Abstract
The human body frequently encounters harmful bacterial pathogens and employs immune defense mechanisms designed to counteract such pathogenic assault. In the adaptive immune system, major histocompatibility complex (MHC)-restricted αβ T cells, along with unconventional αβ or γδ T cells, respond to bacterial antigens to orchestrate persisting protective immune responses and generate immunological memory. Research in the past ten years accelerated our knowledge of how T cells recognize bacterial antigens and how many bacterial species have evolved mechanisms to evade host antimicrobial immune responses. Such escape mechanisms act to corrupt the crosstalk between innate and adaptive immunity, potentially tipping the balance of host immune responses toward pathological rather than protective. This review examines the latest developments in our knowledge of how T cell immunity responds to bacterial pathogens and evaluates some of the mechanisms that pathogenic bacteria use to evade such T cell immunosurveillance, to promote virulence and survival in the host.
1. Introduction
The human body has evolved a complex symbiotic relationship with innocuous, commensal bacteria in which immunological defense mechanisms are tempered, to allow a state of mutualism that is beneficial to both the host and the colonizing microbiota. However, mucosal surfaces of the human body also frequently encounter harmful bacterial pathogens and, in response, employ immune defense mechanisms designed to counteract such pathogenic assault. Recognition of pathogen-associated molecular patterns on the surface of bacteria, such as lipopolysaccharides and endotoxins, initially triggers a rapid, first-line of defense that is driven by the innate immune system. Such protective immunity was originally thought to not be retained in any form of enduring immunological memory. However, this concept was recently challenged, revealing that cellular components of the innate immune system possess “trained immunity” and retain the ability to respond faster, upon pathogen reencounter [1]. Immunological memory is more classically witnessed in the adaptive immune system, composed of T cells and B cells, which respond to foreign antigens from pathogens to orchestrate protective immune responses that persist and are memorized. In the T cell arm of adaptive immunity, CD8+ T cells function to kill infected cells, whilst CD4+ T cells support memory CD8+ T cell responses [2,3] and antibody-generating B cells [4], whilst also possessing direct killing capabilities. T cells use membrane-bound, heterodimeric T cell receptors (TCRs) to detect and respond to infected cells expressing foreign antigen, and are classically sub-divided based on their expression of αβ or γδ TCRs. Conventional αβ T cells typically recognise small peptide antigens (8–13 amino acids) or larger antigenic fragments (>15 amino acids) presented on the cell surface of antigen-presenting cells (APCs), through major histocompatibility complex (MHC) class I or class II molecules. CD8+ T cells typically recognise MHC class I-restricted peptides, whilst CD4+ T cells detect MHC class II-restricted peptides, although it is evident that CD8+ T cells can break these “rules” and bind peptide-MHC (pMHC) class II complexes [5,6]. Classical MHC class I and II molecules are extremely polymorphic (>6000 allelic variants) and can present an immensely diverse array of antigens from pathogens. To respond to such antigenic variance, TCR sequences are also very diverse and TCR α- and β-chain rearrangements can theoretically generate <1021 distinct αβ TCRs [7], although realistically, in humans this is closer to 108 TCRs [8]. To create such clonotypic diversity, somatic recombination of variable (V), diversity (D), and joining (J) gene segments (D regions are only found in TCR β-chains), and junctional adaptations, occur to produce three complementarity determining region (CDR) loops, in each TCR chain that govern recognition of the pMHC complexes. CDR1 and CDR2 loops, germline encoded from 47 TCR α-chain V (TRAV) and 54 TCR β-chain V (TRBV) genes, typically bind the TCR to its target MHC, whilst hypervariable CDR3 loops, defined by unique nucleotide sequences, engage the exposed regions of the MHC bound peptide [7,9]. Such high diversity in the naïve T cell compartment means that the precursor frequency of antigen-specific T cells is very low [10]. Accordingly, mechanisms of clonal expansion are employed to amplify the numbers of antigen-specific effector T cells. Upon antigen exposure, naïve αβ T cells are primed by APCs to clonally expand into effector T cells in a process that is guided by immunological cues delivered by antigen [11], cytokines, and co-stimulatory interactions [12,13]. Antigen clearance leads to effector T cell contraction and conversion into memory T cells [12] that populate the blood, lymphoid organs, and mucosal tissues. Such immunological memory is maintained in the absence of antigen [14] by cytokines, such as IL-7 and IL-15 [15,16], rather than those integral to effector T cell development [17,18,19,20]. Upon re-exposure to antigen, memory T cells can then be rapidly recalled, and evoke immune responses with a greater efficacy.
Conventional αβ T cells were long considered to be only capable of recognising peptidic antigens complexed to classical MHC class I and II molecules. However, it was established that αβ T cells could engage antigens presented by non-classical MHC class Ib molecules. In humans, these include Human Leukocyte Antigen-E (HLA-E) molecules, which possess near allelic monomorphism, in contrast to MHC class I and II [21]. Additionally, αβ T cells can also respond to structurally different antigens (e.g., lipids, metabolites) presented by non-polymorphic MHC-like molecules, such as MHC class I-related gene protein (MR1) and CD1 glycoproteins [22,23,24,25]. Such “unconventional” T cell populations (Figure 1), including mucosal-associated invariant T (MAIT) cells and invariant natural killer T (iNKT) cells, appear to be “innate-like”, as they are poised to respond faster than T cells restricted by classical MHC molecules and display invariance in their TCR repertoire, which limits their clonotypic diversity [24]. Furthermore, these unconventional T cells also include T cells bearing γδ TCRs, which mediate antigen recognition in an MHC-unrestricted process that, to this date, is still poorly understood, since the identity of γδ TCR antigens is largely unclear [26]. However, recent evidence in the past decade provided some clarity and suggests that it involves butyrophilin (BTN) or BTN-like (BTNL) molecules, which are related to the B7 co-stimulatory molecules, CD80 and CD86 [26,27,28]. Furthermore, antigen recognition by γδ T cells also appears to be quite promiscuous, since they can also engage MR1 [29], CD1 variants [30,31,32,33], MHC class I homologs (e.g., MICA [34]) and pMHC class I complexes [35]. γδ T cells, like other unconventional T cell subsets such as MAIT cells, are known to facilitate immunological protection ag ainst Gram-negative and Gram-positive bacterial species [36] and respond to a myriad of structurally different antigens (e.g., lipids, metabolites) generated by bacteria.
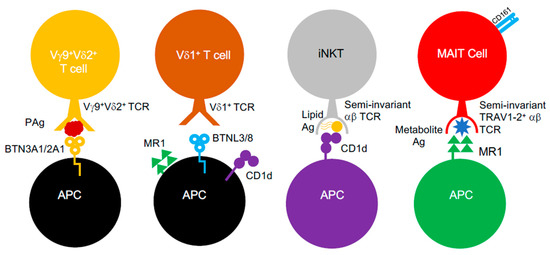
Figure 1.
Unconventional αβ and γδ T cells. Unconventional αβ and γδ T cells populations and known interactions between their TCRs, the antigens they target and the MHC-like molecules involved.
Research in the past ten years accelerated our knowledge of unconventional αβ or γδ T cells and how they recognize bacterial antigens and also how conventional αβ T cells provide anti-bacterial protection. However, many species of bacteria, most likely more than we are currently aware of, have evolved ways to evade host antimicrobial immune responses. Such escape mechanisms act to corrupt the crosstalk between innate and adaptive immunity, thus, potentially tipping the balance of host responses toward pathological rather than protective. Some pathogens cause chronic infections that are difficult to treat and employ virulence mechanisms that might dampen, or in some cases, also inhibit T cell immune responses. Some bacteria, such as Mycobacterium tuberculosis (M. tuberculosis), actively hide from T cells [37], whereas other species, such as Shigella flexneri (S. flexneri), invade T cells and modify their behavior [38]. Other bacteria, such as Staphylococcus aureus (S. aureus) secrete exotoxins called superantigens (SAgs), which evolved to target T cells, in order to evoke cellular dysfunction or deletion [39]. SAgs have received much attention in recent years, due to their profound effect on adaptive immunity and their ability to “anergise” large numbers of antigen-specific T cells that were generated by the immune system in the first place, to combat the SAg-producing bacteria [39]. This review examines the latest developments in the past ten years on T cell immunity to bacterial pathogens and also evaluates the mechanisms bacteria use to evade such T cell immunosurveillance.
2. How T Cells Fight Bacteria
2.1. MHC-Restricted αβ T Cells
To escape the threat of antibody-led defense mechanisms, certain bacteria evolved ways to invade intracellular compartments of mammalian host cells. Bacterial species such as Listeria monocytogenes (L. monocytogenes) and S. flexneri actively target the cytosol of host cells whilst M. tuberculosis and strains of Salmonella are known to reside in vacuolar compartments [37]. Other bacteria, such as S. aureus and Streptococcus pyogenes (S. pyogenes), which were originally classified as extracellular pathogens, also invade intracellular spaces of phagocytic and non-phagocytic cells in order to escape immune recognition [40,41]. Many intracellular bacteria can prosper inside endothelial cells on mucosal surfaces and in phagocytic APCs, such as dendritic cells (DCs) and macrophages [42]. However, such cellular sub-localization exposes these intracellular pathogens to enzymatic degradation and, consequently, to MHC class I and II antigen processing machinery, which elicit the presentation of bacterial fragments as antigens to αβ CD4+ and CD8+ T cells. MHC class I-restricted CD8+ T cells are critical for clearing bacterial infections and are known to provide protective immunity against a range of bacterial species, including L. monocytogenes [43], M. tuberculosis [44], Salmonella enterica serovar typhimurium (S. typhimurium) [45], and S. aureu s [46]. Furthermore, HLA-E restricted CD8+ T cells can also engage antigens from M. tuberculosis and S. typhimurium [21]. MHC class II-restricted CD4+ T cells support memory CD8+ T cell responses [2,3] and are important for protective immunity against bacterial infections, such as M. tuberculosis s [44], S. aureus [46], and S. typhimurium [47]. In response to infection, naïve CD4+ T cells differentiate into distinct “helper” subsets with effector capacity, such as T helper 1 (TH1) and TH2 cells. These effector CD4+ T cell types are distinguished by their roles in cellular or humoral immunity, the cytokines they produce, the transcription factors that control their development, and the types of pathogens they combat. For example, CD4+ TH1 cells utilise T-bet, STAT1, and STAT4 for transcriptional regulation, produce interferon-γ (IFNγ), tumor necrosis factor-α (TNFα), interleukin-2 (IL-2), and drive cellular immune responses against intracellular pathogens, including bacteria. In contrast, CD4+ TH2 cells promote humoral immune responses against large extracellular pathogens (e.g., parasites), are transcriptionally controlled by GATA3, STAT5, and STAT6 and produce cytokines such as IL-4, IL-5, and IL-13 [48]. As such, CD4+ TH cell subsets provide a vital source of effector cytokines that help CD8+ T cells and prime phagocytic cell subsets to kill bacteria. Interestingly, αβ CD4+ T cells can also seize and kill bacteria from infected DCs, in a process more akin to innate immune cells [49]. Such “transphagocytic” CD4+ T cells can also act as bona fide APCs and present bacterial antigens to naïve CD8+ T cells and generate memory CD8+ T cells [50]. These capabilities position CD4+ T cells capable of transcending the boundary between innate and adaptive immunity in a way that is more reminiscent of the unconventional T cell populations. In the following sections, we examine new developments from the past decade in our understanding of how MHC class I restricted CD8+ T cells and MHC class II restricted CD4+ T cells target bacteria and mediate protective immunity.
2.1.1. CD8+ T Cells: The Advent of Tissue Resident Memory T Cell Immunity
Memory CD8+ T cells are crucial for generating rapid recall responses to infection and maintaining long-term immunity. Memory CD8+ T cell responses were defined at the turn of the century to involve two subsets—central memory T (TCM) and effector memory T (TEM) cells, based on their tissue homing patterns [51]. CD8+ TCM and TEM cells both produce effector cytokines (e.g., IFNγ, TNFα, IL-2) and cytotoxic molecules (e.g., perforin) [52] in response to antigen, although CD8+ TEM cells evoke superior protection from bacteria, including L. monocytogenes [53,54]. CD8+ TCM cells express lymph node homing receptors, notably CCR7 and CD62L, which enable them to track from the blood to secondary lymphoid organs. In contrast, CD8+ TEM cells lack CCR7 and preferentially localize in non-lymphoid tissues [55]. However, for many years, it was relatively unknown whether CD8+ TEM cells permanently resided in non-lymphoid tissue, after the resolution of infection or recirculated back to blood. Nonetheless, seminal research between 2008 and 2010 changed the memory T cell landscape and determined that memory CD8+ T cells could remain in non-lymphoid tissues after virus infection, without the need for replenishment [56,57,58], marking the discovery of tissue resident memory T (TRM) cells. Since then, an acceleration of research in humans and mice into CD8+ TRM cells (and CD4+ TRM cells; see Section 2.1.3) in the past ten years, has greatly improved our knowledge of memory T cell immunity in tissues. It highlighted their critical role in facilitating protective immunity against an array of pathogenic microorganisms [59]. CD8+ TRM cells populate mucosal barrier tissues (e.g., lung, intestine, female reproductive tract, salivary glands), which are routinely exposed to infectious pathogens but also inhabit lymph nodes and vital peripheral organs, such as the brain, liver, and kidneys [59]. CD8+ TRM cells are phenotypically, transcriptionally, and metabolically distinct from CD8+ TEM and TCM cells, and are poised in tissue to mount rapid immune responses against invading pathogens [60,61,62,63,64]. A cardinal feature of CD8+ TRM cells is their inability to recirculate through the bloodstream or secondary lymphoid organs. Like CD8+ TEM cells, they lack CD62L and CCR7 [62,65], but typically express CD69, the αE integrin CD103 and downregulated levels of sphingosine-1-phosphate receptors and transcription factors controlling migration, which promote their tissue retention [62,66,67]. CD8+ TRM cell populations are heterogenous, even within the same tissue. Although CD103 and CD69− TRM cells do exist [61,68,69], they can express other phenotypic markers, such as CXCR6, CD11a, CD49a, CD101, and PD-1 [70,71,72,73], which demarcate these cells in a range of anatomical locations. The development and functionality of CD8+ TRM cells does not depend on the presence of antigen in certain tissues [59], but requires the activity of transcription factors, including Hobit [61], Blimp1 [61], Notch1 [71], Runx3 [74], and Bhlhe40 [75]. Additionally, cytokines, such as transforming growth factor-β (TGF-β), IL-15, and IL-33 [20,59,62,67,76,77,78], influence CD8+ TRM cell development and help “program” the TRM phenotype. In response to antigen, CD8+ TRM cells can locally proliferate, produce effector cytokines (IFNγ, TNFα, IL-2, IL-17) [60,63,70,71], and cytotoxic molecules (granzyme B), can also stimulate the recruitment of circulating memory T cells in recall responses, upon antigen re-encounter [79,80].
CD8+ TRM cells provide superior protection against pathogens that invade barrier tissues and mucosal organs, yet the response of CD8+ TRM cells to bacterial infection is less defined than that for viral infection. Nevertheless, these cells provide crucial protective immunity against a number of bacterial species. Studies examining the efficacy of vaccines against M. tuberculosis, notably the Bacillus Calmette–Guérin vaccine [81,82] and those vectored by replication-incompetent [83] or competent [84] viruses (e.g., cytomegalovirus), demonstrated that lung CD8+ TRM cells provide significant protection against tuberculosis, forming a correlate of vaccine-induced immunity. Following infection and re-challenge with L. monocytogenes, a food-borne Gram-positive bacterium, intestinal CD69+CD103+ CD8+ TRM cells, respond to mediate local and protective immunity, and rely on α4β7 integrin to home to the intestine [78]. CD8+ TRM cells provide protection against other food-borne bacterium, Yersinia pseudotuberculosis (Y. pseudotuberculosis), which causes Far East scarlet-like fever. In contrast to CD8+ TRM cells that respond to intestinal L. monocytogenes infection, protection is not dependent on TGF-β. Indeed, in mouse models of Y. pseudotuberculosis infection, intestinal macrophages produce TGF-β-suppressing cytokines (IFN-β, IL-12), which facilitate the development of CD69+CD103− CD8+ TRM cells that mediate protective immunity [85,86]. These differences highlight specific nuances in CD8+ TRM cell driven immunity to bacterial infection, although such protective immunity is not an exclusive property of all bacterial infections as S. typhimurium-driven T cell immunity does not depend on the presence of CD8+ TRM cells [87,88].
