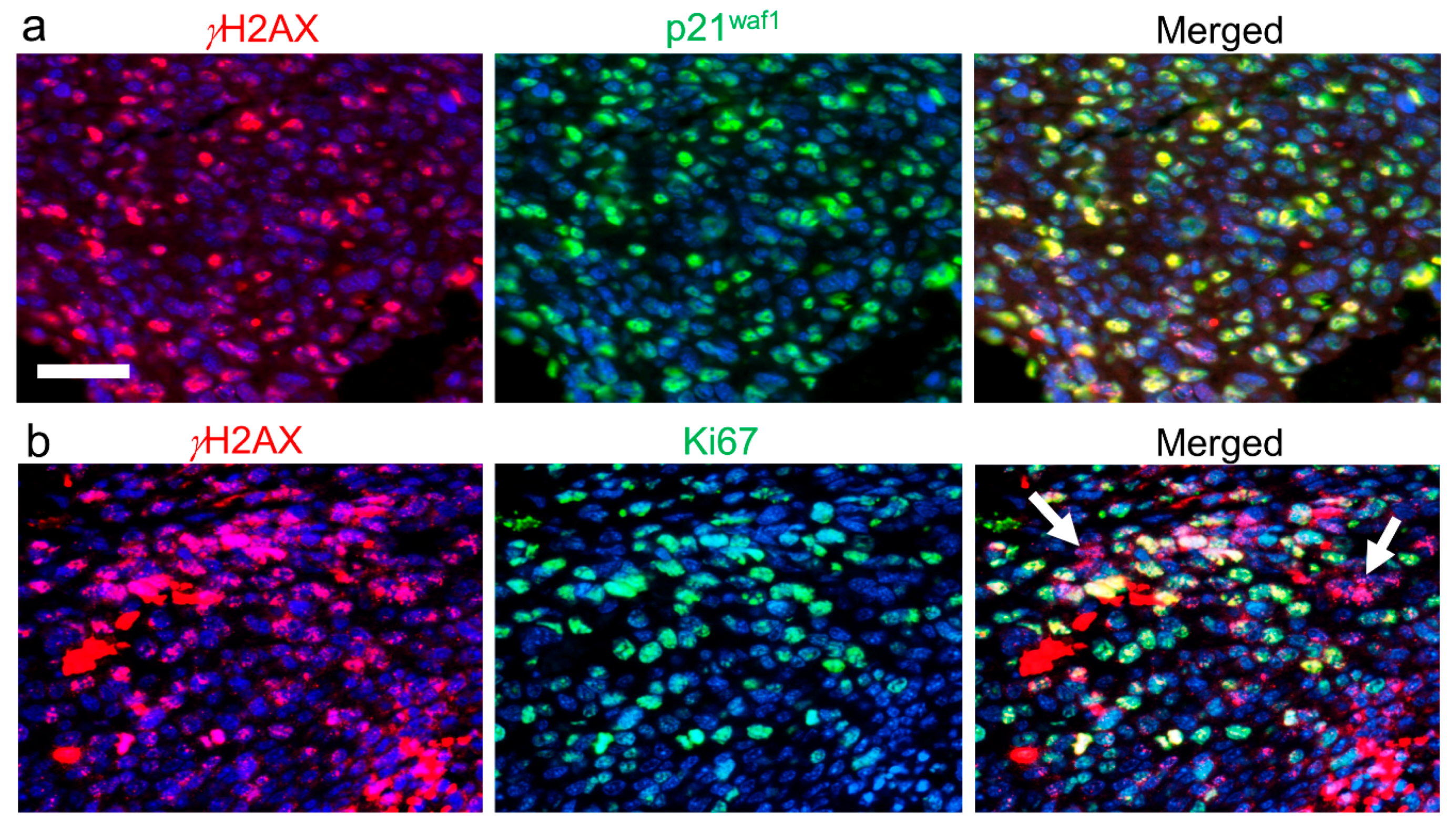
4. mEHT-Induced DNA Double-Strand Breaks and p21waf1-Mediated Senescence
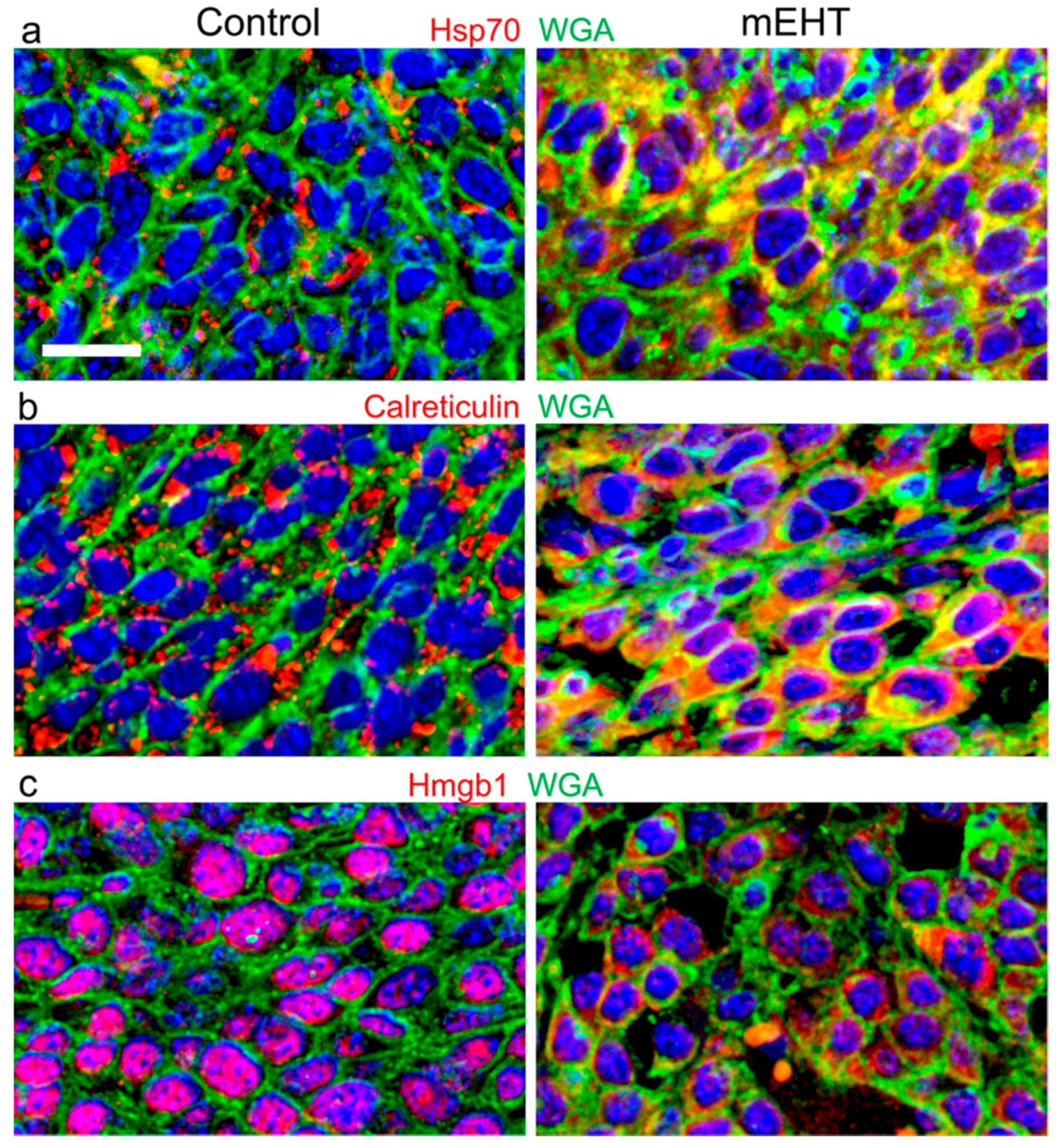
5. mEHT-Induced Cell Stress, Chaperones, and Damage Signaling
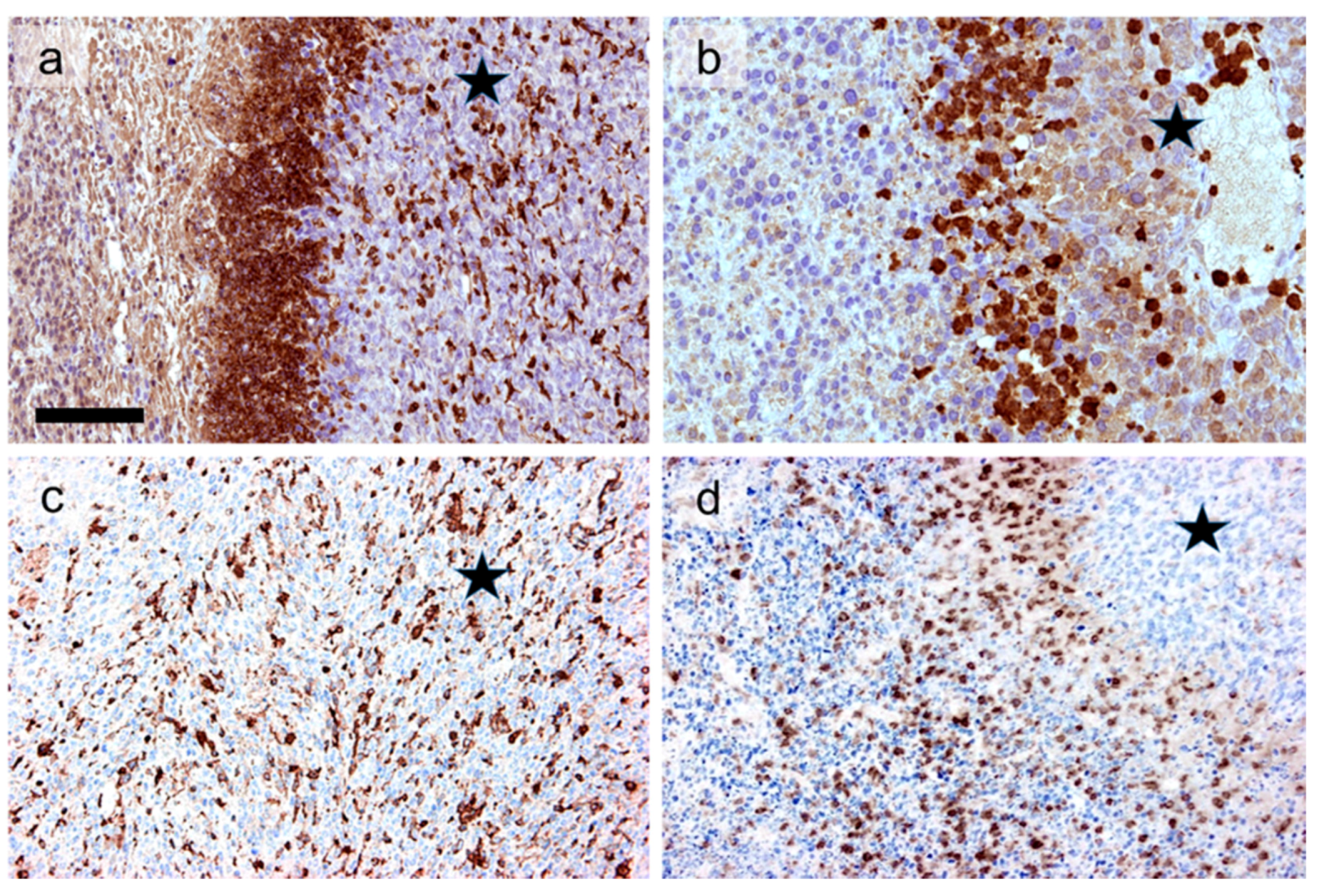
6. Contribution to Systemic Effect and Secondary Immunogenic Cell Death
7. Combination with Dendritic Cell Immunotherapy
8. Combination with Radiotherapy
9. Combination with Chemotherapy and Cancer Thermo-Sensitization
10. Clinical Utilization of mEHT for Upgrading Human Oncotherapy
11. Modulated Electro-hyperthermia Induced Tumor Damage Mechanisms Revealed in Cancer Models
12. Limitations of the Presented Studies
13. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Datta, N.R.; Ordonez, S.G.; Gaipl, U.S.; Paulides, M.M.; Crezee, H.; Gellermann, J.; Marder, D.; Puric, E.; Bodis, S. Local hyperthermia combined with radiotherapy and-/or chemotherapy: Recent advances and promises for the future. Cancer Treat Rev. 2015, 41, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, M.D. Hyperthermia and immunotherapy: Clinical opportunities. Int. J. Hyperth. 2019, 36, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Gas, P.; Wyszkowska, J. Influence of multi-tine electrode configuration in realistic hepatic RF ablative heating. Arch. Elect. Eng. 2019, 68, 521–533. [Google Scholar]
- Gas, P. Essential facts on the history of hyperthermia and their connections with electromedicine. Prz. Elektrotech. 2011, 87, 37–40. [Google Scholar]
- Datta, N.R.; Puric, E.; Klingbiel, D.; Gomez, S.; Bodis, S. Hyperthermia and Radiation Therapy in Locoregional Recurrent Breast Cancers: A Systematic Review and Meta-analysis. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 1073–1087. [Google Scholar] [CrossRef]
- Datta, N.R.; Rogers, S.; Klingbiel, D.; Gomez, S.; Puric, E.; Bodis, S. Hyperthermia and radiotherapy with or without chemotherapy in locally advanced cervical cancer: A systematic review with conventional and network meta-analyses. Int. J. Hyperth. 2016, 32, 809–821. [Google Scholar] [CrossRef]
