Versatility of Induced Pluripotent Stem Cells (iPSCs) for Improving the Knowledge on Musculoskeletal Diseases

 , and
, and 
Abstract
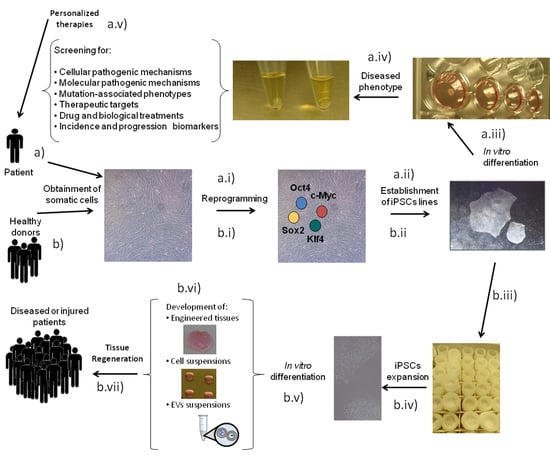
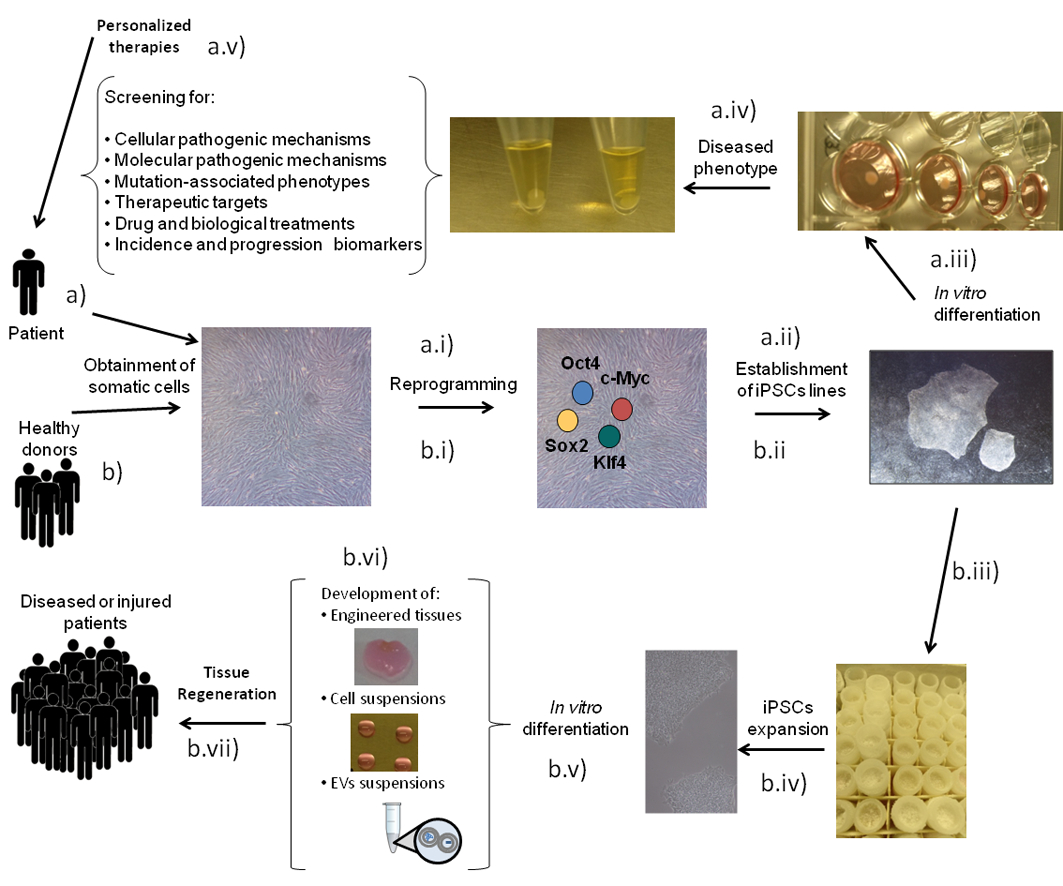
1. Introduction
2. Disease Modeling
2.1. Modeling Bone Diseases with iPSCs
2.2. Modeling Cartilage Diseases with iPSCs
2.3. iPSC-Based Disease Modeling for Skeletal Muscle
2.4. iPSC-Based Disease Modeling for Degenerative Disc Disease
3. iPSCs in Regenerative Medicine
4. Limitations, Challenges and Future Directions of iPSCs Applications
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AS | Andersen’s Syndrome |
| CRISPR/Cas9 | clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 |
| CS | chondroitin sulfate |
| DCM | Dilated cardiomyopathy |
| DMD | Duchenne Muscular Dystrophy |
| EB | embryoid body |
| ECM | extracellular matrix |
| EDMD | Emery-Dreifuss muscular dystrophy |
| ESCs | embryonic stem cells |
| EVs | extracellular vescicles |
| FBN1 | FIBRILLIN-1 gene |
| FOCD | Familial Osteochondritis Disecans |
| FOP | Fibrodysplasia Ossificans Progressiva |
| GAGs | glycosaminoglycans |
| GD | Gaucher Disease |
| HA | hydroxyapatite |
| HLA | human leukocyte antigen |
| IGF-1 | Insulin Growth Factor 1 |
| iPSCs | induced pluripotent stem cells |
| Klf4 | Krüppel-like factor |
| L-CMD | LMNA-related congenital muscular dystrophy |
| LFS | Li-Fraumeni Syndrome |
| LGMD | limb-girdle muscular dystrophy |
| MM | Myoshi Myopathy |
| MFS | Marfan’s Syndrome |
| MSCs | mesenchymal stromal cells |
| NALP3 | NACHT, LRR and PYD domains-containing protein 3 gene |
| NOMID | neonatal-onset multisystem inflammatory disease |
| OA | osteoarthritis |
| Oct3/4 | octamer binding protein 3/4 |
| OI | Osteogenesis Imperfecta |
| PLGA | poly lactic-co-glycolide |
| PLLA | poly-L-lactic acid |
| PEG | Polyethylene Glycol |
| RA | Rheumatoid Arthritis |
| SMA | Spinal Muscular Atrophy |
| Sox2 | SRY (sex determining region Y)-box 2 |
| TALENS | transcription activator-like effector nucleases |
| TCP | beta tricalcium phosphate |
| ZFNs | zinc finger nucleases |
References
- Barruet, E.; Hsiao, E.C. Using Human Induced Pluripotent Stem Cells to Model Skeletal Diseases. Methods Mol. Biol. 2016, 1353, 101–118. [Google Scholar] [PubMed]