2.1.2. HLA-E Restricted CD8+ T Cells: Non-Classical Bacterial Recognition
HLA-E belongs to a family of non-classical MHC class Ib molecules that includes HLA-F, HLA-G, and HFE and possesses two alleles (HLA-E*01:01, HLA-E*01:03) that are encoded in >99% of the global population [21]. HLA-E is found on certain APCs and also in natural killer (NK) cells, where it functions as a ligand for inhibitory CD94/NKG2(A-C) receptors [89] and acts to regulate NK cell activity. Accordingly, HLA-E was thought to be involved in innate immune responses. However, it was determined that HLA-E modulates the activity of CD8+ T cells by binding peptide leader sequences [90], and that viruses could mimic [91] such activity. From here, seminal work between 1998 and 2002 provided the first evidence that HLA-E restricted CD8+ T cells could respond to M. tuberculosis [92,93], heralding a hugely important discovery in the field of non-classical, MHC-restricted T cell recognition of bacteria. It was later found that HLA-E restricted CD8+ T cells could target novel bacterial peptide antigens from S. typhimurium [94], indicating that HLA-E mediated immunity against bacteria was not restricted to M. tuberculosis. In the past decade, a range of approaches including bioinformatics and mass spectrometry discovered a number of HLA-E-specific peptides from M. tuberculosis [95,96,97,98,99]. Furthermore, the crystal structure of HLA-E, in complex with an immunogenic peptide from M. tuberculosis, was recently solved [100], revealing insights into the structural features of HLA-E-specific antigen presentation. HLA-E is enriched on M. tuberculosis phagosomes, enabling the loading of antigenic peptides in HLA-E in infected cells [101] and HLA-E-restricted CD8+ T cells mediate recognition, even when M. tuberculosis resides in late endosomal vacuoles [102]. Immunological investigations in HLA-E-restricted CD8+ T cell responses found that S. typhimurium-reactive cells were polyfunctional [103], were comparable in adults and paediatric individuals vaccinated against S. typhimurium [104], and correlated with clinical outcome in challenge studies [105]. In patients with M. tuberculosis infection, HLA-E restricted CD8+ T cells appeared to be more akin to CD4+ TH2 cells, since they produced TH2-specific cytokines (IL-4, IL-5, IL-13) upon antigen stimulation, expressed the transcription factor GATA3, and could activate B cells [95,99]. Furthermore, these cells inhibited M. tuberculosis growth, challenging conventional wisdom that “TH2-like” cells do not participate in mycobacterial control [99]. These developments in the past decade further indicated that HLA-E drives the protective CD8+ T cell responses to bacteria, embodying an important facet of the adaptive immune response to infection.
2.1.3. CD4+ T Cells: Anti-Bacterial Immunity Requires “Help”
MHC class II-restricted CD4+ T cells are important for protective immunity against a range of bacterial infections and provide a vital source of cytokines that help to facilitate such anti-bacterial immunity. Research on mice deficient in effector cytokines produced by CD4+ TH cell subsets [48] or humans with cytokine receptor deficiencies [106] illustrated the fundamental role that these cytokines and specific CD4+ T cell types play in this process. CD4+ TH1 and TH2 cells are the most categorized subsets in CD4+ T cell biology and were thought to be the only types that existed until the identification of CD4+ regulatory T cells in the early 2000s, and CD4+ TH17 cells during 2005 and 2006 [4]. Unlike CD4+ TH1 or TH2 cells, CD4+ TH17 cells express RORγt and STAT3 transcription factors, which control their lineage commitment and secrete IL-17A, IL-17F, and IL-22 as effector cytokines [48]. Since their identification, it has become well-known that CD4+ TH17 cells are fundamentally important for driving immune responses against bacterial infection, along with CD4+ TH1 subsets. As such, IFNγ and TNFα producing CD4+ TH1 cells provide protective immunity against Francisella tularensis (F. tularensis) [107], whilst CD4+ TH17 cells contribute to immune defense mechanisms directed against Mycobacterium bovis (M. bovis) [108] and Streptococcus pneumoniae (S. pneumoniae) [109]. However, both CD4+ TH1 and TH17 cells elicit immune responses against M. tuberculosis [110,111], L. monocytogenes [112,113], Bordetella pertussis (B. pertussis) [114], and S. aureus [115,116,117] infections. Such anti-bacterial immunity driven by CD4+ T cell appears to be less dependent on CD4+ TH2 cells, which drive humoral responses against pathogens, such as helminths and parasites. However, since 2009, it was established that a distinct subset of CD4+ T cells, called T follicular helper (TFH) cells [118,119,120], helped to support humoral immunity against pathogens that invade intracellular compartments, such as viruses and bacteria. Since then, extensive research into CD4+ TFH cells in humans and mice in the past decade has evolved our knowledge of these cells and the mechanisms of adaptive immunity that they drive. CD4+ TFH cells support antibody production in germinal centers (GCs) of secondary lymphoid organs (e.g., lymph nodes, tonsils), by promoting immunoglobulin class switching, B cell affinity maturation, and memory B cell responses [4,121]. CD4+ TFH cells can also migrate to tertiary lymphoid structures (TLS) in non-lymphoid organs (e.g., lung) and possess a phenotypic, transcriptomic, and functional profile that separates them from other CD4+ Th cell subsets. CD4+ TFH cells express PD-1, ICOS, and high levels of CXCR5 [122], which enables their migration to B-cell-rich follicles of lymphoid organs [123]. GC-resident CD4+ TFH cells depend on Bcl-6 to drive their differentiation [118,119,120,121], which acts to block commitment to non-TFH cell lineages, by repressing transcription factors that govern TH1, TH2, and TH17 cell fate, such as T-bet and GATA3, and genes that are important for their effector function, such as Ifng and Il17a [118,119,120,124,125]. CD4+ TFH cells produce IL-21 and IL-4 [4,121] as effector cytokines and their development and function is influenced by TCR stimulation and other cytokines, such as IL-12 [126] and IL-27 [127]. CD4+ TFH-like cell populations exist in the blood [128], which are clonotypically similar to GC-resident CD4+ TFH cells [129] although these cells lack Bcl-6 [121] and display lower levels of PD-1, CXCR5, and ICOS [121]. CD4+ TFH cells play a vital protective role against viral infection, yet there is evidence that these cells support protective immune responses against bacteria. CD4+ TFH cells promote humoral immune responses against intestinal Citrobacter rodentium infection in mice [130] and mediate protective immunity against M. tuberculosis infection, by accumulating within TLS in lung granulomas in humans, mice, and non-human primates [131]. However, this role for CD4+ TFH cells in anti-bacterial immunity is not fully established and might be specific to distinct bacterial species. GC-resident CD4+ TFH cells in children with recurrent tonsillitis, caused by S. pyogenes infection, can become phenotypically skewed into pathogenic B cell killing effectors, which reduces humoral immunity as a consequence [132]. Thus, further research into the anti-bacterial role for CD4+ TFH cells is of importance.
Much like memory CD8+ T cells, CD4+ T cells form long-lived circulating (TCM, TEM) and tissue-resident (TRM) memory populations, which establish upon the resolution of infection. CD4+ TRM cells share many cardinal features of CD8+ TRM cells—they populate barrier sites and mucosal organs (e.g., lung, skin, salivary glands, intestine), they mediate local protection against infection, they phenotypically express CD69 and CD11a [133] and share a core transcriptional signature [134]. However, CD4+ TRM cells lack, or possess low levels of, CD103 and can depend on receiving different signals (e.g., antigen), even in the same anatomical site [135,136,137,138]. Research in the past decade determined that CD4+ TRM cells are induced at various mucosal sites to mediate protective immunity against an array of bacterial pathogens, including M. tuberculosis [139], B. pertussis [140], S. pneumoniae [141], L. monocytogenes [142], C. rodentium [143], and Chlamydia trachomatis [144]. Despite this, less is known about CD4+ TRM cells in infection settings, in comparison to their CD8+ TRM cell counterparts, or whether CD4+ TRM cells form effector TH cell subtypes, such as TH1, TH2, and TH17, or even TFH cells. Recent evidence provided some insight, indicating that TH1 [145] and TH17-like [146] CD4+ TRM cells can mobilize in response to infection. However, the factors governing the development of these effector CD4+ TRM cell subtypes, especially in the context of memory responses to pathogen re-exposure, requires more definition.
2.2. Unconventional αβ and γδ T Cells
2.2.1. CD1-Restricted T Cells: Lipid-Driven T Cell Immunosurveillance
CD1 molecules are non-polymorphic, antigen-presenting molecules encoded outside of the MHC locus that have evolved to present microbial lipids to T cells. In humans, there are 4 CD1 protein isoforms (CD1a-d), which are capable of presenting antigen that are apportioned into two groups [147]: CD1a, CD1b, and CD1c form group 1, CD1d is the sole member of group 2, as its expression is constitutive rather than inducible [23,24]. These isoforms display distinct patterns of expression on a range of APCs, although all are found on myeloid DCs, and traffic differentially through endosomes and lysosomes to acquire antigens, each with highly hydrophobic antigen binding grooves that are capable of presenting lipids [23,24]. Antigen-specific, CD1-restricted T cells greatly outnumber αβ T cells reactive to a specific pMHC complex [24]. Seminal research between 1989 and 1994 discovered that αβ and γδ T cells were activated by CD1 molecules [148] and that lipid antigens from M. tuberculosis elicited CD1b-restricted T cell responses [22,149]. Many more CD1-restricted lipid antigens of microbial origin were since determined and the identity of T cells that react to them has become clearer. The development of CD1d-specific tetramers in the early 2000s determined that NKT cells could respond to CD1d-presented lipid antigens. Two classes of NKT cells exist—type I iNKT cells and type II “diverse” NKT cells. Type I iNKT cells possess TCR α-chains encoding fixed TRAV–TCR α-chain J (TRAJ) rearrangements (TRAV18/TRAJ10 in humans), which pair with TCR β-chains displaying limited TRBV gene usage, whilst type II NKT cells possess more polyclonal TCR repertoires composed of cells expressing αβ, γδ, or δ/αβ TCRs [24,33,150,151,152,153]. Microbial lipid antigens that are structurally similar to α-galactosylceramide, a glycolipid from the marine sponge Agelas mauritianus [154], are potent ligands for type I iNKT cells [24,153], but not type II NKT cells [150,151]. Type I iNKT cells respond rapidly to antigen and exist in diverse subsets defined by their effector cytokine production and transcription factor profile [24]. Type I iNKT cells play a key role in driving antigen-specific immunity against bacterial pathogens, such as M. bovis [155], Sphingomonas capsulate [156], Borrelia burgdorferi (B. burgdorferi) [157], and S. pneumoniae [158]. However, the contribution of type II NKT cells in anti-bacterial immunity is less defined, although these cells are activated by phospholipid antigens from M. tuberculosis and Corynebacterium glutamicum [159].
Up until the past decade, research into group 1 CD1-restricted T cells was slow, hampered by a lack of reagents that could detect CD1a-, CD1b-, and CD1c-reactive T cells and because mice do not encode group 1 CD1 molecules. However, the advent of tetramers specific to CD1a [160], CD1b [161], and CD1c [162] in the past ten years, changed the landscape and identified group 1 CD1-restricted T cells that are responsive to lipid antigens from bacteria. Two types of CD1b-restricted T cells; those bearing TRBV4-1+ αβ TCRs [161] or those encoding invariant TCR α-chains (“germline-encoded mycolyl lipid-reactive” T cells) [163], were found to be responsive to glucose-6-O-monomycolate from M. tuberculosis. CD1c-restricted T cells exhibiting TRBV7-8 or TRBV7-9+ αβ TCRs engage phosphomycoketide antigens from M. tuberculosis [164] and such CD1c-specific antigen reactivity was not exclusive to αβ TCRs, as bacterial lipid reactive, CD1c-restricted γδ TCRs exist [32]. Studies in human group 1 CD1 transgenic (hCD1tg) mice demonstrated that CD1b-restricted T cells contribute to protective immunity against M. tuberculosis [165], and high numbers of CD1b-restricted T cells are found in humans with active TB infection [166]. CD1-presented lipid antigens were also discovered in other bacterial species, such as B. burgdorferi [167], Brucella melitensis, Salmonella typhimurium, and S. aureus [168]. Recently, an in vivo model of systemic S. aureus infection in hCD1tg mice determined that group 1 CD1-restricted T cells elicit protective immunity, by responding to lipid antigens from S. aureus [169]. As such, these recent developments highlight an integral role for CD1-restricted T cells or NKT cells in anti-bacterial immunity and the key involvement of CD1 molecules is in presenting lipidic antigens to these specialised T cell or NKT cell subsets.
2.2.2. γδ T Cells: A Story of Anti-Bacterial Immunity Bound by Two Divergent TCR δ-Chains
γδ T cells are an unconventional T cell population that represent a small fraction (0.5–10%) of circulating CD3+ T cells in adult humans, and display immunological features common to both innate and adaptive immune systems, rendering them to be often referred to as innate-like lymphocytes [24]. γδ T cells express TCRs composed of rearranged TCR γ- and TCR δ-chains that are distinct from TCRs found on αβ T cells, and are encoded by lower numbers of V, D, and J segments, which limit their clonotypic diversity. Indeed, the restricted repertoire of TCR γ-chain V (TRGV) and TCR δ-chain V (TRDV) gene segments available for rearrangement (e.g., only 9 functional TRGV genes) led to theories that γδ TCRs identify conserved self-proteins of low variability [170]. During embryonic development, γδ T cells formed the first T cell population that was created and these cells established in low numbers in the circulation, but infiltrated peripheral mucosal and epithelial tissues at higher frequency. γδ T cells rapidly produced effector cytokines (e.g., IFNγ, TNFα, IL-17) in response to differing pathologies, including bacterial infection [171], where such cytokine production helped to recruit neutrophils, enhance adaptive immune defenses, and provided protection from bacterial invasion. There is evidence that γδ T cells form long-lived memory populations upon bacterial infection, some of which permanently populate the affected tissues, after pathogen clearance. In vivo studies in rodents and primates showed that γδ T cells provide protective memory responses against a number of bacterial pathogens, such as M. tuberculosis, S. aureus, B. pertussis, L. monocytogenes, and Salmonella enterica (reviewed in [171]). Such immunity was driven by clonotypically distinct γδ T cell subsets possessing effector functionality in different anatomical locations, such as the blood, lungs, peritoneum, skin, and intestine. γδ T cells are often influenced by the microbiota found at barrier sites and the regulation of γδ T cells by commensal bacteria can help drive protective immune responses against pathogenic bacteria, such as Pseudomonas aeruginosa [172].
In humans, γδ T cells can be classified into two main populations, based on their expression of TCR δ-chains encoded by two TRDV genes—TRDV1+ γδ T cells (or Vδ1+ T cells) and TRDV2+ γδ T cells (or Vδ2+ T cells). Vδ1+ T cells are abundant in the skin, intestine, and uterus, whereas Vδ2+ T cells constitute the majority of peripheral blood γδ T cells [24,173,174]. Vδ2+ T cells are dominated by TCR γδ chain rearrangements encoded by the TRGV9 and TRDV2 segments, which almost exclusively pair together, forming the more frequently termed “Vγ9+Vδ2+” T cells that make up large proportions of peripheral blood γδ T cells in humans and other species [175]. Vγ9+Vδ2+ T cells are enriched in fetal peripheral blood, as “pre-programmed”, semi-invariant effector T cells [176] with innate-like capacity, and these cells are known to be responsive to prenyl pyrophosphate metabolites (or “phosphoantigens” (PAgs)), from the eukaryotic mevalonate pathway [177] that are produced by mammalian cells. However, PAgs are also produced by the non-mevalonate pathway in bacterial pathogens, such as M. tuberculosis [178] and Escherichia coli (E. coli) [179], endowing Vγ9+Vδ2+ T cells with the ability to ‘sense’ infected cells [175]. For a while, the mode of PAg presentation to Vγ9+Vδ2+ T cells remained unsolved, yet recent work in the past decade determined that PAgs activate Vγ9+Vδ2+ T cells by binding to BTN3A1 [180,181,182] and that BTN3A2 [183] is an important factor for regulating such activation. The molecular mechanisms governing Vγ9+Vδ2+ TCR recognition of PAgs remained unclear until very recently when it was discovered that BTN2A1 acts a ligand for Vγ9+Vδ2+ T cells, acting to bind germline-encoded regions of TRGV9 and working in concert with BTN3A1, to promote Vγ9+Vδ2+ T cell responses to PAgs [184,185]. Unlike Vγ9+Vδ2+ T cells, peripheral blood Vγ9-Vδ2+ T cells are unresponsive to PAgs, existing as a divergent, adaptive-like subset of Vδ2+ T cells that is found at much lower frequency, but undergoes clonal expansion and effector differentiation in response to viral infection [174]. In humans, Vδ1+ T cells also constitute a minority of the adult peripheral blood γδ T cell population [186], acting more to populate epithelial and mucosal tissues, particularly the intestine [187]. Vδ1+ T cells evolved a distinct biology from Vδ2+ T cell subsets and are shaped by TCR-dependent clonal selection [186]. Murine Vδ1+ T cells display broader TRGV usage patterns and utilize gene segments that are distinctive to individual tissue sites (e.g., TRDV5+ Vδ1+ T cells in the skin, TRGV7+ or TRGV1+ δ1+ T cells in the intestine) [24]. Recent research showed that human intestinal Vδ1+ T cell subsets utilize TCR γ-chains encoding TRGV4 and are regulated by BTNL3 and BTNL8 molecules on the surface of gut epithelia [188]. BTNL3 was shown to bind germline-encoded regions of TRGV4 encoding TCRs [189], providing a mode of antigen recognition for Vδ1+ TCRs. However, Vδ1+ T cells appear to be promiscuous in their antigen-binding capabilities, possessing the ability to engage with pMHC class I complexes [35], and MHC-like molecules or homologs, such as CD1c [32,148], CD1d [30,31,33], and MICA [34]. Indeed, recently it was found that clonally diverse Vδ1+ TCRs could bind MR1 underneath the antigen-binding cleft [29], redefining our understanding of TCR recognition and identifying MR1 as a self-ligand for Vδ1+ T cells. Such MR1-restricted γδ T cells were not found in Vγ9+Vδ2+ T cell populations and populated damaged tissues, suggesting that they possess roles in pathology. Further studies are required to define how MR1-restricted γδ T cells facilitate immune responses in pathological settings, but also to learn more about the molecular determinants of antigen recognition, notably of microbial ligands, through Vδ1+ T cells.