- Datta, N.R.; Rogers, S.; Ordonez, S.G.; Puric, E.; Bodis, S. Hyperthermia and radiotherapy in the management of head and neck cancers: A systematic review and meta-analysis. Int. J. Hyperth. 2016, 32, 31–40. [Google Scholar] [CrossRef]
- Issels, R.D.; Lindner, L.H.; Verweij, J.; Wust, P.; Reichardt, P.; Schem, B.C.; Abdel-Rahman, S.; Daugaard, S.; Salat, C.; Wendtner, C.M.; et al. Neo-adjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft-tissue sarcoma: A randomised phase 3 multicentre study. Lancet Oncol. 2010, 11, 561–570. [Google Scholar] [CrossRef]
- Evans, S.S.; Repasky, E.A.; Fisher, D.T. Fever and the thermal regulation of immunity: The immune system feels the heat. Nat. Rev. Immunol. 2015, 15, 335–349. [Google Scholar] [CrossRef]
- Szasz, A.M.; Minnaar, C.A.; Szentmartoni, G.; Szigeti, G.P.; Dank, M. Review of the Clinical Evidences of Modulated Electro-Hyperthermia (mEHT) Method: An Update for the Practicing Oncologist. Front. Oncol. 2019, 9, 1012. [Google Scholar] [CrossRef]
- Meggyeshazi, N.; Andocs, G.; Balogh, L.; Balla, P.; Kiszner, G.; Teleki, I.; Jeney, A.; Krenacs, T. DNA fragmentation and caspase-independent programmed cell death by modulated electrohyperthermia. Strahlenther. Onkol. 2014, 190, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Vancsik, T.; Kovago, C.; Kiss, E.; Papp, E.; Forika, G.; Benyo, Z.; Meggyeshazi, N.; Krenacs, T. Modulated electro-hyperthermia induced loco-regional and systemic tumor destruction in colorectal cancer allografts. J. Cancer 2018, 9, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.P.; Devilee, P. The Warburg effect in 2012. Curr. Opin. Oncol. 2012, 24, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Kinahan, P.E.; Fletcher, J.W. Positron emission tomography-computed tomography standardized uptake values in clinical practice and assessing response to therapy. Semin. Ultrasound CT MRI 2010, 31, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Andocs, G.; Szasz, O.; Szasz, A. Oncothermia treatment of cancer: From the laboratory to clinic. Electromagn. Biol. Med. 2009, 28, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, T.; Batta, G.; Zakany, F.; Szollosi, J.; Nagy, P. The dipole potential correlates with lipid raft markers in the plasma membrane of living cells. J. Lipid Res. 2017, 58, 1681–1691. [Google Scholar] [CrossRef]
- Andocs, G.; Rehman, M.U.; Zhao, Q.L.; Tabuchi, Y.; Kanamori, M.; Kondo, T. Comparison of biological effects of modulated electro-hyperthermia and conventional heat treatment in human lymphoma U937 cells. Cell Death Discov. 2016, 2, 16039. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Walczak, H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect. Biol. 2013, 5, a008698. [Google Scholar] [CrossRef]
- Dart, C. Lipid microdomains and the regulation of ion channel function. J. Physiol. 2010, 588, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta 2014, 1838, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Andocs, G.; Renner, H.; Balogh, L.; Fonyad, L.; Jakab, C.; Szasz, A. Strong synergy of heat and modulated electromagnetic field in tumor cell killing. Strahlenther. Onkol. 2009, 185, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.L.; Huang, C.C.; Chi, M.S.; Chiang, H.C.; Wang, Y.S.; Hsia, C.C.; Andocs, G.; Wang, H.E.; Chi, K.H. In vitro comparison of conventional hyperthermia and modulated electro-hyperthermia. Oncotarget 2016, 7, 84082–84092. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zeng, G. Cancer and innate immune system interactions: Translational potentials for cancer immunotherapy. J. Immunother. 2012, 35, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Expasy-Cellosaurus. Available online: https://web.expasy.org/cellosaurus. (accessed on 26 August 2020).