2.2.3. MAIT Cells: Masters of Riboflavin-Driven T Cell Immunity
MAIT cells are an innate-like, unconventional T cells that respond to MR1-restricted bacterial metabolite antigens and provide a powerful source of pro-inflammatory cytokines, relative to their frequency in T cell populations [24,36,190]. In 1993, MAIT cells were first identified as a CD4-CD8- (DN) T cell population, expressing an invariant, TRAV1-2/TRAJ33 encoded a TCR α-chain [191] that was later found to pair with a limited repertoire of TCR β-chains (TRBV20-1 or TRBV6 family genes in humans) that are conserved across mammals [192]. In 2003, a landmark study that devised the term “MAIT cells”, demonstrated the specificity of MAIT cells to MR1 and their enrichment in mucosal locations, such as the gut lamina propria [193]. For many years after, the function of MAIT cells and the antigens they respond to, remained unknown. However, the development of monoclonal antibodies that could detect TRAV1-2 [194] or block MR1-facilitated seminal discoveries into the function of MAIT cells, which in 2010 was proved to be capable of responding to a range of Gram-negative and Gram-positive bacterial species, including Klebsiella pneumoniae (K. pneumoniae), E. coli, and S. aureus [195,196]. Such reactivity was not exclusive to all bacteria, which indicated that such microbial sensing was potentially driven by recognition of common antigen(s). Since then, the past decade witnessed an exceptional surge in research centered around understanding the role and function of MAIT cells in combating bacterial infections and also on the process through which these cells develop in the periphery, the phenotypic and transcriptomic signatures they possess, the antigens they engage, and the activation profiles they generate. In 2012, an important breakthrough was made in the identity of MAIT cell reactive antigens, which determined that MR1 could present metabolites of vitamin B2 (riboflavin) and B9 (folic acid) from bacterial species to MAIT cells [25]. This significant discovery paved way for the identification of key ribityllumazine- and pyrimidine-based metabolite antigens of the microbial riboflavin biosynthesis pathway, including 5-OP-RU (5-(2-oxopropylideneamino)-6-D-ribitylaminouracil), which could potently activate MAIT cells [25,197]. Bacterial species that activate MAIT cells possess intact riboflavin biosynthesis, and such dependency on antigens from this pathway was verified in experiments where deletion of key pathway enzymes rendered bacteria unresponsive to MAIT cells [25,190,197]. Furthermore, many structural-based studies examined the precise mechanism involved in the recognition of MR1-bound ribityllumazine- and pyrimidine-based antigens through MAIT-specific TCRs [190] and suggested that these metabolite antigens did not fill the MR1 antigen-binding cleft, which opened up the possibility that other antigens could be presented by MR1 to MAIT cells. Indeed, further research determined that MR1 could present non-riboflavin-derived microbial antigens [198,199] and possessed enough flexibility to accommodate the binding of anti-inflammatory drugs [200]. Nonetheless, such identification of bacterial metabolite antigens led to the development of human and mouse MR1 tetramers [201,202], loaded with antigens such as 5-OP-RU. Such a technological advancement helped revolutionize our understanding of these cells, how they develop in the thymus, and also the bacterial infections they are mobilized to fight against.
In humans, MAIT cells are now typically defined as 5-OP-RU/MR1 tetramer binding T cells that bear a TRAV1-2/TRAJ33 encoded TCR α-chain [190], although “non-classical” MAIT cells that possess TRAV1-2- TCR α-chains but are found at lower frequency, do exist. 5-OP-RU/MR1 tetramer+ MAIT cells also encode TCR α-chains with alternative TRAJ genes (e.g., TRAJ20, 12) in humans [202] and mice, notably those deficient in TRAJ33 [203]. The majority of MAIT cells represent CD8αα, CD8αβ+, or DN T cell subsets, which make up 1–10% of all blood T cells and up to 50% of T cells found in mucosal organs [190]. MAIT cells display an effector memory (CD45RO+CCR7-CD62L-) phenotype and typically express high levels of CD161, found to be more than a 100-fold higher than that expressed by many other T cell types. MAIT cells express many chemokine receptors, including CCR5 and CCR6, which enable them to recirculate between the circulation and tissues [204], and upon TCR-mediated recognition of antigen, produce effector cytokines (IFNγ, TNFα, IL-2, IL-17, IL-22) as part of defined effector subsets (e.g., MAIT1 cells produce IFNγ, MAIT17 cells produce IL-17), and gain cytotoxic capabilities [190]. MAIT cells express transcription factors, such as PZLF1, T-bet, Eomes, RORγt, and Helios, which drive functionality (e.g., T-bet and MAIT1 cells, RORγt and MAIT17 cells) and share many phenotypic similarities with NK cells [190]. Many studies showed that in the absence of MR1-presented antigen, MAIT cells can be activated by cytokines, such as IL-12 and IL-18 [205], whose receptors are found at high levels on the cell surface and produce efficient activation in synergy, rather than alone. Such TCR-independent activation is observed during bacterial [190] and also viral [206] infection, broadening the role that MAIT cells play in immune responses to bacteria carrying intact riboflavin biosynthesis to infectious situations where IL-12 or IL-18 is produced [205]. More recently, it was shown that IL-23 co-stimulates antigen-specific MAIT cells during pulmonary bacterial infection and drives their proliferation and differentiation [207]. Interestingly, CD80/86-CD28 signaling was found to be not instrumental for MAIT cell expansion, in contravention of the rules for conventional T cells and iNKT cells [207]. A prominent feature of MAIT cells is their capability to exhibit effector functions, prior to exiting the thymus. The use of 5-OP-RU/MR1 tetramers helped to determine that MAIT cells follow a three-stage intrathymic developmental pathway in humans and mice [208]. A recent study determined that commensal bacteria are vital for intrathymic development of MAIT cells in mice, providing a source of riboflavin metabolites that are transferred to the thymus to help direct MAIT cell production [209]. Such synergy between MAIT cells and riboflavin-producing commensals, during early life, depends on a small temporal window that functions to imprint MAIT cell abundance in the periphery [210]. In response to bacterial pathogens, MAIT cells provide a fundamental cellular population that is designed to limit infection. Studies using human blood samples, Mr1-/- mice and other mouse models (e.g., MAIT TCR transgenic mice) generated for studying MAIT cell control of bacterial infection in vivo, revealed the true landscape of infections that MAIT cells mobilize against, to provide protective immunity. MAIT cells were found to be induced or activated in response to infection from a large repertoire of bacterial species that included Clostridium difficile, E. coli, F. tularensis, Haemophilius influenzae, K. pneumoniae, Legionella longbeachae, Mycobacterium abscessus, M. tuberculosis, S. flexneri, S. typhimurium, and S. pneumoniae (reviewed in [190]). Whilst this protective role was highly apparent, MAIT cells were showed to also have a pathogenic role, notably during Helicobacter pylori infection [211]. As such, it is important to learn more about the protective or pathogenic roles of MAIT cells in response to bacterial infection.
3. Bacterial Immune Evasion Strategies
3.1. A Pathogenic Game of ‘Hide and Seek’ and T Cell Evasion by M. tuberculosis
A key characteristic of many pathogens is persistence—the continued presence of pathogen in environments that are considered stressful or hostile conditions, which might have limited nutrients and might be shared with antimicrobial regents or threatening immune cells. During persistence, the pathogen is non-infectious, having stopped progressive activities, such as cell development and reproduction, and thus remains undetected by the host, while it ‘hides’ in a non-replicating state [212]. Until a more comfortable environment can be secured, such persistence will continue where the pathogen remains viable but does not thrive. However, the pathogen can play ‘hide and seek’ and re-appear once the immune system is evaded and possibly deceived at the infection or colonization site. One pathogenic bacterium that excels at this is M. tuberculosis [213], which evades host immune responses in order to establish a chronic infection. M. tuberculosis is a facultative intracellular pathogen that is known to reside inside a variety of APCs, including macrophage and DC subsets. Following inhalation of M. tuberculosis droplets, the bacteria specifically targets alveolar macrophages that do not robustly respond or detect the infection, resulting in a dampened innate immune response and delayed activation of adaptive immunity [214]. Indeed, murine models established that the triggering of antigen-specific T cells following aerosol infection with M. tuberculosis is delayed, relative to that of other pathogens such as the influenza virus [215]. As such, the delay in immune triggering that is manufactured by M. tuberculosis is unique amongst lung bacterial pathogens, suggesting that M. tuberculosis actively uses the alveolar macrophages to avoid rapid detection. Upon invading host cells through phagocytosis, M. tuberculosis can replicate within the infected cells by arresting phagosome maturation [216]. This is accomplished by M. tuberculosis changing its composition, as the structure of the cell wall and specific molecules on its surface serve as a barrier that allows the macrophages to maintain a neutral pH [217]. This mechanism allows the pathogen to avoid exposure to lysosomal hydrolases, unfavorable low pH conditions produced by the immune response, and other bactericidal lysosomal components [212]. Additionally, M. tuberculosis is capable of producing factors that modulate the expression of pro-apoptotic and anti-apoptotic genes in macrophages [218], which has implications for innate immune responses. Inhibition of apoptosis might be a major mechanism, whereby M. tuberculosis delays the acquisition of bacteria by DCs and the onset of adaptive immunity. It was also established that M. tuberculosis-infected macrophages preferentially synthesize lipoxin A4 [219,220], which helps create an anti-apoptotic environment that delays the onset of CD4+ and CD8+ T cell responses.
M. tuberculosis not only hides from the immune system but can also modulate adaptive immune responses by inhibiting T cell activities. M. tuberculosis can chronically stimulate antigen-specific CD4+ T cells (i.e., ESAT6-specific) to drive functional exhaustion [221], whilst equally reducing the expression of antigens targeted by other CD4+ T cell populations to evade detection [222,223]. M. tuberculosis can suppress T cell responses by recruiting mesenchymal stem cells to the site of infection, which produce nitric oxide to inhibit T cell activity [224]. M. tuberculosis possesses antigens that are recognised by FoxP3+CD4+ regulatory T cells, which expand in patients with an active infection and act to suppress the effector T cell responses against the bacterium, enabling the infection to expand and prolonging the bacterial burden [225,226]. M. tuberculosis does not evolve rapidly and, as a result, the antigens that drive M. tuberculosis-specific T cell responses are conserved across the human population [227]—a striking contrast to influenza or HIV that actively evades T cell immunity by mutating antigens to avoid detection [212]. It is also thought that defective interactions between T cells and cells harbouring M. tuberculosis contribute to the failure of T cells to remove infection. The ability to image M. tuberculosis-induced T cells and their real-time interactions with infected cells in tissues is likely to guide the design and selection of TB vaccines [228]. No global approaches were previously employed to systemically define the repertoire of M. tuberculosis immune evasion genes, especially those that enable escape from T cell surveillance. The distinct mechanisms used by M. tuberculosis to avoid detection, and thus, extinction in a host, can now be explored in great detail, using global approaches to modify host genetics or to integrate diversity into host models. In particular, the development of CRISPR-Cas gene editing mechanisms that generate “loss of function” and “gain of function” systems, might be particularly useful in understanding the mechanisms used by M. tuberculosis for immune evasion [229] and is a likely avenue of future research.
3.2. Type III Secretion Systems and Pore-Forming Toxins: Secretion-Driven Bacterial Immune Defense
Pathogenic bacteria evolved a wide repertoire of virulence mechanisms that promote immune evasion and bacterial persistence, including the use of type III secretion systems (T3SSs). T3SSs are complex, macromolecular transport machines found on many pathogenic Gram-negative bacteria [230], including members of Yersinia, Shigella, and Salmonella species, which subvert host cell immune responses by injecting bacterial effector proteins directly into the host cell, in order to modify their functionality [231]. It was originally thought that the species of Shigella and Salmonella used T3SSs to gain entry to cells. However, research evolved to show that T3SSs work at a different level, by altering the phagocytic properties of macrophages and possibly their killing capacities [232]. As such, proteins secreted by T3SSs can be classed as manipulators of innate defense mechanisms [233], endowing bacterial pathogens with the ability to alter inflammatory responses from within phagocytic cells. Shigella use T3SSs to secrete effector proteins, such as IpaB [234], which binds to and activates Caspase-1 [235] in macrophages, through a process involving the IPAF/ASC inflammasome, which enables them to evade the phagosome and induce pyroptosis [236]. Such exploitation of the IPAF/ASC inflammasome is thought to help Shigella to escape macrophages, enabling them to invade the intestinal epithelium [237]. The evasive techniques employed here by Shigella, enable it to establish infectious processes [238]. Moreover, Shigella evolved to inhibit the production of certain antimicrobial peptides, which are key effector molecules in bacterial host defense. Indeed, early in Shigella infections, expression of peptides LL-37 and human β-defensin-1 were found to be dramatically reduced or turned off [239]. This downregulation of immediate defense effectors might encourage bacterial adherence and invasion into host epithelium, and could be an important virulence parameter. Such T3SS-driven evasion strategies act to guard against innate immunity, but in terms of defending against adaptive immune responses, bacteria can use T3SSs to modify T cell behavior. S. flexneri evolved to use T3SSs to invade CD4+ T cells in order to “paralyze” their migratory patterns and utilizes injected effector proteins to induce inhibitory signals that alter cellular dynamics [38]. Salmonella enterica serovars use a unique T3SS that is capable of injecting up to 30 effector proteins with the ability to disrupt cell signaling pathways, interfere with MHC-dependent antigen presentation in DCs [240], and slow the migration of infected DCs [241] with a downstream effect on T cell activation. Salmonella-encoded T3SSs can also directly contact T cells, inhibit their proliferation [242] and augment co-inhibitory signaling (e.g., PD-1/PD-L1) between T cells and APCs [243]. S. aureus, which displays various levels of virulence, can manipulate host T cell responses that limit bacterial growth but do not eliminate the pathogen during persistent infections. Along with producing potent, T-cell-targeting SAgs (reviewed in Section 3.3), S. aureus produces extracellular, pore-forming toxins that lyse T cells upon cellular engagement. S. aureus α-toxin forms heptameric pores that destroy T cells [244], whilst leukocidin DE binds to CCR5 to kill T cells [245]. Furthermore, during persistent S. aureus infection, T cells can become anergic through a failure in TCR signaling events, which render the T cells unable to respond to antigenic stimulation [246]. The presence of T3SSs and pore-forming toxins, arms bacterial pathogens, such as Shigella and S. aureus, with an attack and evasion “arsenal”, which acts to dampen host innate and adaptive immune responses and aids their virulence and survival.
3.3. SAgs: Microbial Super-Agents That Manipulate T Cell Immunity
SAgs are potent enterotoxins secreted by distinct Gram-positive bacteria, including S. aureus and S. pyogenes, which target T cells, evoking mechanisms of cellular dysfunction and the initiation of a cytokine storm (involving large amounts of IFNγ, TNFα, and IL-2), which can activate up to 20% of the T cell population [39]. In the late 1960s, the first SAg was identified from the secreted toxins of S. aureus and was named staphylococcal enterotoxin A (SEA) for its strong enterotoxic properties. Staphylococcal enterotoxins (SEs) are the causative agents in staphylococcal food poisoning and induce vomiting and diarrhea shortly after ingestion; an effect which is normally self-limiting. However, it is now known that SAgs from both S. aureus and S. pyogenes are strongly associated with life-threatening conditions, such as pneumonia, sepsis, and toxic shock syndrome (TSS) [39]. Following their initial identification, the mitogenic activity of SEs was not uncovered for some time, with the term ‘superantigen’ not being coined until 1989 when it was discovered that the profound effect of SAgs on the immune system was a result of an immense expansion of T cells bearing the same TCR Vβ domain [247]. This suggested that the interaction between SAgs and TCR Vβ domains was not completely indiscriminate, as only specific TCR Vβ domains could be engaged, leaving only a minority of T cells susceptible to triggering by any given SAg [248]. However, such an act exposes “holes” in the repertoire of T cells available to protect against infection [247], which helps to enhance bacterial colonization [249], providing a bonafide mechanism for bacterial persistence and immune evasion. Since then, many research studies used a range of techniques, including x-ray crystallography, to confirm that SAgs are able to bind and cross-link human αβ TCRs on CD4+ or CD8+ T cells to HLA class II molecules on APCs, through a variety of structural mechanisms [247,250,251,252,253,254] that bypass the laws of conventional TCR-pMHC interactions. Such a physical interaction between TCRs and SAgs, which exists outside of the peptide-binding groove, involves a large number of TRBV region genes that are targeted in distinct patterns by bacterial SAgs [39,255]. At present, 26 different SAgs from S. aureus are described, comprising toxic shock syndrome toxin-1 (TSST-1), 11 SEs (SEA–SEE, SEG–SEI, SER–SET), and 14 SE-like (SEl) proteins (SElJ–SElQ, SElU–SElZ) [256,257]. There are also 14 Streptococcal pyrogenic exotoxins (Spe), which include SpeA, SpeC, SpeG, SpeH, streptococcal mitogenic exotoxin Z, and streptococcal superantigen A [258]. Such TCR Vβ region-SAg interactions are indicated in Figure 2, highlighting the known relationships between specific TRBV genes and a number (but not all) of SAgs from S. aureus and S. pyogenes. To date, all SAgs appear to specifically target TRBV genes, with the exception of SEH, which interacts with TRAV27+ TCR α-chains, whilst also mediating contact with TRBV19 [253,254,259]. It is plausible that SEH might also bind other TRAV genes, however, this is yet to be confirmed.