- Wust, P.; Ghadjar, P.; Nadobny, J.; Beck, M.; Kaul, D.; Winter, L.; Zschaeck, S. Physical analysis of temperature-dependent effects of amplitude-modulated electromagnetic hyperthermia. Int. J. Hyperth. 2019, 36, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Szasz, A.; Szasz, N.; Szasz, O. Oncothermia: Principles and Practices; Springer Ltd.: Heidelberg, Germany, 2011. [Google Scholar]
- Vancsik, T.; Forika, G.; Balogh, A.; Kiss, E.; Krenacs, T. Modulated electro-hyperthermia induced p53 driven apoptosis and cell cycle arrest additively support doxorubicin chemotherapy of colorectal cancer in vitro. Cancer Med. 2019, 8, 4292–4303. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Nygaard, T.G.; Burlett, M. Response of cells to hyperthermia under acute and chronic hypoxic conditions. Cancer Res. 1979, 39, 966–972. [Google Scholar]
- Kalamida, D.; Karagounis, I.V.; Mitrakas, A.; Kalamida, S.; Giatromanolaki, A.; Koukourakis, M.I. Fever-range hyperthermia vs. hypothermia effect on cancer cell viability, proliferation and HSP90 expression. PLoS ONE 2015, 10, e0116021. [Google Scholar] [CrossRef]
- Meggyesházi, N.; Andócs, G.; Spisák, S.; Krenács, T. Early Changes in mRNA and Protein Expression Related to Cancer Treatment by Modulated Electrohyperthermia. Conf. Pap. Med. 2013. [Google Scholar] [CrossRef]
- Qin, W.; Akutsu, Y.; Andocs, G.; Suganami, A.; Hu, X.; Yusup, G.; Komatsu-Akimoto, A.; Hoshino, I.; Hanari, N.; Mori, M.; et al. Modulated electro-hyperthermia enhances dendritic cell therapy through an abscopal effect in mice. Oncol. Rep. 2014, 32, 2373–2379. [Google Scholar] [CrossRef] [PubMed]
- Andocs, G.; Meggyeshazi, N.; Balogh, L.; Spisak, S.; Maros, M.E.; Balla, P.; Kiszner, G.; Teleki, I.; Kovago, C.; Krenacs, T. Upregulation of heat shock proteins and the promotion of damage-associated molecular pattern signals in a colorectal cancer model by modulated electrohyperthermia. Cell Stress Chaperones 2015, 20, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Andocs, G.; Rehman, M.U.; Zhao, Q.L.; Papp, E.; Kondo, T.; Szasz, A. Nanoheating without Artificial Nanoparticles Part II. Experimental Support of the Nanoheating Concept of the Modulated Electro-Hyperthermia Method, Using U937 Cell Suspension Model. Biol. Med. 2015, 7. [Google Scholar] [CrossRef]
- Tsang, Y.W.; Huang, C.C.; Yang, K.L.; Chi, M.S.; Chiang, H.C.; Wang, Y.S.; Andocs, G.; Szasz, A.; Li, W.T.; Chi, K.H. Improving immunological tumor microenvironment using electro-hyperthermia followed by dendritic cell immunotherapy. BMC Cancer 2015, 15, 708. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Jeon, T.W.; Lee, C.G.; Oh, S.T.; Yang, H.B.; Choi, K.J.; Seo, D.; Yun, I.; Baik, I.H.; Park, K.R.; et al. Electro-hyperthermia inhibits glioma tumorigenicity through the induction of E2F1-mediated apoptosis. Int. J. Hyperth. 2015, 31, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Jeon, T.W.; Yang, H.; Lee, C.G.; Oh, S.T.; Seo, D.; Baik, I.H.; Lee, E.H.; Yun, I.; Park, K.R.; Lee, Y.H. Electro-hyperthermia up-regulates tumour suppressor Septin 4 to induce apoptotic cell death in hepatocellular carcinoma. Int. J. Hyperth. 2016, 32, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, M.S.; Kim, H.J.; Lee, E.; Jeong, J.H.; Park, I.; Jeong, Y.K.; Jang, W.I. Role of HIF-1α in response of tumors to a combination of hyperthermia and radiation in vivo. Int. J. Hyperth. 2018, 34, 276–283. [Google Scholar] [CrossRef]
- Tsang, Y.W.; Chi, K.H.; Huang, C.C.; Chi, M.S.; Chiang, H.C.; Yang, K.L.; Li, W.T.; Wang, Y.S. Modulated electro-hyperthermia-enhanced liposomal drug uptake by cancer cells. Int. J. Nanomed. 2019, 14, 1269–1279. [Google Scholar] [CrossRef]
- Besztercei, B.; Vancsik, T.; Benedek, A.; Major, E.; Thomas, M.J.; Schvarcz, C.A.; Krenacs, T.; Benyo, Z.; Balogh, A. Stress-Induced, p53-Mediated Tumor Growth Inhibition of Melanoma by Modulated Electrohyperthermia in Mouse Models without Major Immunogenic Effects. Int. J. Mol. Sci. 2019, 20, 4019. [Google Scholar] [CrossRef]
- McDonald, M.; Corde, S.; Lerch, M.; Rosenfeld, A.; Jackson, M.; Tehei, M. First in vitro evidence of modulated electro-hyperthermia treatment performance in combination with megavoltage radiation by clonogenic assay. Sci. Rep. 2018, 8, 16608. [Google Scholar] [CrossRef]
- Yang, W.; Han, G.H.; Shin, H.Y.; Lee, E.J.; Cho, H.; Chay, D.B.; Kim, J.H. Combined treatment with modulated electro-hyperthermia and an autophagy inhibitor effectively inhibit ovarian and cervical cancer growth. Int. J. Hyperth. 2019, 36, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Kim, S.; Cho, W.; Kim, J.K.; Kim, Y.A.; Kim, S.; Wu, H.G. Quantitative Estimation of the Equivalent Radiation Dose Escalation using Radiofrequency Hyperthermia in Mouse Xenograft Models of Human Lung Cancer. Sci. Rep. 2019, 9, 3942. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.-J.; Kim, H.-J.; Hong, S.-T. Iron-dextran as a thermosensitizer in radiofrequency hyperthermia for cancer treatment. Appl. Biol. Chem. 2019, 62, 1–9. [Google Scholar] [CrossRef]
- Chen, C.C.; Chen, C.L.; Li, J.J.; Chen, Y.Y.; Wang, C.Y.; Wang, Y.S.; Chi, K.H.; Wang, H.E. Presence of Gold Nanoparticles in Cells Associated with the CellKilling Effect of Modulated Electro-Hyperthermia. Acs Appl. Bio. Mater. 2019, 2, 3573–3581. [Google Scholar] [CrossRef]