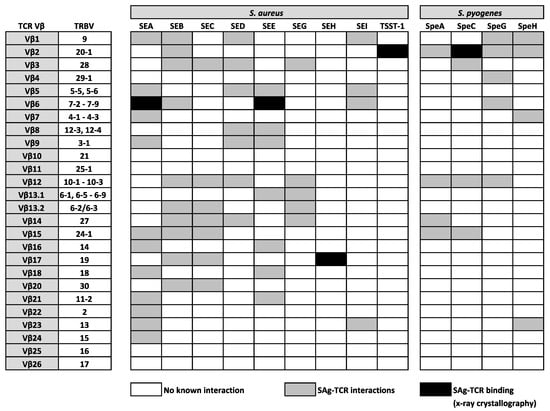
Figure 2.
Established interactions between well characterised SAgs from S. aureus and S. pyogenes and human TCR β-chain variable (TRBV) regions.
The ability for SAgs to drive cellular dysfunction or deletion in T cell populations is highly affected by HLA class II polymorphisms [260], which influence patient outcome in infectious diseases driven by these toxins [261]. Some SAgs display a stronger binding affinity to specific HLA class II alleles (e.g., SpeA and HLA-DQB1*06:02) and susceptibility to TSS and invasive forms of group A streptococcal (GAS) infection (e.g., S. pyogenes) appears to be correlative with HLA-DQB1*06:02 expression [132]. The physical binding relationship between SAgs, TCRs, and HLA class II alleles was universally accepted for over two decades, until the discovery that SAgs were capable of binding T cell co-stimulatory molecules, notably CD28 [262], and their ligands (i.e., B7-2 [263]) on APCs. Prior to this, co-stimulatory pathways were not thought to bind pathogens or virulence factors [264], and such engagement potently enhanced the avidity between B7-2 and CD28, thereby inducing T-cell hyperactivation, which is a cardinal feature of SAg activity [263]. These binding features position superantigens as biological molecules that are capable of forming quaternary complexes between T cells and APCs, providing evidence of further complexity that was originally unknown. Thus, it provides the necessary scope for further research that examines the structural mechanics behind how these biological toxins interact with T cells.
SAg-mediated illnesses, such as TSS, are associated with immunosuppression upon bacterial invasion, threatening the ability of the host to fight the invading pathogens. It is well-established that the introduction of SAgs leads to an initial expansion of SAg-reactive T cells, yet following this, a large percentage of these T cells are eliminated by apoptosis or become anergic [265]. Such events appear to occur more in memory T cells, consequently reducing the available pool of antigen-experienced T cells [266,267], which creates a state of immune suppression that enables opportunistic infections to thrive. Staphylococcal SAgs are a causative agent of severe pneumonia, acting to induce damage to the pulmonary epithelium and to disrupt mechanisms of neutrophil activation that results in severe immunopathology [268]. The ability of S. aureus to form biofilms, combined with the emergence of multidrug-resistant strains and its ability to produce Sags, amplifies the challenge faced when treating infections. As such, the frequency of SAg-producing infections is increasing. S. aureus and GAS infections routinely cause recurrent tonsillitis (RT) or tonsillar hyperplasia and drive TCR repertoire skewing patterns that are associated to SAg activity [269]. Children with RT caused by GAS infections display lower titres of antibodies against SpeA, compared to those with singular bouts of tonsillitis, and possess GC-resident CD4+ TFH cells that have become phenotypically skewed into pathogenic B cell killing effectors in the tonsil [132]. Studies showed that SAgs can function to subvert T cell immunity to viruses (e.g., influenza) [270,271], which has profound implications for patients with viral/bacterial co-infections. However, recent evidence opposing this view has emerged, suggesting that SAgs (i.e., SEB) could expand influenza-specific CD8+ T cells when administered in vivo in mice, prior to virus infection, can re-organize the hierarchical pattern of primary CD8+ T cell responses, and promote improved recall immunity [272]. Such an effect was dependent on SAg pre-exposure, as a contrasting phenotype was seen when SEB was administered following a pre-existing influenza infection, highlighting the potential core differences in how SAgs can function when exposed to naïve or memory virus-specific CD8+ T cells. SAgs possess the ability to bind αβ TCRs on MHC-restricted CD4+ or CD8+ T cells, but also target unconventional, antimicrobial T cell populations bearing αβ TCRs, such as MAIT cells [273,274] and iNKT cells [275,276] (Figure 3). However, SAgs can also recognise γδ T cells, demonstrating that these toxins can bind TCRs lacking TCR α- or β-chains, and such interaction by certain SAgs involves TRGV9+ γδ TCRs [277,278,279,280]. MAIT cells typically encode TCRs with biased TRBV20-1 and TRBV6-2/6-3 gene usage [192], exposing them to SAgs such as SEB, TSST-1, and SpeC, which was proven in recent studies [273,274]. Indeed, SAg-triggered MAIT cells produce substantially high levels of effector cytokines (e.g., IFNγ), through an IL-12/IL-18-dependent mechanism that does not involve MR1. Such hyperactivation induces MAIT cell anergy both in vitro and in vivo, in humanized mouse models, impeding their anti-bacterial capabilities [273,274]. Highly activated MAIT cells form major contributors of cytokines, produced in the acute phase of streptococcal TSS (STSS), despite the GAS species lacking de novo riboflavin synthesis, indicating that SAg-driven MAIT cell activation underlies the cytokine storm seen in STSS patients [274]. All of this demonstrates the considerable impact SAgs have in clinical settings and the direct effects they have on both conventional αβ T cells and unconventional αβ or γδ T cells. It highlights the breadth of T cell populations that are targets for these toxins, and further research will define if SAg-targeting of T cells is more intricate than initially thought.
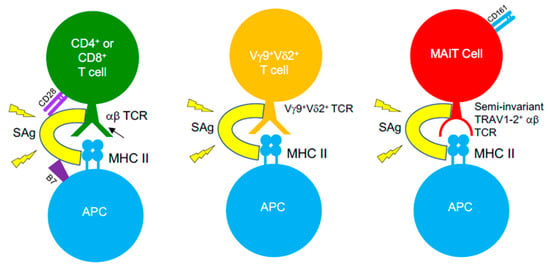
Figure 3.
SAgs target αβ and γδ TCRs on different T cell subsets. SAgs bypass conventional TCR–pMHC interactions by binding TCRs from αβ and γδ T cells, outside the peptide-binding groove and cross-linking them to MHC class II molecules. Such an interaction with certain T cell subsets also involves binding interactions with co-stimulatory molecules on T cells (CD28) and their ligands on APCs (B7-2), indicating that SAgs are capable of forming quaternary complexes between T cells and APCs, in order to induce T cell hyperactivation.
4. Conclusions
Cellular components of the adaptive immune system, notably T cells, evolved a multitude of ways to detect and respond to pathogenic bacterial infections, utilizing MHC and MHC-like molecules to present structurally and chemically different antigens from bacteria. MHC-restricted αβ CD4+ and CD8+ T cells, provide immune defense in response to peptidic antigens, whilst unconventional αβ (MAIT cells, CD1-restricted T cells) and γδ T cells, utilize MR1, CD1 molecules and BTN/BTNL molecules to respond to lipidic, metabolic, or undiscovered antigens of bacterial origin. Seminal research, notably in the past decade, helped to unravel how key antimicrobial T cell populations, notably unconventional subsets, recognize bacteria and the intrathymic developmental processes that are involved to seed populations into the periphery. In the case of γδ T cells, the process of identifying γδ T cell antigens is complicated and, as such, the nature of bonafide antigens that respond to γδ TCRs is largely unclear [26]. New developments shed light on γδ TCR-driven antigen recognition and further work could theoretically uncover novel antigens from bacterial sources. Similarly, the repertoire of MAIT cell antigens is expanding and the flexibility observed in the MR1-binding cleft suggest that many more antigens could be presented to MAIT cells or T cells that can react to MR1-restricted antigens. The next ten years will inevitably provide new discoveries that will help to further define the fundamental process underlying T cell control of bacterial infections. Such information will be incredibly informative for novel therapies and vaccination strategies. However, T cell immunity is not always sterilizing and bacteria have evolved ways to evade T cell immunosurveillance, utilizing strategies to hide in T cells or APCs or approaches that inhibit or exhaust T cell function. Furthermore, bacteria can release pore-forming toxins that kill T cells and potent exotoxins (SAgs) that bind TCRs, re-wiring the cells into a hyperactive state that induces anergy and the production of a cytokine storm that could be lethal to the host. Subsequently, strategies to vaccinate against bacterial toxins are equally as important as those that elicit protective, antimicrobial immunity. The production of SAgs is strongly associated with many life-threatening conditions, such as pneumonia, sepsis, and TSS, and these toxins accordingly have become pivotal targets for vaccination strategies in recent clinical trials. As such, it is important to learn more about how bacteria evade the adaptive immune system but also the breadth of interactions that bacterial toxins have evolved, in order to design therapies that neutralize their activity.
Author Contributions
Conceptualization: J.E.M. Investigation, F.R.S. and J.E.M. Writing—original draft preparation: F.R.S. and J.E.M. Writing—review and editing: F.R.S. and J.E.M. Visualization: F.R.S. and J.E.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the Welsh Government (Sêr Cymru) and the Wellcome Trust (grant 217096/Z/19/Z).
Conflicts of Interest
The authors have no conflict of interest.
References
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, D.J.; Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003, 300, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 2015, 15, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.; Farenc, C.; Mukhopadhyay, M.; Jayasinghe, D.; Decroos, A.; Benati, D.; Tan, L.L.; Ciacchi, L.; Reid, H.H.; Rossjohn, J.; et al. CD4(+) T cell-mediated HLA class II cross-restriction in HIV controllers. Sci. Immunol. 2018, 3, eaat0687. [Google Scholar] [CrossRef]
- Ranasinghe, S.; Lamothe, P.A.; Soghoian, D.Z.; Kazer, S.W.; Cole, M.B.; Shalek, A.K.; Yosef, N.; Jones, R.B.; Donaghey, F.; Nwonu, C.; et al. Antiviral CD8(+) T Cells Restricted by Human Leukocyte Antigen Class II Exist during Natural HIV Infection and Exhibit Clonal Expansion. Immunity 2016, 45, 917–930. [Google Scholar] [CrossRef]
- La Gruta, N.L.; Gras, S.; Daley, S.R.; Thomas, P.G.; Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 2018, 18, 467–478. [Google Scholar] [CrossRef]
- Arstila, T.P.; Casrouge, A.; Baron, V.; Even, J.; Kanellopoulos, J.; Kourilsky, P. A direct estimate of the human alphabeta T cell receptor diversity. Science 1999, 286, 958–961. [Google Scholar] [CrossRef]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Jenkins, M.K.; Moon, J.J. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J. Immunol. 2012, 188, 4135–4140. [Google Scholar] [CrossRef]
- Kaech, S.M.; Ahmed, R. Memory CD8+ T cell differentiation: Initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2001, 2, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.L.; Jamieson, B.D.; Somasundaram, T.; Ahmed, R. Cytotoxic T-cell memory without antigen. Nature 1994, 369, 648–652. [Google Scholar] [CrossRef]
- Surh, C.D.; Sprent, J. Homeostasis of naive and memory T cells. Immunity 2008, 29, 848–862. [Google Scholar] [CrossRef]
- Goldrath, A.W.; Sivakumar, P.V.; Glaccum, M.; Kennedy, M.K.; Bevan, M.J.; Benoist, C.; Mathis, D.; Butz, E.A. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J. Exp. Med. 2002, 195, 1515–1522. [Google Scholar] [CrossRef]
- Schmidt, C.S.; Mescher, M.F. Peptide antigen priming of naive, but not memory, CD8 T cells requires a third signal that can be provided by IL-12. J. Immunol. 2002, 168, 5521–5529. [Google Scholar] [CrossRef]
- Hosking, M.P.; Flynn, C.T.; Whitton, J.L. Type I IFN Signaling Is Dispensable during Secondary Viral Infection. PLoS. Pathog. 2016, 12, e1005861. [Google Scholar] [CrossRef]
- Bonilla, W.V.; Frohlich, A.; Senn, K.; Kallert, S.; Fernandez, M.; Johnson, S.; Kreutzfeldt, M.; Hegazy, A.N.; Schrick, C.; Fallon, P.G.; et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science 2012, 335, 984–989. [Google Scholar] [CrossRef]
- McLaren, J.E.; Clement, M.; Marsden, M.; Miners, K.L.; Llewellyn-Lacey, S.; Grant, E.J.; Rubina, A.; Gimeno Brias, S.; Gostick, E.; Stacey, M.A.; et al. IL-33 Augments Virus-Specific Memory T Cell Inflation and Potentiates the Efficacy of an Attenuated Cytomegalovirus-Based Vaccine. J. Immunol. 2019, 202, 943–955. [Google Scholar] [CrossRef]
- Grant, E.J.; Nguyen, A.T.; Lobos, C.A.; Szeto, C.; Chatzileontiadou, D.S.M.; Gras, S. The unconventional role of HLA-E: The road less traveled. Mol. Immunol. 2020, 120, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Beckman, E.M.; Porcelli, S.A.; Morita, C.T.; Behar, S.M.; Furlong, S.T.; Brenner, M.B. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature 1994, 372, 691–694. [Google Scholar] [CrossRef]
- Van Rhijn, I.; Godfrey, D.I.; Rossjohn, J.; Moody, D.B. Lipid and small-molecule display by CD1 and MR1. Nat. Rev. Immunol. 2015, 15, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; Uldrich, A.P.; McCluskey, J.; Rossjohn, J.; Moody, D.B. The burgeoning family of unconventional T cells. Nat. Immunol. 2015, 16, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.E.; Willcox, C.R. gammadelta TCR ligands: The quest to solve a 500-million-year-old mystery. Nat. Immunol. 2019, 20, 121–128. [Google Scholar] [CrossRef]
- Hayday, A.C.; Vantourout, P. The Innate Biologies of Adaptive Antigen Receptors. Annu. Rev. Immunol. 2020, 38, 487–510. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Reith, W.; Trowsdale, J. Regulation of Immunity by Butyrophilins. Annu. Rev. Immunol. 2016, 34, 151–172. [Google Scholar] [CrossRef]
- Le Nours, J.; Gherardin, N.A.; Ramarathinam, S.H.; Awad, W.; Wiede, F.; Gully, B.S.; Khandokar, Y.; Praveena, T.; Wubben, J.M.; Sandow, J.J.; et al. A class of gammadelta T cell receptors recognize the underside of the antigen-presenting molecule MR1. Science 2019, 366, 1522–1527. [Google Scholar] [CrossRef]
- Bai, L.; Picard, D.; Anderson, B.; Chaudhary, V.; Luoma, A.; Jabri, B.; Adams, E.J.; Savage, P.B.; Bendelac, A. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vdelta1 TCR. Eur. J. Immunol. 2012, 42, 2505–2510. [Google Scholar] [CrossRef]