- Forika, G.; Balogh, A.; Vancsik, T.; Zalatnai, A.; Petovari, G.; Benyo, Z.; Krenacs, T. Modulated Electro-Hyperthermia Resolves Radioresistance of Panc1 Pancreas Adenocarcinoma and Promotes DNA Damage and Apoptosis In Vitro. Int. J. Mol. Sci. 2020, 21, 5100. [Google Scholar] [CrossRef]
- Wust, P.; Kortüm, B.; Strauss, U.; Nadobny, J.; Zschaeck, S.; Beck, M.; Stein, U.; Ghadjar, P. Non-thermal effects of radiofrequency electromagnetic fields. Sci. Rep. 2020, 10, 13488. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef]
- Castle, J.C.; Loewer, M.; Boegel, S.; de Graaf, J.; Bender, C.; Tadmor, A.D.; Boisguerin, V.; Bukur, T.; Sorn, P.; Paret, C.; et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genom. 2014, 15, 190. [Google Scholar] [CrossRef]
- Kwan, R.; Looi, K.S.; Omary, M.B. Absence of keratins 8 and 18 in rodent epithelial cell lines associates with keratin gene mutation and DNA methylation: Cell line selective effects on cell invasion. Exp. Cell Res. 2015, 335, 12–22. [Google Scholar] [CrossRef][Green Version]
- McIlroy, D.; Sakahira, H.; Talanian, R.V.; Nagata, S. Involvement of caspase 3-activated DNase in internucleosomal DNA cleavage induced by diverse apoptotic stimuli. Oncogene 1999, 18, 4401–4408. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Leal, J.F.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, N.; Zhang, H.; Durden, D.L.; Feng, Y.; Zhou, M. Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell 2009, 15, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- Kumari, A.; Iwasaki, T.; Pyndiah, S.; Cassimere, E.K.; Palani, C.D.; Sakamuro, D. Regulation of E2F1-induced apoptosis by poly(ADP-ribosyl)ation. Cell Death Differ. 2015, 22, 311–322. [Google Scholar] [CrossRef]
- Engelmann, D.; Pützer, B.M. The Dark Side of E2F1: In Transit beyond Apoptosis. Cancer Res. 2012, 72, 571–575. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Barras, D. BRAF Mutation in Colorectal Cancer: An Update. Biomark Cancer 2015, 7, 9–12. [Google Scholar] [CrossRef]
- Foster, R.; Griffin, S.; Grooby, S.; Feltell, R.; Christopherson, C.; Chang, M.; Sninsky, J.; Kwok, S.; Torrance, C. Multiple metabolic alterations exist in mutant PI3K cancers, but only glucose is essential as a nutrient source. PLoS ONE 2012, 7, e45061. [Google Scholar] [CrossRef]
- Baritaud, M.; Boujrad, H.; Lorenzo, H.K.; Krantic, S.; Susin, S.A. Histone H2AX: The missing link in AIF-mediated caspase-independent programmed necrosis. Cell Cycle 2010, 9, 3166–3173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Jurvansuu, J.; Beard, P. H2AX is required for cell cycle arrest via the p53/p21 pathway. Mol. Cell Biol. 2009, 29, 2828–2840. [Google Scholar] [CrossRef]
- Tabasso, A.F.S.; Jones, D.J.L.; Jones, G.D.D.; Macip, S. Radiotherapy-Induced Senescence and its Effects on Responses to Treatment. Clin. Oncol. R. Coll. Radiol. 2019, 31, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Guillon, J.; Petit, C.; Toutain, B.; Guette, C.; Lelievre, E.; Coqueret, O. Chemotherapy-induced senescence, an adaptive mechanism driving resistance and tumor heterogeneity. Cell Cycle 2019, 18, 2385–2397. [Google Scholar] [CrossRef]
- Harnicek, D.; Kampmann, E.; Lauber, K.; Hennel, R.; Cardoso Martins, A.S.; Guo, Y.; Belka, C.; Mortl, S.; Gallmeier, E.; Kanaar, R.; et al. Hyperthermia adds to trabectedin effectiveness and thermal enhancement is associated with BRCA2 degradation and impairment of DNA homologous recombination repair. Int. J. Cancer 2016, 139, 467–479. [Google Scholar] [CrossRef]
- Oei, A.L.; Vriend, L.E.; Crezee, J.; Franken, N.A.; Krawczyk, P.M. Effects of hyperthermia on DNA repair pathways: One treatment to inhibit them all. Radiat. Oncol. 2015, 10, 165. [Google Scholar] [CrossRef]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Shevtsov, M.; Huile, G.; Multhoff, G. Membrane heat shock protein 70: A theranostic target for cancer therapy. Philos. Trans. R. Soc. B Biol. Sci. 2018, 372. [Google Scholar] [CrossRef]
- Shevtsov, M.; Multhoff, G. Heat Shock Protein-Peptide and HSP-Based Immunotherapies for the Treatment of Cancer. Front. Immunol. 2016, 7, 171. [Google Scholar] [CrossRef]
- Gutierrez, T.; Simmen, T. Endoplasmic reticulum chaperones and oxidoreductases: Critical regulators of tumor cell survival and immunorecognition. Front. Oncol. 2014, 4, 291. [Google Scholar] [CrossRef]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Martin, S.; Golab, J.; Agostinis, P. Danger signalling during cancer cell death: Origins, plasticity and regulation. Cell Death Differ. 2014, 21, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L.; Tang, D.; Lotze, M.T. Life after death: Targeting high mobility group box 1 in emergent cancer therapies. Am. J. Cancer Res. 2013, 3, 1–20. [Google Scholar] [PubMed]
- Kang, T.Y.; Yang, H.R.; Zhang, J.; Li, D.; Lin, J.; Wang, L.; Xu, X. The studies of chlorogenic Acid antitumor mechanism by gene chip detection: The immune pathway gene expression. J. Anal. Methods Chem. 2013, 2013, 617243. [Google Scholar] [CrossRef] [PubMed]
- Ladoire, S.; Hannani, D.; Vetizou, M.; Locher, C.; Aymeric, L.; Apetoh, L.; Kepp, O.; Kroemer, G.; Ghiringhelli, F.; Zitvogel, L. Cell-death-associated molecular patterns as determinants of cancer immunogenicity. Antioxid. Redox Signal. 2014, 20, 1098–1116. [Google Scholar] [CrossRef]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef]