- Luoma, A.M.; Castro, C.D.; Mayassi, T.; Bembinster, L.A.; Bai, L.; Picard, D.; Anderson, B.; Scharf, L.; Kung, J.E.; Sibener, L.V.; et al. Crystal structure of Vdelta1 T cell receptor in complex with CD1d-sulfatide shows MHC-like recognition of a self-lipid by human gammadelta T cells. Immunity 2013, 39, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Ly, D.; Castro, C.D.; Li, N.S.; Hawk, A.J.; Altman, J.D.; Meredith, S.C.; Piccirilli, J.A.; Moody, D.B.; Adams, E.J. Molecular Analysis of Lipid-Reactive Vdelta1 gammadelta T Cells Identified by CD1c Tetramers. J. Immunol. 2016, 196, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Uldrich, A.P.; Le Nours, J.; Pellicci, D.G.; Gherardin, N.A.; McPherson, K.G.; Lim, R.T.; Patel, O.; Beddoe, T.; Gras, S.; Rossjohn, J.; et al. CD1d-lipid antigen recognition by the gammadelta TCR. Nat. Immunol. 2013, 14, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Pizarro, J.C.; Holmes, M.A.; McBeth, C.; Groh, V.; Spies, T.; Strong, R.K. Crystal structure of a gammadelta T-cell receptor specific for the human MHC class I homolog MICA. Proc. Natl. Acad. Sci. USA 2011, 108, 2414–2419. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, P.M.; Roy, S.; Nakatsugawa, M.; Chen, E.L.Y.; Nguyen, L.; Millar, D.G.; Ohashi, P.S.; Hirano, N.; Adams, E.J.; Zuniga-Pflucker, J.C. Generation and molecular recognition of melanoma-associated antigen-specific human gammadelta T cells. Sci. Immunol. 2018, 3. [Google Scholar]
- Liuzzi, A.R.; McLaren, J.E.; Price, D.A.; Eberl, M. Early innate responses to pathogens: Pattern recognition by unconventional human T-cells. Curr. Opin. Immunol. 2015, 36, 31–37. [Google Scholar] [CrossRef]
- Kerksiek, K.M.; Pamer, E.G. T cell responses to bacterial infection. Curr. Opin. Immunol. 1999, 11, 400–405. [Google Scholar] [CrossRef]
- Salgado-Pabon, W.; Celli, S.; Arena, E.T.; Nothelfer, K.; Roux, P.; Sellge, G.; Frigimelica, E.; Bousso, P.; Sansonetti, P.J.; Phalipon, A. Shigella impairs T lymphocyte dynamics in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 4458–4463. [Google Scholar] [CrossRef]
- Tuffs, S.W.; Haeryfar, S.M.M.; McCormick, J.K. Manipulation of Innate and Adaptive Immunity by Staphylococcal Superantigens. Pathogens 2018, 7, 53. [Google Scholar] [CrossRef]
- Fraunholz, M.; Sinha, B. Intracellular Staphylococcus aureus: Live-in and let die. Front. Cell Infect. Microbiol. 2012, 2, 43. [Google Scholar] [CrossRef]
- Silva, M.T. Classical labeling of bacterial pathogens according to their lifestyle in the host: Inconsistencies and alternatives. Front. Microbiol. 2012, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H. Intracellular pathogens: Living in an extreme environment. Immunol. Rev. 2011, 240, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Khairallah, C.; Sheridan, B.S. Listeria Monocytogenes: A Model Pathogen Continues to Refine Our Knowledge of the CD8 T Cell Response. Pathogens 2018, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Flynn, J.L. CD8 T cells and Mycobacterium tuberculosis infection. Semin Immunopathol. 2015, 37, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Dunmire, S.; McSorley, S.J. MHC class-I-restricted CD8 T cells play a protective role during primary Salmonella infection. Immunol. Lett. 2012, 148, 138–143. [Google Scholar] [CrossRef]
- O’Brien, E.C.; McLoughlin, R.M. Considering the ‘Alternatives’ for Next-Generation Anti-Staphylococcus aureus Vaccine Development. Trends Mol. Med. 2019, 25, 171–184. [Google Scholar] [CrossRef]
- Hess, J.; Ladel, C.; Miko, D.; Kaufmann, S.H. Salmonella typhimurium aroA- infection in gene-targeted immunodeficient mice: Major role of CD4+ TCR-alpha beta cells and IFN-gamma in bacterial clearance independent of intracellular location. J. Immunol. 1996, 156, 3321–3326. [Google Scholar]
- Thakur, A.; Mikkelsen, H.; Jungersen, G. Intracellular Pathogens: Host Immunity and Microbial Persistence Strategies. J. Immunol. Res. 2019, 2019, 1356540. [Google Scholar] [CrossRef]
- Cruz-Adalia, A.; Ramirez-Santiago, G.; Calabia-Linares, C.; Torres-Torresano, M.; Feo, L.; Galan-Diez, M.; Fernandez-Ruiz, E.; Pereiro, E.; Guttmann, P.; Chiappi, M.; et al. T cells kill bacteria captured by transinfection from dendritic cells and confer protection in mice. Cell Host Microbe 2014, 15, 611–622. [Google Scholar] [CrossRef]
- Cruz-Adalia, A.; Ramirez-Santiago, G.; Osuna-Perez, J.; Torres-Torresano, M.; Zorita, V.; Martinez-Riano, A.; Boccasavia, V.; Borroto, A.; Martinez Del Hoyo, G.; Gonzalez-Granado, J.M.; et al. Conventional CD4(+) T cells present bacterial antigens to induce cytotoxic and memory CD8(+) T cell responses. Nat. Commun 2017, 8, 1591. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Teichgraber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Huster, K.M.; Koffler, M.; Stemberger, C.; Schiemann, M.; Wagner, H.; Busch, D.H. Unidirectional development of CD8+ central memory T cells into protective Listeria-specific effector memory T cells. Eur. J. Immunol. 2006, 36, 1453–1464. [Google Scholar] [CrossRef]
- Olson, J.A.; McDonald-Hyman, C.; Jameson, S.C.; Hamilton, S.E. Effector-like CD8(+) T cells in the memory population mediate potent protective immunity. Immunity 2013, 38, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Vezys, V.; Marzo, A.L.; Lefrancois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 2001, 291, 2413–2417. [Google Scholar] [CrossRef]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef]
- Masopust, D.; Choo, D.; Vezys, V.; Wherry, E.J.; Duraiswamy, J.; Akondy, R.; Wang, J.; Casey, K.A.; Barber, D.L.; Kawamura, K.S.; et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 2010, 207, 553–564. [Google Scholar] [CrossRef]
- Wakim, L.M.; Waithman, J.; van Rooijen, N.; Heath, W.R.; Carbone, F.R. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 2008, 319, 198–202. [Google Scholar] [CrossRef]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Woodward-Davis, A.; Liu, R.; Hu, Y.; Villadangos, J.; Smyth, G.; Bevan, M.J. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 2012, 189, 3462–3471. [Google Scholar] [CrossRef]
- Mackay, L.K.; Braun, A.; Macleod, B.L.; Collins, N.; Tebartz, C.; Bedoui, S.; Carbone, F.R.; Gebhardt, T. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 2015, 194, 2059–2063. [Google Scholar] [CrossRef]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef]
- Ma, C.; Mishra, S.; Demel, E.L.; Liu, Y.; Zhang, N. TGF-beta Controls the Formation of Kidney-Resident T Cells via Promoting Effector T Cell Extravasation. J. Immunol. 2017, 198, 749–756. [Google Scholar] [CrossRef]
- Steinert, E.M.; Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Manlove, L.S.; Igyarto, B.Z.; Southern, P.J.; Masopust, D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 2015, 161, 737–749. [Google Scholar] [CrossRef]
- Cheuk, S.; Schlums, H.; Gallais Serezal, I.; Martini, E.; Chiang, S.C.; Marquardt, N.; Gibbs, A.; Detlofsson, E.; Introini, A.; Forkel, M.; et al. CD49a Expression Defines Tissue-Resident CD8(+) T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017, 46, 287–300. [Google Scholar] [CrossRef]
- Hombrink, P.; Helbig, C.; Backer, R.A.; Piet, B.; Oja, A.E.; Stark, R.; Brasser, G.; Jongejan, A.; Jonkers, R.E.; Nota, B.; et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat. Immunol. 2016, 17, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Kratchmarov, R.; Miron, M.; Carpenter, D.J.; Senda, T.; Lerner, H.; Friedman, A.; Reiner, S.L.; Farber, D.L. Functional heterogeneity of human tissue-resident memory T cells based on dye efflux capacities. JCI Insight 2018, 3, e123568. [Google Scholar] [CrossRef] [PubMed]
- Pallett, L.J.; Davies, J.; Colbeck, E.J.; Robertson, F.; Hansi, N.; Easom, N.J.W.; Burton, A.R.; Stegmann, K.A.; Schurich, A.; Swadling, L.; et al. IL-2(high) tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017, 214, 1567–1580. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.J.; Toma, C.; Yu, B.; Zhang, K.; Omilusik, K.; Phan, A.T.; Wang, D.; Getzler, A.J.; Nguyen, T.; Crotty, S.; et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 2017, 552, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, B.; Son, Y.M.; Wang, Z.; Jiang, L.; Xiang, M.; Ye, Z.; Beckermann, K.E.; Wu, Y.; Jenkins, J.W.; et al. The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8(+) T Cell Fitness and Functionality. Immunity 2019, 51, 491–507 e7. [Google Scholar] [CrossRef]
- Casey, K.A.; Fraser, K.A.; Schenkel, J.M.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef]
- Zhang, N.; Bevan, M.J. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 2013, 39, 687–696. [Google Scholar] [CrossRef]
- Sheridan, B.S.; Pham, Q.M.; Lee, Y.T.; Cauley, L.S.; Puddington, L.; Lefrancois, L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 2014, 40, 747–757. [Google Scholar] [CrossRef]
- Beura, L.K.; Mitchell, J.S.; Thompson, E.A.; Schenkel, J.M.; Mohammed, J.; Wijeyesinghe, S.; Fonseca, R.; Burbach, B.J.; Hickman, H.D.; Vezys, V.; et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol. 2018, 19, 173–182. [Google Scholar] [CrossRef]
- Park, S.L.; Zaid, A.; Hor, J.L.; Christo, S.N.; Prier, J.E.; Davies, B.; Alexandre, Y.O.; Gregory, J.L.; Russell, T.A.; Gebhardt, T.; et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 2018, 19, 183–191. [Google Scholar] [CrossRef]
- Perdomo, C.; Zedler, U.; Kuhl, A.A.; Lozza, L.; Saikali, P.; Sander, L.E.; Vogelzang, A.; Kaufmann, S.H.; Kupz, A. Mucosal BCG Vaccination Induces Protective Lung-Resident Memory T Cell Populations against Tuberculosis. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Connor, L.M.; Harvie, M.C.; Rich, F.J.; Quinn, K.M.; Brinkmann, V.; Le Gros, G.; Kirman, J.R. A key role for lung-resident memory lymphocytes in protective immune responses after BCG vaccination. Eur. J. Immunol. 2010, 40, 2482–2492. [Google Scholar] [CrossRef] [PubMed]
- Haddadi, S.; Thanthrige-Don, N.; Afkhami, S.; Khera, A.; Jeyanathan, M.; Xing, Z. Expression and role of VLA-1 in resident memory CD8 T cell responses to respiratory mucosal viral-vectored immunization against tuberculosis. Sci. Rep. 2017, 7, 9525. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Zak, D.E.; Xu, G.; Ford, J.C.; Marshall, E.E.; Malouli, D.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ainslie, E.; et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat. Med. 2018, 24, 130–143. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Bevan, M.J.; Fink, P.J. Local Inflammatory Cues Regulate Differentiation and Persistence of CD8(+) Tissue-Resident Memory T Cells. Cell Rep. 2017, 19, 114–124. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Bevan, M.J. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat. Immunol. 2015, 16, 406–414. [Google Scholar] [CrossRef]
- Benoun, J.M.; Peres, N.G.; Wang, N.; Pham, O.H.; Rudisill, V.L.; Fogassy, Z.N.; Whitney, P.G.; Fernandez-Ruiz, D.; Gebhardt, T.; Pham, Q.M.; et al. Optimal protection against Salmonella infection requires noncirculating memory. Proc. Natl. Acad. Sci. USA 2018, 115, 10416–10421. [Google Scholar] [CrossRef]
- Lee, S.J.; Benoun, J.; Sheridan, B.S.; Fogassy, Z.; Pham, O.; Pham, Q.M.; Puddington, L.; McSorley, S.J. Dual Immunization with SseB/Flagellin Provides Enhanced Protection against Salmonella Infection Mediated by Circulating Memory Cells. J. Immunol. 2017, 199, 1353–1361. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Braud, V.; Jones, E.Y.; McMichael, A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur. J. Immunol. 1997, 27, 1164–1169. [Google Scholar] [CrossRef]
- Tomasec, P.; Braud, V.M.; Rickards, C.; Powell, M.B.; McSharry, B.P.; Gadola, S.; Cerundolo, V.; Borysiewicz, L.K.; McMichael, A.J.; Wilkinson, G.W. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 2000, 287, 1031. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, A.S.; Grotzke, J.E.; Lines, R.A.; Lewinsohn, D.A.; McNabb, A.L.; Streblow, D.N.; Braud, V.M.; Grieser, H.J.; Belisle, J.T.; Lewinsohn, D.M. HLA-E-dependent presentation of Mtb-derived antigen to human CD8+ T cells. J. Exp. Med. 2002, 196, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Lewinsohn, D.M.; Alderson, M.R.; Briden, A.L.; Riddell, S.R.; Reed, S.G.; Grabstein, K.H. Characterization of human CD8+ T cells reactive with Mycobacterium tuberculosis-infected antigen-presenting cells. J. Exp. Med. 1998, 187, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Salerno-Goncalves, R.; Fernandez-Vina, M.; Lewinsohn, D.M.; Sztein, M.B. Identification of a human HLA-E-restricted CD8+ T cell subset in volunteers immunized with Salmonella enterica serovar Typhi strain Ty21a typhoid vaccine. J. Immunol. 2004, 173, 5852–5862. [Google Scholar] [CrossRef]
- Caccamo, N.; Pietra, G.; Sullivan, L.C.; Brooks, A.G.; Prezzemolo, T.; La Manna, M.P.; Di Liberto, D.; Joosten, S.A.; van Meijgaarden, K.E.; Di Carlo, P.; et al. Human CD8 T lymphocytes recognize Mycobacterium tuberculosis antigens presented by HLA-E during active tuberculosis and express type 2 cytokines. Eur. J. Immunol. 2015, 45, 1069–1081. [Google Scholar] [CrossRef]
- Harriff, M.J.; Wolfe, L.M.; Swarbrick, G.; Null, M.; Cansler, M.E.; Canfield, E.T.; Vogt, T.; Toren, K.G.; Li, W.; Jackson, M.; et al. HLA-E Presents Glycopeptides from the Mycobacterium tuberculosis Protein MPT32 to Human CD8(+) T cells. Sci. Rep. 2017, 7, 4622. [Google Scholar] [CrossRef]
- Joosten, S.A.; van Meijgaarden, K.E.; van Weeren, P.C.; Kazi, F.; Geluk, A.; Savage, N.D.; Drijfhout, J.W.; Flower, D.R.; Hanekom, W.A.; Klein, M.R.; et al. Mycobacterium tuberculosis peptides presented by HLA-E molecules are targets for human CD8 T-cells with cytotoxic as well as regulatory activity. PLoS. Pathog. 2010, 6, e1000782. [Google Scholar] [CrossRef]
- Prezzemolo, T.; van Meijgaarden, K.E.; Franken, K.; Caccamo, N.; Dieli, F.; Ottenhoff, T.H.M.; Joosten, S.A. Detailed characterization of human Mycobacterium tuberculosis specific HLA-E restricted CD8(+) T cells. Eur. J. Immunol. 2018, 48, 293–305. [Google Scholar] [CrossRef]
- van Meijgaarden, K.E.; Haks, M.C.; Caccamo, N.; Dieli, F.; Ottenhoff, T.H.; Joosten, S.A. Human CD8+ T-cells recognizing peptides from Mycobacterium tuberculosis (Mtb) presented by HLA-E have an unorthodox Th2-like, multifunctional, Mtb inhibitory phenotype and represent a novel human T-cell subset. PLoS. Pathog. 2015, 11, e1004671. [Google Scholar] [CrossRef]
- Walters, L.C.; Harlos, K.; Brackenridge, S.; Rozbesky, D.; Barrett, J.R.; Jain, V.; Walter, T.S.; O’Callaghan, C.A.; Borrow, P.; Toebes, M.; et al. Pathogen-derived HLA-E bound epitopes reveal broad primary anchor pocket tolerability and conformationally malleable peptide binding. Nat. Commun. 2018, 9, 3137. [Google Scholar] [CrossRef]
- Grotzke, J.E.; Harriff, M.J.; Siler, A.C.; Nolt, D.; Delepine, J.; Lewinsohn, D.A.; Lewinsohn, D.M. The Mycobacterium tuberculosis phagosome is a HLA-I processing competent organelle. PLoS. Pathog. 2009, 5, e1000374. [Google Scholar] [CrossRef] [PubMed]
- Harriff, M.J.; Cansler, M.E.; Toren, K.G.; Canfield, E.T.; Kwak, S.; Gold, M.C.; Lewinsohn, D.M. Human lung epithelial cells contain Mycobacterium tuberculosis in a late endosomal vacuole and are efficiently recognized by CD8(+) T cells. PLoS. ONE 2014, 9, e97515. [Google Scholar] [CrossRef]
- Salerno-Goncalves, R.; Wahid, R.; Sztein, M.B. Ex Vivo kinetics of early and long-term multifunctional human leukocyte antigen E-specific CD8+ cells in volunteers immunized with the Ty21a typhoid vaccine. Clin. Vaccine Immunol. 2010, 17, 1305–1314. [Google Scholar] [CrossRef]
- Rudolph, M.E.; McArthur, M.A.; Magder, L.S.; Barnes, R.S.; Chen, W.H.; Sztein, M.B. Age-Associated Heterogeneity of Ty21a-Induced T Cell Responses to HLA-E Restricted Salmonella Typhi Antigen Presentation. Front. Immunol. 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Fresnay, S.; McArthur, M.A.; Magder, L.; Darton, T.C.; Jones, C.; Waddington, C.S.; Blohmke, C.J.; Angus, B.; Levine, M.M.; Pollard, A.J.; et al. Salmonella Typhi-specific multifunctional CD8+ T cells play a dominant role in protection from typhoid fever in humans. J. Transl Med. 2016, 14, 62. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Barricarte, R.; Markle, J.G.; Ma, C.S.; Deenick, E.K.; Ramirez-Alejo, N.; Mele, F.; Latorre, D.; Mahdaviani, S.A.; Aytekin, C.; Mansouri, D.; et al. Human IFN-gamma immunity to mycobacteria is governed by both IL-12 and IL-23. Sci. Immunol. 2018, 3. [Google Scholar]
- Elkins, K.L.; Colombini, S.M.; Meierovics, A.I.; Chu, M.C.; Chou, A.Y.; Cowley, S.C. Survival of secondary lethal systemic Francisella LVS challenge depends largely on interferon gamma. Microbes Infect. 2010, 12, 28–36. [Google Scholar] [CrossRef]
- Wozniak, T.M.; Saunders, B.M.; Ryan, A.A.; Britton, W.J. Mycobacterium bovis BCG-specific Th17 cells confer partial protection against Mycobacterium tuberculosis infection in the absence of gamma interferon. Infect. Immun 2010, 78, 4187–4194. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, B.; Guo, Y.; Li, W.; Tian, Y.; Sonnenberg, G.F.; Weiser, J.N.; Ni, X.; Shen, H. Cross-protective mucosal immunity mediated by memory Th17 cells against Streptococcus pneumoniae lung infection. Mucosal Immunol. 2017, 10, 250–259. [Google Scholar] [CrossRef]
- Khader, S.A.; Pearl, J.E.; Sakamoto, K.; Gilmartin, L.; Bell, G.K.; Jelley-Gibbs, D.M.; Ghilardi, N.; deSauvage, F.; Cooper, A.M. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 2005, 175, 788–795. [Google Scholar] [CrossRef]
- Cooper, A.M.; Dalton, D.K.; Stewart, T.A.; Griffin, J.P.; Russell, D.G.; Orme, I.M. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 1993, 178, 2243–2247. [Google Scholar] [CrossRef] [PubMed]
- Meeks, K.D.; Sieve, A.N.; Kolls, J.K.; Ghilardi, N.; Berg, R.E. IL-23 is required for protection against systemic infection with Listeria monocytogenes. J. Immunol. 2009, 183, 8026–8034. [Google Scholar] [CrossRef] [PubMed]
- Pepper, M.; Linehan, J.L.; Pagan, A.J.; Zell, T.; Dileepan, T.; Cleary, P.P.; Jenkins, M.K. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat. Immunol. 2010, 11, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.J.; Sutton, C.E.; Higgins, S.; Allen, A.C.; Walsh, K.; Misiak, A.; Lavelle, E.C.; McLoughlin, R.M.; Mills, K.H. Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: Towards the rational design of an improved acellular pertussis vaccine. PLoS. Pathog. 2013, 9, e1003264. [Google Scholar] [CrossRef]
- Joshi, A.; Pancari, G.; Cope, L.; Bowman, E.P.; Cua, D.; Proctor, R.A.; McNeely, T. Immunization with Staphylococcus aureus iron regulated surface determinant B (IsdB) confers protection via Th17/IL17 pathway in a murine sepsis model. Hum. Vaccin. Immunother 2012, 8, 336–346. [Google Scholar] [CrossRef]
- Lin, L.; Ibrahim, A.S.; Xu, X.; Farber, J.M.; Avanesian, V.; Baquir, B.; Fu, Y.; French, S.W.; Edwards, J.E., Jr.; Spellberg, B. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS. Pathog. 2009, 5, e1000703. [Google Scholar] [CrossRef]
- Brown, A.F.; Murphy, A.G.; Lalor, S.J.; Leech, J.M.; O’Keeffe, K.M.; Mac Aogain, M.; O’Halloran, D.P.; Lacey, K.A.; Tavakol, M.; Hearnden, C.H.; et al. Memory Th1 Cells Are Protective in Invasive Staphylococcus aureus Infection. PLoS. Pathog. 2015, 11, e1005226. [Google Scholar] [CrossRef]
- Johnston, R.J.; Poholek, A.C.; DiToro, D.; Yusuf, I.; Eto, D.; Barnett, B.; Dent, A.L.; Craft, J.; Crotty, S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009, 325, 1006–1010. [Google Scholar] [CrossRef]
- Nurieva, R.I.; Chung, Y.; Martinez, G.J.; Yang, X.O.; Tanaka, S.; Matskevitch, T.D.; Wang, Y.H.; Dong, C. Bcl6 mediates the development of T follicular helper cells. Science 2009, 325, 1001–1005. [Google Scholar] [CrossRef]
- Yu, D.; Rao, S.; Tsai, L.M.; Lee, S.K.; He, Y.; Sutcliffe, E.L.; Srivastava, M.; Linterman, M.; Zheng, L.; Simpson, N.; et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 2009, 31, 457–468. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef] [PubMed]
- Breitfeld, D.; Ohl, L.; Kremmer, E.; Ellwart, J.; Sallusto, F.; Lipp, M.; Forster, R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 2000, 192, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Ansel, K.M.; Ngo, V.N.; Hyman, P.L.; Luther, S.A.; Forster, R.; Sedgwick, J.D.; Browning, J.L.; Lipp, M.; Cyster, J.G. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 2000, 406, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hatzi, K.; Nance, J.P.; Kroenke, M.A.; Bothwell, M.; Haddad, E.K.; Melnick, A.; Crotty, S. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J. Exp. Med. 2015, 212, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Kusam, S.; Toney, L.M.; Sato, H.; Dent, A.L. Inhibition of Th2 differentiation and GATA-3 expression by BCL-6. J. Immunol. 2003, 170, 2435–2441. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, N.; Morita, R.; Bourdery, L.; Bentebibel, S.E.; Zurawski, S.M.; Banchereau, J.; Ueno, H. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity 2009, 31, 158–169. [Google Scholar] [CrossRef]
- Batten, M.; Ramamoorthi, N.; Kljavin, N.M.; Ma, C.S.; Cox, J.H.; Dengler, H.S.; Danilenko, D.M.; Caplazi, P.; Wong, M.; Fulcher, D.A.; et al. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J. Exp. Med. 2010, 207, 2895–2906. [Google Scholar] [CrossRef]
- Hale, J.S.; Ahmed, R. Memory T follicular helper CD4 T cells. Front. Immunol. 2015, 6, 16. [Google Scholar] [CrossRef]
- Brenna, E.; Davydov, A.N.; Ladell, K.; McLaren, J.E.; Bonaiuti, P.; Metsger, M.; Ramsden, J.D.; Gilbert, S.C.; Lambe, T.; Price, D.A.; et al. CD4(+) T Follicular Helper Cells in Human Tonsils and Blood Are Clonally Convergent but Divergent from Non-Tfh CD4(+) Cells. Cell Rep. 2020, 30, 137–152.e5. [Google Scholar] [CrossRef]
- Bai, X.; Chi, X.; Qiao, Q.; Xie, S.; Wan, S.; Ni, L.; Wang, P.; Jin, W.; Dong, C. T Follicular Helper Cells Regulate Humoral Response for Host Protection against Intestinal Citrobacter rodentium Infection. J. Immunol. 2020, 204, 2754–2761. [Google Scholar] [CrossRef]
- Slight, S.R.; Rangel-Moreno, J.; Gopal, R.; Lin, Y.; Fallert Junecko, B.A.; Mehra, S.; Selman, M.; Becerril-Villanueva, E.; Baquera-Heredia, J.; Pavon, L.; et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J. Clin. Investig. 2013, 123, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Havenar-Daughton, C.; Kendric, K.; Al-Kolla, R.; Kaushik, K.; Rosales, S.L.; Anderson, E.L.; LaRock, C.N.; Vijayanand, P.; Seumois, G.; et al. Recurrent group A Streptococcus tonsillitis is an immunosusceptibility disease involving antibody deficiency and aberrant TFH cells. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Kallies, A. Transcriptional Regulation of Tissue-Resident Lymphocytes. Trends Immunol. 2017, 38, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Thom, J.T.; Weber, T.C.; Walton, S.M.; Torti, N.; Oxenius, A. The Salivary Gland Acts as a Sink for Tissue-Resident Memory CD8(+) T Cells, Facilitating Protection from Local Cytomegalovirus Infection. Cell Rep. 2015, 13, 1125–1136. [Google Scholar] [CrossRef]
- Collins, N.; Jiang, X.; Zaid, A.; Macleod, B.L.; Li, J.; Park, C.O.; Haque, A.; Bedoui, S.; Heath, W.R.; Mueller, S.N.; et al. Skin CD4(+) memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat. Commun 2016, 7, 11514. [Google Scholar] [CrossRef]
- Gebhardt, T.; Whitney, P.G.; Zaid, A.; Mackay, L.K.; Brooks, A.G.; Heath, W.R.; Carbone, F.R.; Mueller, S.N. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 2011, 477, 216–219. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Turner, D.; Pham, Q.; Wherry, E.J.; Lefrancois, L.; Farber, D.L. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol. 2011, 187, 5510–5514. [Google Scholar] [CrossRef]
- Sakai, S.; Kauffman, K.D.; Schenkel, J.M.; McBerry, C.C.; Mayer-Barber, K.D.; Masopust, D.; Barber, D.L. Cutting edge: Control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J. Immunol. 2014, 192, 2965–2969. [Google Scholar] [CrossRef]
- Wilk, M.M.; Misiak, A.; McManus, R.M.; Allen, A.C.; Lynch, M.A.; Mills, K.H.G. Lung CD4 Tissue-Resident Memory T Cells Mediate Adaptive Immunity Induced by Previous Infection of Mice with Bordetella pertussis. J. Immunol. 2017, 199, 233–243. [Google Scholar] [CrossRef]
- Smith, N.M.; Wasserman, G.A.; Coleman, F.T.; Hilliard, K.L.; Yamamoto, K.; Lipsitz, E.; Malley, R.; Dooms, H.; Jones, M.R.; Quinton, L.J.; et al. Regionally compartmentalized resident memory T cells mediate naturally acquired protection against pneumococcal pneumonia. Mucosal Immunol. 2018, 11, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, P.A.; Fu, H.H.; Qiu, Z.; Khairallah, C.; Pham, Q.M.; Puddington, L.; Khanna, K.M.; Lefrancois, L.; Sheridan, B.S. Differentiation of distinct long-lived memory CD4 T cells in intestinal tissues after oral Listeria monocytogenes infection. Mucosal Immunol. 2017, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Bishu, S.; Hou, G.; El Zaatari, M.; Bishu, S.R.; Popke, D.; Zhang, M.; Grasberger, H.; Zou, W.; Stidham, R.W.; Higgins, P.D.R.; et al. Citrobacter rodentium Induces Tissue-Resident Memory CD4(+) T Cells. Infect. Immun. 2019, 87, e00295-19. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Yu, H.; Strank, N.O.; Karunakaran, K.; Zhu, Y.; Brunham, R.C. B Cell Presentation of Chlamydia Antigen Selects Out Protective CD4gamma13 T Cells: Implications for Genital Tract Tissue-Resident Memory Lymphocyte Clusters. Infect. Immun 2018, 86. [Google Scholar] [CrossRef]
- Schreiner, D.; King, C.G. CD4+ Memory T Cells at Home in the Tissue: Mechanisms for Health and Disease. Front. Immunol. 2018, 9, 2394. [Google Scholar] [CrossRef]
- Park, C.O.; Fu, X.; Jiang, X.; Pan, Y.; Teague, J.E.; Collins, N.; Tian, T.; O’Malley, J.T.; Emerson, R.O.; Kim, J.H.; et al. Staged development of long-lived T-cell receptor alphabeta TH17 resident memory T-cell population to Candida albicans after skin infection. J. Allergy Clin. Immunol. 2018, 142, 647–662. [Google Scholar] [CrossRef]
- Calabi, F.; Jarvis, J.M.; Martin, L.; Milstein, C. Two classes of CD1 genes. Eur. J. Immunol. 1989, 19, 285–292. [Google Scholar] [CrossRef]
- Porcelli, S.; Brenner, M.B.; Greenstein, J.L.; Balk, S.P.; Terhorst, C.; Bleicher, P.A. Recognition of cluster of differentiation 1 antigens by human CD4-CD8-cytolytic T lymphocytes. Nature 1989, 341, 447–450. [Google Scholar] [CrossRef]
- Porcelli, S.; Morita, C.T.; Brenner, M.B. CD1b restricts the response of human CD4-8- T lymphocytes to a microbial antigen. Nature 1992, 360, 593–597. [Google Scholar] [CrossRef]
- Girardi, E.; Maricic, I.; Wang, J.; Mac, T.T.; Iyer, P.; Kumar, V.; Zajonc, D.M. Type II natural killer T cells use features of both innate-like and conventional T cells to recognize sulfatide self antigens. Nat. Immunol. 2012, 13, 851–856. [Google Scholar] [CrossRef]
- Patel, O.; Pellicci, D.G.; Gras, S.; Sandoval-Romero, M.L.; Uldrich, A.P.; Mallevaey, T.; Clarke, A.J.; Le Nours, J.; Theodossis, A.; Cardell, S.L.; et al. Recognition of CD1d-sulfatide mediated by a type II natural killer T cell antigen receptor. Nat. Immunol. 2012, 13, 857–863. [Google Scholar] [CrossRef]
- Pellicci, D.G.; Uldrich, A.P.; Le Nours, J.; Ross, F.; Chabrol, E.; Eckle, S.B.; de Boer, R.; Lim, R.T.; McPherson, K.; Besra, G.; et al. The molecular bases of delta/alphabeta T cell-mediated antigen recognition. J. Exp. Med. 2014, 211, 2599–2615. [Google Scholar] [CrossRef]
- Gras, S.; Van Rhijn, I.; Shahine, A.; Le Nours, J. Molecular recognition of microbial lipid-based antigens by T cells. Cell Mol. Life Sci. 2018, 75, 1623–1639. [Google Scholar] [CrossRef]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Motoki, K.; Ueno, H.; Nakagawa, R.; Sato, H.; Kondo, E.; et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 1997, 278, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Scotet, E.; Niemeyer, M.; Koebernick, H.; Zerrahn, J.; Maillet, S.; Hurwitz, R.; Kursar, M.; Bonneville, M.; Kaufmann, S.H.; et al. Mycobacterial phosphatidylinositol mannoside is a natural antigen for CD1d-restricted T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10685–10690. [Google Scholar] [CrossRef] [PubMed]
- Mattner, J.; Debord, K.L.; Ismail, N.; Goff, R.D.; Cantu, C., 3rd; Zhou, D.; Saint-Mezard, P.; Wang, V.; Gao, Y.; Yin, N.; et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 2005, 434, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.M., Jr.; Bates, T.C.; Izadi, H.; Radolf, J.D.; Huber, S.A.; Boyson, J.E.; Anguita, J. Local production of IFN-gamma by invariant NKT cells modulates acute Lyme carditis. J. Immunol. 2009, 182, 3728–3734. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, Y.; Illarionov, P.; Vela, J.L.; Pei, B.; Girardi, E.; Li, X.; Li, Y.; Imamura, M.; Kaneko, Y.; Okawara, A.; et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat. Immunol. 2011, 12, 966–974. [Google Scholar] [CrossRef]
- Tatituri, R.V.; Watts, G.F.; Bhowruth, V.; Barton, N.; Rothchild, A.; Hsu, F.F.; Almeida, C.F.; Cox, L.R.; Eggeling, L.; Cardell, S.; et al. Recognition of microbial and mammalian phospholipid antigens by NKT cells with diverse TCRs. Proc. Natl. Acad. Sci. USA 2013, 110, 1827–1832. [Google Scholar] [CrossRef]
- Kasmar, A.G.; Van Rhijn, I.; Magalhaes, K.G.; Young, D.C.; Cheng, T.Y.; Turner, M.T.; Schiefner, A.; Kalathur, R.C.; Wilson, I.A.; Bhati, M.; et al. Cutting Edge: CD1a tetramers and dextramers identify human lipopeptide-specific T cells ex vivo. J. Immunol. 2013, 191, 4499–4503. [Google Scholar] [CrossRef]
- Kasmar, A.G.; van Rhijn, I.; Cheng, T.Y.; Turner, M.; Seshadri, C.; Schiefner, A.; Kalathur, R.C.; Annand, J.W.; de Jong, A.; Shires, J.; et al. CD1b tetramers bind alphabeta T cell receptors to identify a mycobacterial glycolipid-reactive T cell repertoire in humans. J. Exp. Med. 2011, 208, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Ly, D.; Kasmar, A.G.; Cheng, T.Y.; de Jong, A.; Huang, S.; Roy, S.; Bhatt, A.; van Summeren, R.P.; Altman, J.D.; Jacobs, W.R.; et al. CD1c tetramers detect ex vivo T cell responses to processed phosphomycoketide antigens. J. Exp. Med. 2013, 210, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Van Rhijn, I.; Kasmar, A.; de Jong, A.; Gras, S.; Bhati, M.; Doorenspleet, M.E.; de Vries, N.; Godfrey, D.I.; Altman, J.D.; de Jager, W.; et al. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat. Immunol. 2013, 14, 706–713. [Google Scholar] [CrossRef]