- Inoue, Y.; Hazama, S.; Suzuki, N.; Tokumitsu, Y.; Kanekiyo, S.; Tomochika, S.; Tsunedomi, R.; Tokuhisa, Y.; Iida, M.; Sakamoto, K.; et al. Cetuximab strongly enhances immune cell infiltration into liver metastatic sites in colorectal cancer. Cancer Sci. 2017, 108, 455–460. [Google Scholar] [CrossRef]
- Serrano-Del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic Cell Death and Immunotherapy of Multiple Myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef]
- Mace, T.A.; Zhong, L.; Kilpatrick, C.; Zynda, E.; Lee, C.T.; Capitano, M.; Minderman, H.; Repasky, E.A. Differentiation of CD8+ T cells into effector cells is enhanced by physiological range hyperthermia. J. Leukoc. Biol. 2011, 90, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Ostberg, J.R.; Repasky, E.A. Emerging evidence indicates that physiologically relevant thermal stress regulates dendritic cell function. Cancer Immunol. Immunother. 2006, 55, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Ostberg, J.R.; Dayanc, B.E.; Yuan, M.; Oflazoglu, E.; Repasky, E.A. Enhancement of natural killer (NK) cell cytotoxicity by fever-range thermal stress is dependent on NKG2D function and is associated with plasma membrane NKG2D clustering and increased expression of MICA on target cells. J. Leukoc. Biol. 2007, 82, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Dammeijer, F.; Aerts, J.; Vroman, H. Current State of Dendritic Cell-Based Immunotherapy: Opportunities for in vitro Antigen Loading of Different DC Subsets? Front. Immunol. 2018, 9, 2804. [Google Scholar] [CrossRef] [PubMed]
- Moeller, B.J.; Dewhirst, M.W. HIF-1 and tumour radiosensitivity. Br. J. Cancer 2006, 95, 1–5. [Google Scholar] [CrossRef]
- Elming, P.B.; Sorensen, B.S.; Oei, A.L.; Franken, N.A.P.; Crezee, J.; Overgaard, J.; Horsman, M.R. Hyperthermia: The Optimal Treatment to Overcome Radiation Resistant Hypoxia. Cancers 2019, 11, 60. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, J.H.; Han, Y.H.; Cho, D.H. The effect of modulated electro-hyperthermia on temperature and blood flow in human cervical carcinoma. Int. J. Hyperth. 2018, 34, 953–960. [Google Scholar] [CrossRef]
- Prasad, B.; Kim, S.; Cho, W.; Kim, S.; Kim, J.K. Effect of tumor properties on energy absorption, temperature mapping, and thermal dose in 13.56-MHz radiofrequency hyperthermia. J. Therm. Biol. 2018, 74, 281–289. [Google Scholar] [CrossRef]
- Rajendran, V.; Jain, M.V. In Vitro Tumorigenic Assay: Colony Forming Assay for Cancer Stem Cells. Methods Mol. Biol. 2018, 1692, 89–95. [Google Scholar] [CrossRef]
- Falcone, S.; Cocucci, E.; Podini, P.; Kirchhausen, T.; Clementi, E.; Meldolesi, J. Macropinocytosis: Regulated coordination of endocytic and exocytic membrane traffic events. J. Cell Sci. 2006, 119, 4758–4769. [Google Scholar] [CrossRef]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.; Polyák, A.; Pöstényi, Z.; Kovács-Haász, V.; Gyöngy, M.; Thuróczy, J. Temperature increase induced by modulated electrohyperthermia (oncothermia) in the anesthetized pig liver. J. Cancer Res. 2016, 12, 1153–1159. [Google Scholar]
- Minnaar, C.A.; Kotzen, J.A.; Ayeni, O.A.; Vangu, M.D.T.; Baeyens, A. Potentiation of the Abscopal Effect by Modulated Electro-Hyperthermia in Locally Advanced Cervical Cancer Patients. Front. Oncol. 2020, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, G.; Sarti, D.; Casadei, V.; Milandri, C.; Dentico, P.; Mambrini, A.; Nani, R.; Fiorentini, C.; Guadagni, S. Modulated Electro-Hyperthermia as Palliative Treatment for Pancreatic Cancer: A Retrospective Observational Study on 106 Patients. Integr. Cancer Ther. 2019, 18, 1534735419878505. [Google Scholar] [CrossRef]
- Fiorentini, G.; Sarti, D.; Milandri, C.; Dentico, P.; Mambrini, A.; Fiorentini, C.; Mattioli, G.; Casadei, V.; Guadagni, S. Modulated Electrohyperthermia in Integrative Cancer Treatment for Relapsed Malignant Glioblastoma and Astrocytoma: Retrospective Multicenter Controlled Study. Integr. Cancer Ther. 2018, 1534735418812691. [Google Scholar] [CrossRef]
- Roussakow, S.V. Clinical and economic evaluation of modulated electrohyperthermia concurrent to dose-dense temozolomide 21/28 days regimen in the treatment of recurrent glioblastoma: A retrospective analysis of a two-centre German cohort trial with systematic comparison and effect-to-treatment analysis. BMJ Open 2017, 7, e017387. [Google Scholar] [CrossRef]
- Minnaar, C.A.; Kotzen, J.A.; Ayeni, O.A.; Naidoo, T.; Tunmer, M.; Sharma, V.; Vangu, M.D.; Baeyens, A. The effect of modulated electro-hyperthermia on local disease control in HIV-positive and -negative cervical cancer women in South Africa: Early results from a phase III randomised controlled trial. PLoS ONE 2019, 14, e0217894. [Google Scholar] [CrossRef]
- Papp, E.; Vancsik, T.; Kiss, E.; Szasz, O. Energy Absorption by the Membrane Rafts in the Modulated Electro-Hyperthermia (mEHT). Open J. Biophys. 2017, 7, 216–229. [Google Scholar] [CrossRef][Green Version]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Fatehi, D.; Van Der Zee, J.; Van der Wal, E.; Van Wieringen, W.N.; Fatehi, D.; Van der Zee, J.; Van der Wal, E.; Van Wieringen, W.N.; Van Rhoon, G.C. Temperature data analysis for 22 patients with advanced cervical carcinoma treated in Rotterdam using radiotherapy, hyperthermia and chemotherapy: A reference point is needed. Int. J. Hyperth. 2006, 22, 353–363. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Vujaskovic, Z.; Jones, E.; Thrall, D. Re-setting the biologic rationale for thermal therapy. Int. J. Hyperth. 2005, 21, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, M.; Mulder, H.T.; van Holthe, J.M.; Aangeenbrug, A.A.; Mens, J.W.M.; van Doorn, H.C.; Paulides, M.M.; Oomen-de Hoop, E.; Vernhout, R.M.; Lutgens, L.C.; et al. Confirmation of thermal dose as a predictor of local control in cervical carcinoma patients treated with state-of-the-art radiation therapy and hyperthermia. Radiother. Oncol. 2019, 140, 150–158. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, C.M.; Oei, A.L.; Chin, K.W.; Crezee, J.; Bel, A.; Westermann, A.M.; Buist, M.R.; Franken, N.A.; Stalpers, L.J.; Kok, H.P. A short time interval between radiotherapy and hyperthermia reduces in-field recurrence and mortality in women with advanced cervical cancer. Radiat. Oncol. 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Reference No. | Dilution | Antigen Retrieval | Vendor |
|---|---|---|---|---|
| Calreticulin | #12238 | 1:200 | T-E | Cell Signaling |
| CD3 | #IS503 | (RTU)1:2 | T-E | Dako |
| *F4/80 | #16-4801-86 | 1:100 | T-E | Thermo |
| GLUT1 | #355A-14 | 1:100 | T-E | Cell Marque |
| H2AXγ | #9718 | 1:50 | T-E | Cell Signaling |
| HMGB1 | #6893 | 1:200 | Citrate | Cell Signaling |
| HSP70 | #4872 | 1:50 | T-E | Cell Signaling |
| Ki67 | #MA5-14520 | 1:400 | T-E | Thermo |
| p21waf1 | #MA5-14949 | 1:50 | T-E | Thermo |
| S100 | #RB-9018 | 1:500 | T-E | Thermo |
| Tumor Type (Mice) | Treatment/Model | Major mEHT Effect | Main Mechanism | Publication |
|---|---|---|---|---|
| HT29 human CRC xenograft (BALB/c nude mice) | single mEHT shot, 42 °C, 30 min; compared with infrared heating to 42 °C | ≈3 times higher tumor damage than after radiation heating | synergy between the temperature-dependent (heat) and -independent (electromagnetic field) effects | Andocs et al., 2009 [23] |
| HT29 human CRC xenograft (BALB/c nude mice) | single mEHT shot, 42 °C, 30 min | significant tumor apoptosis | extrinsic pathway activation; up-regulation of TRAIL-R2, FAS, FADD | Meggyeshazi et al., 2013 [32] |
| HT29 human CRC xenograft (BALB/c nude mice) | single mEHT shot, 42 °C, 30 min | significant tumor apoptosis | DNA fragmentation; AIF1-mediated apoptosis | Meggyeshazi et al., 2014 [11] |
| SCCVII mouse head and neck squamous cell carcinoma (C3H/He mice) | intratumoral DC injection + 3 × mEHT: 9, 11, and 13 days after inoculation | reduced tumor sizes, both at the treated and distant locations | elevated DC activity, Hsp gp96 levels, CD3+ and CD8+, and S100+ cells; reduced Foxp3+ cells | Qin et al., 2014 [33] |
| HT29 human CRC xenograft (BALB/c -nu/nu mice) | single mEHT shot, 42 °C, 30 min | significant cell stress and apoptosis | upregulation and cell membrane translocation of DAMP signals (HSP70, HSP90, calreticulin, HMGB1) | Andocs et al., 2015 [34] |
| U937 human myelomonocytic lymphoma cells | single mEHT shot, 39–46 °C vs. WB, 30 min.; in vitro and in silico modeling of mEHT on membrane lipid rafts | similar apoptotic cell death with mEHT at 39 °C to that of WB at 44 °C | selective energy absorption (hot spots) focused on membrane rafts | Andocs et al., 2015 [35] |
| CT26 mouse CRC allograft (BALB/c immunocompetent mice) | single mEHT shot, 42 °C, 30 min + intratumoral injection of DCs | significant apoptosis; additive effect of mEHT on DC therapy | Hsp70 release and elevated cytotoxic T cell number and activity; prevention of tumor seeding after tumor re-challenge | Tsang et al., 2015 [36] |
| U87-MG and A172 human glioma cells (BALB/c nude mice) | 3 × mEHT, 42 °C, 60 min (every other day) in vitro and in vivo | significant apoptotic cell death; reduced tumor cell migration | increased E2F1 and p53, reduced PARP1 mRNA levels; reduced proportion of CD133+ stem cell fraction | Cha et al., 2015 [37] |
| U937 human myelomonocytic lymphoma cells | single mEHT shot, 42 °C, 30 min; comparison with WB heating, in vitro | significant apoptotic cell death (mainly protective effect of WB) | caspase-mediated apoptosis; FAS, c-JUN N-terminal kinases (JNK), and ERK signaling upregulation | Andocs et al., 2016 [18] |
| Human Huh7 hepatocellular cc. and HepG2 hepatoblastoma cells (BALB/c nude mice) | 3 × mEHT, 42 °C, 60 min (every other day) in vitro and in vivo | significant apoptotic cell death in both cell types in vitro; growth inhibition of HepG2 in vivo | suppressed cell proliferation and long term colony formation; upregulation of septin-4, p53, and p21waf1 | Jeon et al., 2016 [38] |
| HepG2 human hepatoblastoma cells | single mEHT shot, compared to capacitive coupling HT (cCHT) and WB heating at 42 °C, 30 min, in vitro | similar range of apoptosis by mEHT at 42 °C to that by WB at 46 °C | HSP70 upregulation by all three treatments; caspase-dependent apoptosis only by mEHT | Yang et al., 2016 [24] |
| C26 mouse CRC allograft (BALB/c immunocompetent mice) | single mEHT shot, 42 °C, 30 min | significant apoptotic tumor damage | blockade of cell cycle progression; caspase-mediated apoptosis, DAMP signaling, ICD | Vancsik et al., 2018 [12] |