- Roy, S.; Ly, D.; Li, N.S.; Altman, J.D.; Piccirilli, J.A.; Moody, D.B.; Adams, E.J. Molecular basis of mycobacterial lipid antigen presentation by CD1c and its recognition by alphabeta T cells. Proc. Natl. Acad. Sci. USA 2014, 111, E4648–E4657. [Google Scholar] [CrossRef]
- Zhao, J.; Siddiqui, S.; Shang, S.; Bian, Y.; Bagchi, S.; He, Y.; Wang, C.R. Mycolic acid-specific T cells protect against Mycobacterium tuberculosis infection in a humanized transgenic mouse model. Elife 2015, 4, e08525. [Google Scholar] [CrossRef]
- Montamat-Sicotte, D.J.; Millington, K.A.; Willcox, C.R.; Hingley-Wilson, S.; Hackforth, S.; Innes, J.; Kon, O.M.; Lammas, D.A.; Minnikin, D.E.; Besra, G.S.; et al. A mycolic acid-specific CD1-restricted T cell population contributes to acute and memory immune responses in human tuberculosis infection. J. Clin. Investig. 2011, 121, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Reinink, P.; Souter, M.N.T.; Cheng, T.Y.; van Gorkom, T.; Lenz, S.; Kubler-Kielb, J.; Strle, K.; Kremer, K.; Thijsen, S.F.T.; Steere, A.C.; et al. CD1b presents self and Borrelia burgdorferi diacylglycerols to human T cells. Eur. J. Immunol. 2019, 49, 737–746. [Google Scholar] [CrossRef]
- Van Rhijn, I.; van Berlo, T.; Hilmenyuk, T.; Cheng, T.Y.; Wolf, B.J.; Tatituri, R.V.; Uldrich, A.P.; Napolitani, G.; Cerundolo, V.; Altman, J.D.; et al. Human autoreactive T cells recognize CD1b and phospholipids. Proc. Natl. Acad. Sci. USA 2016, 113, 380–385. [Google Scholar] [CrossRef]
- Visvabharathy, L.; Genardi, S.; Cao, L.; He, Y.; Alonzo, F., 3rd; Berdyshev, E.; Wang, C.R. Group 1 CD1-restricted T cells contribute to control of systemic Staphylococcus aureus infection. PLoS. Pathog. 2020, 16, e1008443. [Google Scholar] [CrossRef]
- Adams, E.J.; Gu, S.; Luoma, A.M. Human gamma delta T cells: Evolution and ligand recognition. Cell Immunol. 2015, 296, 31–40. [Google Scholar] [CrossRef]
- Khairallah, C.; Chu, T.H.; Sheridan, B.S. Tissue Adaptations of Memory and Tissue-Resident Gamma Delta T Cells. Front. Immunol. 2018, 9, 2636. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, Y.; Li, H.; Ai, Q.; Yu, J. The microbiota protects against Pseudomonas aeruginosa pneumonia via gammadelta T cell-neutrophil axis in mice. Microbes Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chennupati, V.; Worbs, T.; Liu, X.; Malinarich, F.H.; Schmitz, S.; Haas, J.D.; Malissen, B.; Forster, R.; Prinz, I. Intra- and intercompartmental movement of gammadelta T cells: Intestinal intraepithelial and peripheral gammadelta T cells represent exclusive nonoverlapping populations with distinct migration characteristics. J. Immunol. 2010, 185, 5160–5168. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.S.; Willcox, C.R.; Hunter, S.; Kasatskaya, S.A.; Remmerswaal, E.B.M.; Salim, M.; Mohammed, F.; Bemelman, F.J.; Chudakov, D.M.; Oo, Y.H.; et al. The human Vdelta2(+) T-cell compartment comprises distinct innate-like Vgamma9(+) and adaptive Vgamma9(-) subsets. Nat. Commun. 2018, 9, 1760. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, M.M.; Gobel, T.W.; Starick, L.; Walter, L.; Herrmann, T. Vgamma9 and Vdelta2 T cell antigen receptor genes and butyrophilin 3 (BTN3) emerged with placental mammals and are concomitantly preserved in selected species like alpaca (Vicugna pacos). Immunogenetics 2014, 66, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Dimova, T.; Brouwer, M.; Gosselin, F.; Tassignon, J.; Leo, O.; Donner, C.; Marchant, A.; Vermijlen, D. Effector Vgamma9Vdelta2 T cells dominate the human fetal gammadelta T-cell repertoire. Proc. Natl. Acad. Sci. USA 2015, 112, E556–E565. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Sano, S.; Nieves, E.; De Libero, G.; Rosa, D.; Modlin, R.L.; Brenner, M.B.; Bloom, B.R.; Morita, C.T. Nonpeptide ligands for human gamma delta T cells. Proc. Natl. Acad. Sci. USA 1994, 91, 8175–8179. [Google Scholar] [CrossRef]
- Morita, C.T.; Jin, C.; Sarikonda, G.; Wang, H. Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vgamma2Vdelta2 T cells: Discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol. Rev. 2007, 215, 59–76. [Google Scholar] [CrossRef]
- Hintz, M.; Reichenberg, A.; Altincicek, B.; Bahr, U.; Gschwind, R.M.; Kollas, A.K.; Beck, E.; Wiesner, J.; Eberl, M.; Jomaa, H. Identification of (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate as a major activator for human gammadelta T cells in Escherichia coli. Febs. Lett. 2001, 509, 317–322. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Chen, H.C.; Price, A.J.; Keeble, A.H.; Davey, M.S.; James, L.C.; Eberl, M.; Trowsdale, J. Activation of human gammadelta T cells by cytosolic interactions of BTN3A1 with soluble phosphoantigens and the cytoskeletal adaptor periplakin. J. Immunol. 2015, 194, 2390–2398. [Google Scholar] [CrossRef]
- Sandstrom, A.; Peigne, C.M.; Leger, A.; Crooks, J.E.; Konczak, F.; Gesnel, M.C.; Breathnach, R.; Bonneville, M.; Scotet, E.; Adams, E.J. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vgamma9Vdelta2 T cells. Immunity 2014, 40, 490–500. [Google Scholar] [CrossRef]
- Vavassori, S.; Kumar, A.; Wan, G.S.; Ramanjaneyulu, G.S.; Cavallari, M.; El Daker, S.; Beddoe, T.; Theodossis, A.; Williams, N.K.; Gostick, E.; et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human gammadelta T cells. Nat. Immunol. 2013, 14, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, P.; Laing, A.; Woodward, M.J.; Zlatareva, I.; Apolonia, L.; Jones, A.W.; Snijders, A.P.; Malim, M.H.; Hayday, A.C. Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing gammadelta T cell biology. Proc. Natl. Acad. Sci. USA 2018, 115, 1039–1044. [Google Scholar] [CrossRef]
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nohren, A.; Begley, C.R.; Berwick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vgamma9Vdelta2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 2020, 52, 487–498.e6. [Google Scholar] [CrossRef] [PubMed]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, A.K.; et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by gammadelta T cells. Science 2020, 367, eaay5516. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.S.; Willcox, C.R.; Joyce, S.P.; Ladell, K.; Kasatskaya, S.A.; McLaren, J.E.; Hunter, S.; Salim, M.; Mohammed, F.; Price, D.A.; et al. Clonal selection in the human Vdelta1 T cell repertoire indicates gammadelta TCR-dependent adaptive immune surveillance. Nat. Commun. 2017, 8, 14760. [Google Scholar] [CrossRef]
- Deusch, K.; Luling, F.; Reich, K.; Classen, M.; Wagner, H.; Pfeffer, K. A major fraction of human intraepithelial lymphocytes simultaneously expresses the gamma/delta T cell receptor, the CD8 accessory molecule and preferentially uses the V delta 1 gene segment. Eur. J. Immunol. 1991, 21, 1053–1059. [Google Scholar] [CrossRef]
- Di Marco Barros, R.; Roberts, N.A.; Dart, R.J.; Vantourout, P.; Jandke, A.; Nussbaumer, O.; Deban, L.; Cipolat, S.; Hart, R.; Iannitto, M.L.; et al. Epithelia Use Butyrophilin-like Molecules to Shape Organ-Specific gammadelta T Cell Compartments. Cell 2016, 167, 203–218.e17. [Google Scholar] [CrossRef]
- Willcox, C.R.; Vantourout, P.; Salim, M.; Zlatareva, I.; Melandri, D.; Zanardo, L.; George, R.; Kjaer, S.; Jeeves, M.; Mohammed, F.; et al. Butyrophilin-like 3 Directly Binds a Human Vgamma4(+) T Cell Receptor Using a Modality Distinct from Clonally-Restricted Antigen. Immunity 2019, 51, 813–825.e4. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Koay, H.F.; McCluskey, J.; Gherardin, N.A. The biology and functional importance of MAIT cells. Nat. Immunol. 2019, 20, 1110–1128. [Google Scholar] [CrossRef]
- Porcelli, S.; Yockey, C.E.; Brenner, M.B.; Balk, S.P. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J. Exp. Med. 1993, 178, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tilloy, F.; Treiner, E.; Park, S.H.; Garcia, C.; Lemonnier, F.; de la Salle, H.; Bendelac, A.; Bonneville, M.; Lantz, O. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J. Exp. Med. 1999, 189, 1907–1921. [Google Scholar] [CrossRef] [PubMed]
- Treiner, E.; Duban, L.; Bahram, S.; Radosavljevic, M.; Wanner, V.; Tilloy, F.; Affaticati, P.; Gilfillan, S.; Lantz, O. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 2003, 422, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Treiner, E.; Duban, L.; Guerri, L.; Laude, H.; Toly, C.; Premel, V.; Devys, A.; Moura, I.C.; Tilloy, F.; et al. Stepwise development of MAIT cells in mouse and human. PLoS. Biol. 2009, 7, e54. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.C.; Cerri, S.; Smyk-Pearson, S.; Cansler, M.E.; Vogt, T.M.; Delepine, J.; Winata, E.; Swarbrick, G.M.; Chua, W.J.; Yu, Y.Y.; et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS. Biol. 2010, 8, e1000407. [Google Scholar] [CrossRef]
- Le Bourhis, L.; Martin, E.; Peguillet, I.; Guihot, A.; Froux, N.; Core, M.; Levy, E.; Dusseaux, M.; Meyssonnier, V.; Premel, V.; et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat. Immunol. 2010, 11, 701–708. [Google Scholar] [CrossRef]
- Corbett, A.J.; Eckle, S.B.; Birkinshaw, R.W.; Liu, L.; Patel, O.; Mahony, J.; Chen, Z.; Reantragoon, R.; Meehan, B.; Cao, H.; et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 2014, 509, 361–365. [Google Scholar] [CrossRef]
- Harriff, M.J.; McMurtrey, C.; Froyd, C.A.; Jin, H.; Cansler, M.; Null, M.; Worley, A.; Meermeier, E.W.; Swarbrick, G.; Nilsen, A.; et al. MR1 displays the microbial metabolome driving selective MR1-restricted T cell receptor usage. Sci. Immunol. 2018, 3, eaao2556. [Google Scholar] [CrossRef]
- Meermeier, E.W.; Laugel, B.F.; Sewell, A.K.; Corbett, A.J.; Rossjohn, J.; McCluskey, J.; Harriff, M.J.; Franks, T.; Gold, M.C.; Lewinsohn, D.M. Human TRAV1-2-negative MR1-restricted T cells detect S. pyogenes and alternatives to MAIT riboflavin-based antigens. Nat. Commun. 2016, 7, 12506. [Google Scholar] [CrossRef]
- Keller, A.N.; Eckle, S.B.; Xu, W.; Liu, L.; Hughes, V.A.; Mak, J.Y.; Meehan, B.S.; Pediongco, T.; Birkinshaw, R.W.; Chen, Z.; et al. Drugs and drug-like molecules can modulate the function of mucosal-associated invariant T cells. Nat. Immunol. 2017, 18, 402–411. [Google Scholar] [CrossRef]
- Rahimpour, A.; Koay, H.F.; Enders, A.; Clanchy, R.; Eckle, S.B.; Meehan, B.; Chen, Z.; Whittle, B.; Liu, L.; Fairlie, D.P.; et al. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J. Exp. Med. 2015, 212, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Reantragoon, R.; Corbett, A.J.; Sakala, I.G.; Gherardin, N.A.; Furness, J.B.; Chen, Z.; Eckle, S.B.; Uldrich, A.P.; Birkinshaw, R.W.; Patel, O.; et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J. Exp. Med. 2013, 210, 2305–2320. [Google Scholar] [CrossRef] [PubMed]
- Koay, H.F.; Gherardin, N.A.; Xu, C.; Seneviratna, R.; Zhao, Z.; Chen, Z.; Fairlie, D.P.; McCluskey, J.; Pellicci, D.G.; Uldrich, A.P.; et al. Diverse MR1-restricted T cells in mice and humans. Nat. Commun. 2019, 10, 2243. [Google Scholar] [CrossRef]
- Dusseaux, M.; Martin, E.; Serriari, N.; Peguillet, I.; Premel, V.; Louis, D.; Milder, M.; Le Bourhis, L.; Soudais, C.; Treiner, E.; et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 2011, 117, 1250–1259. [Google Scholar] [CrossRef]
- Ussher, J.E.; Bilton, M.; Attwod, E.; Shadwell, J.; Richardson, R.; de Lara, C.; Mettke, E.; Kurioka, A.; Hansen, T.H.; Klenerman, P.; et al. CD161++ CD8+ T cells, including the MAIT cell subset, are specifically activated by IL-12+IL-18 in a TCR-independent manner. Eur. J. Immunol. 2014, 44, 195–203. [Google Scholar] [CrossRef]
- van Wilgenburg, B.; Scherwitzl, I.; Hutchinson, E.C.; Leng, T.; Kurioka, A.; Kulicke, C.; de Lara, C.; Cole, S.; Vasanawathana, S.; Limpitikul, W.; et al. MAIT cells are activated during human viral infections. Nat. Commun. 2016, 7, 11653. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kjer-Nielsen, L.; Shi, M.; D’Souza, C.; Pediongco, T.J.; Cao, H.; Kostenko, L.; Lim, X.Y.; Eckle, S.B.G.; Meehan, B.S.; et al. IL-23 costimulates antigen-specific MAIT cell activation and enables vaccination against bacterial infection. Sci. Immunol. 2019, 4, eaaw0402. [Google Scholar] [CrossRef]
- Koay, H.F.; Gherardin, N.A.; Enders, A.; Loh, L.; Mackay, L.K.; Almeida, C.F.; Russ, B.E.; Nold-Petry, C.A.; Nold, M.F.; Bedoui, S.; et al. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat. Immunol. 2016, 17, 1300–1311. [Google Scholar] [CrossRef]
- Legoux, F.; Bellet, D.; Daviaud, C.; El Morr, Y.; Darbois, A.; Niort, K.; Procopio, E.; Salou, M.; Gilet, J.; Ryffel, B.; et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science 2019, 366, 494–499. [Google Scholar] [CrossRef]
- Constantinides, M.G.; Link, V.M.; Tamoutounour, S.; Wong, A.C.; Perez-Chaparro, P.J.; Han, S.J.; Chen, Y.E.; Li, K.; Farhat, S.; Weckel, A.; et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science 2019, 366, eaax6624. [Google Scholar] [CrossRef]
- D’Souza, C.; Pediongco, T.; Wang, H.; Scheerlinck, J.Y.; Kostenko, L.; Esterbauer, R.; Stent, A.W.; Eckle, S.B.G.; Meehan, B.S.; Strugnell, R.A.; et al. Mucosal-Associated Invariant T Cells Augment Immunopathology and Gastritis in Chronic Helicobacter pylori Infection. J. Immunol. 2018, 200, 1901–1916. [Google Scholar] [PubMed]
- Ankley, L.; Thomas, S.; Olive, A.J. Fighting persistence; how chronic infections with Mycobacterium tuberculosis evade T cell-mediated clearance and new strategies to defeat them. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef] [PubMed]
- Hingley-Wilson, S.M.; Sambandamurthy, V.K.; Jacobs, W.R., Jr. Survival perspectives from the world’s most successful pathogen, Mycobacterium tuberculosis. Nat. Immunol. 2003, 4, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; van der Most, R.G.; Akondy, R.S.; Glidewell, J.T.; Albott, S.; Masopust, D.; Murali-Krishna, K.; Mahar, P.L.; Edupuganti, S.; Lalor, S.; et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 2008, 28, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, C.W.; Braciale, T.J. Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J. Immunol. 2004, 173, 1209–1218. [Google Scholar] [CrossRef]
- Zhai, W.; Wu, F.; Zhang, Y.; Fu, Y.; Liu, Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, T.; Liu, Z.; Zhang, G.; Wang, J.; Feng, S.; Liang, J. Inhibition of Autophagy by MiR-30A Induced by Mycobacteria tuberculosis as a Possible Mechanism of Immune Escape in Human Macrophages. Jpn. J. Infect. Dis. 2015, 68, 420–424. [Google Scholar] [CrossRef]