| FSaII mouse fibrosarcoma allograft (C3H mice) | single or 3 × mEHT shot(s), 41 °C, 30 min + RT 15 Gy 60Co irradiation | enhanced tumor apoptosis and reduced tumor growth by mEHT; additive effect of mEHT on RT apoptosis | increased blood perfusion and tumor oxygenation; reduced Hif-1a, CaIX, and Vegf levels | Kim et al., 2018 [39] |
| C26 mouse CRC cells | single mEHT shot, 42 °C, 30 min, in vitro | significant cell stress and apoptosis; additive effect on doxorubicin | upregulated γH2ax and p-p53(Ser15), and downregulated p-Akt (Ser473); inhibited long-term colony formation | Vancsik et al., 2019 [29] |
| HepG2 human hepatoblastoma, A549 (lung cc), and U-87MG (glioblastoma) cells, and CT26 mouse CRC cells (BALB/c mice) | liposomal doxorubicin (Lipodox) + single mEHT shot, 42 °C, 30 min, in vivo (CT26 only) and in vitro (all cell lines) | mEHT significantly increased Lipodox (and 70 kDa dextran-FITC) uptake in HerG2 >A549 >U87MG >CT26; Lipodox + mEHT: most significant tumor (CT26) reduction in vivo | macropinocytosis indicated by the prevention of Lipodox uptake (and mEHT effect) by wortmannin | Tsang et al., 2019 [40] |
| B16F10 melanoma cell culture and allograft (C57Bl/6 mice) | 3 × mEHT, 42 °C, 30 min; 4, 6, and 8 days after inoculation, in vivo and in vitro | reduced tumor size and induction of γH2AX | upregulation and release of DAMPs (Hsp70, Hmgb1, ATP); elevated p53, p21waf1, and p27kip1 (senescence) and NK cell number; reduced Mhc-I and melan-A | Besztercei et al., 2019 [41] |
| 9 L human gliosarcoma, MCF-7 (breast cc), and MDKC (canine kidney epithelial cells) | single mEHT shot, 42 °C, 30 min + RT 10 MV 5 gyirradiation in vitro | supra additive tumor damage in L9 radioresistant glioma cells | significantly reduced tumor precursor cell fraction in IL9 and MCF-7 by clonogenic assay | McDonald et al., 2018 [42] |
| Human ovarian (OVCAR-3, and SK-OV-3) and cervical (HeLa and SNU-17) cancer cell lines (BALB/c nude mice) | single mEHT shot, 42 °C, 30 min + autophagy inhibitor 3-methyladenine (3-MA); in vitro and in vivo | significant apoptosis (increased sub-G1-phase fraction) reduced tumor xenograft weight and volume; combined treatment caused additive tumor damage | phosphorylation of p38, elevated caspase-3, and PARP, and mEHT-induced cellular damage recovery (autophagy) | Yang et al., 2019 [43] |
| A549 and NCI-H1299 human lung adenocarcinoma cell lines (BALB/c nude mice) | 2 x mEHT shot, 42 °C, 30 min + 2 x RT 2–8 Gy; in vitro and in vivo | significant radiosensitizing effect, apoptosis, and tumor volume reduction | increased equivalent radiation dose by mEHT; dependence on tumor dielectric properties | Prasad et al., 2019 [44] |
| NCI-H460-luc2 human lung adenocarcinoma cell line (BALB/c nude mice) | mEHT every third day for 5 weeks, combined with i.v. iron-dextran solution (mEHT + IronD) in vitro and in vivo | significantly higher tumor necrosis in mEHT + IronD than after paclitaxel monotherapy | mEHT + IronD increased tumor temperature to 47°C compared to 42 °C after mEHT monotherapy, resulting in increased tumor sensitization | Chung et al., 2019 [45] |
| HepG2 human hepatoblastoma cell line | mEHT, 15W for 10 min plus 50 nm size spherical-, urchin-, or rod-shape gold nanoparticles (AuNP) in vitro | AuNP in the medium: no tumor damage; cell-incorporated AuNP: tumor protection | AuNPs absorbed mEHT energy at this power without temperature increase, independent of particle shape | Chen et al., 2019 [46] |
| Panc1 human pancreas ductal adenocarecinoma cell line | single mEHT shot, 42 °C, 60 min, combined with RT (Cs-137/2 Gy) in vitro | significant apoptosis in mEHT + RT combination, resolved tumor radioresistance | upregulated caspase-3 and p21waf1, reduced p-Akt levels; DNA double-strand breaks | Forika et al., 2020 [47] |
| HT29 and SW480 human colorectal adenocarcinoma cell lines | mEHT 42 °C, 60 min, in comparison with WB heating; in vitro | mEHT induced significantly reduced proliferation and clonogenicity | induction of ion fluxes through tumor cell membrane channels; disequilibrium of most ions; tumor damage | Wust et al., 2020 [48] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krenacs, T.; Meggyeshazi, N.; Forika, G.; Kiss, E.; Hamar, P.; Szekely, T.; Vancsik, T. Modulated Electro-Hyperthermia-Induced Tumor Damage Mechanisms Revealed in Cancer Models. Int. J. Mol. Sci. 2020, 21, 6270. https://doi.org/10.3390/ijms21176270
Krenacs T, Meggyeshazi N, Forika G, Kiss E, Hamar P, Szekely T, Vancsik T. Modulated Electro-Hyperthermia-Induced Tumor Damage Mechanisms Revealed in Cancer Models. International Journal of Molecular Sciences. 2020; 21(17):6270. https://doi.org/10.3390/ijms21176270
Chicago/Turabian StyleKrenacs, Tibor, Nora Meggyeshazi, Gertrud Forika, Eva Kiss, Peter Hamar, Tamas Szekely, and Tamas Vancsik. 2020. "Modulated Electro-Hyperthermia-Induced Tumor Damage Mechanisms Revealed in Cancer Models" International Journal of Molecular Sciences 21, no. 17: 6270. https://doi.org/10.3390/ijms21176270
APA StyleKrenacs, T., Meggyeshazi, N., Forika, G., Kiss, E., Hamar, P., Szekely, T., & Vancsik, T. (2020). Modulated Electro-Hyperthermia-Induced Tumor Damage Mechanisms Revealed in Cancer Models. International Journal of Molecular Sciences, 21(17), 6270. https://doi.org/10.3390/ijms21176270