- Briken, V.; Porcelli, S.A.; Besra, G.S.; Kremer, L. Mycobacterial lipoarabinomannan and related lipoglycans: From biogenesis to modulation of the immune response. Mol. Microbiol. 2004, 53, 391–403. [Google Scholar] [CrossRef]
- Chen, M.; Divangahi, M.; Gan, H.; Shin, D.S.; Hong, S.; Lee, D.M.; Serhan, C.N.; Behar, S.M.; Remold, H.G. Lipid mediators in innate immunity against tuberculosis: Opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 2008, 205, 2791–2801. [Google Scholar] [CrossRef]
- Urdahl, K.B.; Shafiani, S.; Ernst, J.D. Initiation and regulation of T-cell responses in tuberculosis. Mucosal Immunol. 2011, 4, 288–293. [Google Scholar] [CrossRef]
- Moguche, A.O.; Musvosvi, M.; Penn-Nicholson, A.; Plumlee, C.R.; Mearns, H.; Geldenhuys, H.; Smit, E.; Abrahams, D.; Rozot, V.; Dintwe, O.; et al. Antigen Availability Shapes T Cell Differentiation and Function during Tuberculosis. Cell Host Microbe 2017, 21, 695–706.e5. [Google Scholar] [CrossRef] [PubMed]
- Bold, T.D.; Banaei, N.; Wolf, A.J.; Ernst, J.D. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS. Pathog. 2011, 7, e1002063. [Google Scholar] [CrossRef] [PubMed]
- Egen, J.G.; Rothfuchs, A.G.; Feng, C.G.; Horwitz, M.A.; Sher, A.; Germain, R.N. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity 2011, 34, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Raghuvanshi, S.; Sharma, P.; Singh, S.; Van Kaer, L.; Das, G. Mycobacterium tuberculosis evades host immunity by recruiting mesenchymal stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 21653–21658. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, B.; Li, M.; Deng, Q.; Wu, X.; Le, X.; Wu, C.; Larmonier, N.; Zhang, W.; Zhang, H.; et al. CD4(+)CD25(+)FoxP3(+) regulatory T cells suppress Mycobacterium tuberculosis immunity in patients with active disease. Clin. Immunol. 2007, 123, 50–59. [Google Scholar] [CrossRef]
- Shafiani, S.; Tucker-Heard, G.; Kariyone, A.; Takatsu, K.; Urdahl, K.B. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J. Exp. Med. 2010, 207, 1409–1420. [Google Scholar] [CrossRef]
- Comas, I.; Chakravartti, J.; Small, P.M.; Galagan, J.; Niemann, S.; Kremer, K.; Ernst, J.D.; Gagneux, S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 2010, 42, 498–503. [Google Scholar] [CrossRef]
- Ernst, J.D. Mechanisms of M. tuberculosis Immune Evasion as Challenges to TB Vaccine Design. Cell Host Microbe 2018, 24, 34–42. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Coburn, B.; Sekirov, I.; Finlay, B.B. Type III secretion systems and disease. Clin. Microbiol. Rev. 2007, 20, 535–549. [Google Scholar] [CrossRef]
- Deng, W.; Marshall, N.C.; Rowland, J.L.; McCoy, J.M.; Worrall, L.J.; Santos, A.S.; Strynadka, N.C.J.; Finlay, B.B. Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microbiol. 2017, 15, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J.; Di Santo, J.P. Debugging how bacteria manipulate the immune response. Immunity 2007, 26, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, G.R. The type III secretion injectisome. Nat. Rev. Microbiol. 2006, 4, 811–825. [Google Scholar] [CrossRef]
- Ashida, H.; Ogawa, M.; Mimuro, H.; Sasakawa, C. Shigella Infection of Intestinal Epithelium and Circumvention of the Host Innate Defense System. In Molecular Mechanisms of Bacterial Infection via the Gut; Sasakawa, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 231–255. [Google Scholar]
- Hilbi, H.; Moss, J.E.; Hersh, D.; Chen, Y.; Arondel, J.; Banerjee, S.; Flavell, R.A.; Yuan, J.; Sansonetti, P.J.; Zychlinsky, A. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 1998, 273, 32895–32900. [Google Scholar] [CrossRef]
- Senerovic, L.; Tsunoda, S.P.; Goosmann, C.; Brinkmann, V.; Zychlinsky, A.; Meissner, F.; Kolbe, M. Spontaneous formation of IpaB ion channels in host cell membranes reveals how Shigella induces pyroptosis in macrophages. Cell Death Dis. 2012, 3, e384. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Kim, M.; Sasakawa, C. Manipulation of the host cell death pathway by Shigella. Cell Microbiol. 2014, 16, 1757–1766. [Google Scholar] [CrossRef]
- Phalipon, A.; Sansonetti, P.J. Shigella’s ways of manipulating the host intestinal innate and adaptive immune system: A tool box for survival? Immunol. Cell Biol. 2007, 85, 119–129. [Google Scholar] [CrossRef]
- Islam, D.; Bandholtz, L.; Nilsson, J.; Wigzell, H.; Christensson, B.; Agerberth, B.; Gudmundsson, G. Downregulation of bactericidal peptides in enteric infections: A novel immune escape mechanism with bacterial DNA as a potential regulator. Nat. Med. 2001, 7, 180–185. [Google Scholar] [CrossRef]
- Cerny, O.; Holden, D.W. Salmonella SPI-2 type III secretion system-dependent inhibition of antigen presentation and T cell function. Immunol. Lett. 2019, 215, 35–39. [Google Scholar] [CrossRef]
- Carden, S.E.; Walker, G.T.; Honeycutt, J.; Lugo, K.; Pham, T.; Jacobson, A.; Bouley, D.; Idoyaga, J.; Tsolis, R.M.; Monack, D. Pseudogenization of the Secreted Effector Gene sseI Confers Rapid Systemic Dissemination of S. Typhimurium ST313 within Migratory Dendritic Cells. Cell Host Microbe 2017, 21, 182–194. [Google Scholar] [CrossRef]
- van der Velden, A.W.; Copass, M.K.; Starnbach, M.N. Salmonella inhibit T cell proliferation by a direct, contact-dependent immunosuppressive effect. Proc. Natl. Acad. Sci. USA 2005, 102, 17769–17774. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Kim, K.; Shutinoski, B.; Wachholz, K.; Krishnan, L.; Sad, S. Culling of APCs by inflammatory cell death pathways restricts TIM3 and PD-1 expression and promotes the survival of primed CD8 T cells. Cell Death Differ. 2017, 24, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Broker, B.M.; Mrochen, D.; Peton, V. The T Cell Response to Staphylococcus aureus. Pathogens 2016, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F., 3rd; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; DuMont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef]
- Ziegler, C.; Goldmann, O.; Hobeika, E.; Geffers, R.; Peters, G.; Medina, E. The dynamics of T cells during persistent Staphylococcus aureus infection: From antigen-reactivity to in vivo anergy. EMBO Mol. Med. 2011, 3, 652–666. [Google Scholar] [CrossRef]
- White, J.; Herman, A.; Pullen, A.M.; Kubo, R.; Kappler, J.W.; Marrack, P. The V-Beta-Specific Superantigen Staphylococcal Enterotoxin-B-Stimulation of Mature T-Cells and Clonal Deletion in Neonatal Mice. Cell 1989, 56, 27–35. [Google Scholar] [CrossRef]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef]
- Zeppa, J.J.; Kasper, K.J.; Mohorovic, I.; Mazzuca, D.M.; Haeryfar, S.M.M.; McCormick, J.K. Nasopharyngeal infection by Streptococcus pyogenes requires superantigen-responsive Vbeta-specific T cells. Proc. Natl. Acad. Sci. USA 2017, 114, 10226–10231. [Google Scholar] [CrossRef]
- Dellabona, P.; Peccoud, J.; Kappler, J.; Marrack, P.; Benoist, C.; Mathis, D. Superantigens interact with MHC class II molecules outside of the antigen groove. Cell 1990, 62, 1115–1121. [Google Scholar] [CrossRef]
- Gunther, S.; Varma, A.K.; Moza, B.; Kasper, K.J.; Wyatt, A.W.; Zhu, P.; Rahman, A.K.M.N.U.; Li, Y.L.; Mariuzza, R.A.; McCormick, J.K.; et al. A novel loop domain in superantigens extends their T cell receptor recognition site. J. Mol. Biol. 2007, 371, 210–221. [Google Scholar] [CrossRef]
- Moza, B.; Varma, A.K.; Buonpane, R.A.; Zhu, P.; Herfst, C.A.; Nicholson, M.J.; Wilbuer, A.K.; Seth, N.P.; Wucherpfennig, K.W.; McCormick, J.K.; et al. Structural basis of T-cell specificity and activation by the bacterial superantigen TSST-1. Embo J. 2007, 26, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Pumphrey, N.; Vuidepot, A.; Jakobsen, B.; Forsberg, G.; Walse, B.; Lindkvist-Petersson, K. Cutting edge: Evidence of direct TCR alpha-chain interaction with superantigen. J. Immunol. 2007, 179, 2700–2704. [Google Scholar] [CrossRef] [PubMed]
- Saline, M.; Rodstrom, K.E.; Fischer, G.; Orekhov, V.Y.; Karlsson, B.G.; Lindkvist-Petersson, K. The structure of superantigen complexed with TCR and MHC reveals novel insights into superantigenic T cell activation. Nat. Commun 2010, 1, 119. [Google Scholar] [CrossRef]
- Thomas, D.; Dauwalder, O.; Brun, V.; Badiou, C.; Ferry, T.; Etienne, J.; Vandenesch, F.; Lina, G. Staphylococcus aureus superantigens elicit redundant and extensive human Vbeta patterns. Infect. Immun 2009, 77, 2043–2050. [Google Scholar] [CrossRef]
- Abdurrahman, G.; Schmiedeke, F.; Bachert, C.; Broker, B.M.; Holtfreter, S. Allergy-A New Role for T Cell Superantigens of Staphylococcus aureus? Toxins 2020, 12, 176. [Google Scholar] [CrossRef]
- Seo, K.S.; Park, J.Y.; Terman, D.S.; Bohach, G.A. A quantitative real time PCR method to analyze T cell receptor Vbeta subgroup expansion by staphylococcal superantigens. J. Transl Med. 2010, 8, 2. [Google Scholar] [CrossRef]
- Commons, R.J.; Smeesters, P.R.; Proft, T.; Fraser, J.D.; Robins-Browne, R.; Curtis, N. Streptococcal superantigens: Categorization and clinical associations. Trends Mol. Med. 2014, 20, 48–62. [Google Scholar] [CrossRef]
- Petersson, K.; Pettersson, H.; Skartved, N.J.; Walse, B.; Forsberg, G. Staphylococcal enterotoxin H induces V alpha-specific expansion of T cells. J. Immunol. 2003, 170, 4148–4154. [Google Scholar] [CrossRef]
- Llewelyn, M.; Sriskandan, S.; Peakman, M.; Ambrozak, D.R.; Douek, D.C.; Kwok, W.W.; Cohen, J.; Altmann, D.M. HLA class II polymorphisms determine responses to bacterial superantigens. J. Immunol. 2004, 172, 1719–1726. [Google Scholar] [CrossRef]
- Kotb, M.; Norrby-Teglund, A.; McGeer, A.; El-Sherbini, H.; Dorak, M.T.; Khurshid, A.; Green, K.; Peeples, J.; Wade, J.; Thomson, G.; et al. An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat. Med. 2002, 8, 1398–1404. [Google Scholar] [CrossRef]
- Arad, G.; Levy, R.; Nasie, I.; Hillman, D.; Rotfogel, Z.; Barash, U.; Supper, E.; Shpilka, T.; Minis, A.; Kaempfer, R. Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS. Biol. 2011, 9, e1001149. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Rotfogel, Z.; Hillman, D.; Popugailo, A.; Arad, G.; Supper, E.; Osman, F.; Kaempfer, R. Superantigens hyperinduce inflammatory cytokines by enhancing the B7-2/CD28 costimulatory receptor interaction. Proc. Natl. Acad. Sci. USA 2016, 113, E6437–E6446. [Google Scholar] [CrossRef] [PubMed]
- Kaempfer, R. Bacterial Superantigen Toxins, CD28, and Drug Development. Toxins 2018, 10, 459. [Google Scholar] [CrossRef]
- Kawabe, Y.; Ochi, A. Selective anergy of V beta 8+,CD4+ T cells in Staphylococcus enterotoxin B-primed mice. J. Exp. Med. 1990, 172, 1065–1070. [Google Scholar] [CrossRef]
- Janik, D.K.; Lee, W.T. Staphylococcal Enterotoxin B (SEB) Induces Memory CD4 T Cell Anergy in vivo and Impairs Recall Immunity to Unrelated Antigens. J. Clin. Cell Immunol. 2015, 6, 1–8. [Google Scholar]
- Watson, A.R.; Janik, D.K.; Lee, W.T. Superantigen-induced CD4 memory T cell anergy. I. Staphylococcal enterotoxin B induces Fyn-mediated negative signaling. Cell Immunol. 2012, 276, 16–25. [Google Scholar] [CrossRef]
- Spaulding, A.R.; Salgado-Pabon, W.; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447. [Google Scholar] [CrossRef] [PubMed]
- Radcliff, F.J.; Clow, F.; Mahadevan, M.; Johnston, J.; Proft, T.; Douglas, R.G.; Fraser, J.D. A potential role for staphylococcal and streptococcal superantigens in driving skewing of TCR Vbeta subsets in tonsillar hyperplasia. Med. Microbiol. Immunol. 2017, 206, 337–346. [Google Scholar] [CrossRef]
- Huang, C.C.; Shah, S.; Nguyen, P.; Altman, J.D.; Blackman, M.A. Bacterial superantigen exposure after resolution of influenza virus infection perturbs the virus-specific memory CD8(+)-T-cell repertoire. J. Virol. 2002, 76, 6852–6856. [Google Scholar] [CrossRef]
- Zhang, W.J.; Sarawar, S.; Nguyen, P.; Daly, K.; Rehg, J.E.; Doherty, P.C.; Woodland, D.L.; Blackman, M.A. Lethal synergism between influenza infection and staphylococcal enterotoxin B in mice. J. Immunol. 1996, 157, 5049–5060. [Google Scholar]
- Meilleur, C.E.; Memarnejadian, A.; Shivji, A.N.; Benoit, J.M.; Tuffs, S.W.; Mele, T.S.; Singh, B.; Dikeakos, J.D.; Topham, D.J.; Mu, H.H.; et al. Discordant rearrangement of primary and anamnestic CD8+ T cell responses to influenza A viral epitopes upon exposure to bacterial superantigens: Implications for prophylactic vaccination, heterosubtypic immunity and superinfections. PLoS. Pathog. 2020, 16, e1008393. [Google Scholar] [CrossRef] [PubMed]
- Shaler, C.R.; Choi, J.; Rudak, P.T.; Memarnejadian, A.; Szabo, P.A.; Tun-Abraham, M.E.; Rossjohn, J.; Corbett, A.J.; McCluskey, J.; McCormick, J.K.; et al. MAIT cells launch a rapid, robust and distinct hyperinflammatory response to bacterial superantigens and quickly acquire an anergic phenotype that impedes their cognate antimicrobial function: Defining a novel mechanism of superantigen-induced immunopathology and immunosuppression. PLoS. Biol. 2017, 15, e2001930. [Google Scholar]
- Emgard, J.; Bergsten, H.; McCormick, J.K.; Barrantes, I.; Skrede, S.; Sandberg, J.K.; Norrby-Teglund, A. MAIT Cells Are Major Contributors to the Cytokine Response in Group A Streptococcal Toxic Shock Syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 25923–25931. [Google Scholar] [CrossRef]
- Szabo, P.A.; Rudak, P.T.; Choi, J.; Xu, S.X.; Schaub, R.; Singh, B.; McCormick, J.K.; Haeryfar, S.M.M. Invariant Natural Killer T Cells Are Pathogenic in the HLA-DR4-Transgenic Humanized Mouse Model of Toxic Shock Syndrome and Can Be Targeted to Reduce Morbidity. J. Infect. Dis. 2017, 215, 824–829. [Google Scholar]
- Hayworth, J.L.; Mazzuca, D.M.; Maleki Vareki, S.; Welch, I.; McCormick, J.K.; Haeryfar, S.M. CD1d-independent activation of mouse and human iNKT cells by bacterial superantigens. Immunol. Cell Biol. 2012, 90, 699–709. [Google Scholar] [CrossRef]
- Kalyan, S.; Chow, A.W. Human peripheral gamma delta T cells potentiate the early proinflammatory cytokine response to staphylococcal toxic shock syndrome toxin-1. J. Infect. Dis. 2004, 189, 1892–1896. [Google Scholar] [CrossRef][Green Version]
- Morita, C.T.; Li, H.M.; Lamphear, J.G.; Rich, R.R.; Fraser, J.D.; Mariuzza, R.A.; Lee, H.K. Superantigen recognition by gamma delta T cells: SEA recognition site for human V gamma 2 T cell receptors. Immunity 2001, 14, 331–344. [Google Scholar] [CrossRef]
- Rust, C.J.J.; Verreck, F.; Vietor, H.; Koning, F. Specific Recognition of Staphylococcal Enterotoxin-a by Human T-Cells Bearing Receptors with the V-Gamma-9 Region. Nature 1990, 346, 572–574. [Google Scholar] [CrossRef]
- Stinissen, P.; Vandevyver, C.; Raus, J.; Zhang, J.W. Superantigen Reactivity of Gamma-Delta T-Cell Clones Isolated from Patients with Multiple-Sclerosis and Controls. Cell Immunol. 1995, 166, 227–235. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).