- Briggs, A.M.; Cross, M.J.; Hoy, D.G.; Sànchez-Riera, L.; Blyth, F.M.; Woolf, A.D.; March, L. Musculoskeletal Health Conditions Represent a Global Threat to Healthy Aging: A Report for the 2015 World Health Organization World Report on Ageing and Health. Gerontologist 2016, 56, S243–S255. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-J.; Jiao, H.; Walczak, B.E. Emerging Opportunities for Induced Pluripotent Stem Cells in Orthopaedics. J. Orthop. Transl. 2019, 17, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Schofield, D.J.; Kelly, S.J.; Shrestha, R.N.; Callander, E.; Passey, M.E.; Percival, R. The Impact of Back Problems on Retirement Wealth. Pain 2012, 153, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Vitali, J.; Darabi, R. IPSCs As a Platform for Disease Modeling, Drug Screening, and Personalized Therapy in Muscular Dystrophies. Cells 2019, 8, 20. [Google Scholar] [CrossRef]
- Ricci, F.; Vacchetti, M.; Brusa, C.; Vercelli, L.; Davico, C.; Vitiello, B.; Mongini, T. New Pharmacotherapies for Genetic Neuromuscular Disorders: Opportunities and Challenges. Expert Rev. Clin. Pharmacol. 2019, 12, 757–770. [Google Scholar] [CrossRef]
- Bannuru, R.R.; Osani, M.; Vaysbrot, E.; Arden, N.; Bennell, K.; Bierma-Zeinstra, S.; Kraus, V.; Lohmander, L.; Abbott, J.; Bhandari, M.; et al. OARSI Guidelines for the Non-Surgical Management of Knee, Hip, and Polyarticular Osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1578–1589. [Google Scholar] [CrossRef]
- National Research Council. Opportunities in Biology; The National Academies Press: Washington, DC, USA, 1989; p. 464. [Google Scholar]
- Thysen, S.; Luyten, F.P.; Lories, R.J. Targets, Models and Challenges in Osteoarthritis Research. Dis. Model. Mech. 2015, 8, 17–30. [Google Scholar] [CrossRef]
- Passier, R.; Orlova, V.V.; Mummery, C. Complex Tissue and Disease Modeling Using HiPSCs. Cell Stem Cell 2016, 18, 309–321. [Google Scholar] [CrossRef]
- Liu, H.; Yang, L.; Yu, F.F.; Wang, S.; Wu, C.; Qu, C.; Lammi, M.J.; Guo, X. The Potential of Induced Pluripotent Stem Cells as a Tool to Study Skeletal Dysplasias and Cartilage-Related Pathologic Conditions. Osteoarthr. Cartil. 2017, 25, 616–624. [Google Scholar] [CrossRef]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced Pluripotent Stem Cells: Applications in Regenerative Medicine, Disease Modeling, and Drug Discovery. Front. Cell Dev. Boil. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Desnuelle, C.; Dib, M.; Garrel, C.; Favier, A. A Double-Blind, Placebo-Controlled Randomized Clinical Trial of α-Tocopherol (vitamin E) in the Treatment of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. 2001, 2, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Cudkowicz, M.E.; Schoenfeld, D.; Conrad, T.; Taft, J.; Chilton, M.; Urbinelli, L.; Qureshi, M.; Zhang, H.; Pestronk, A.; et al. A Clinical Trial of Creatine in ALS. Neurology 2004, 63, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Blangero, J.; Curran, J.E. Induced Pluripotent Stem Cells in Disease Modeling and Gene Identification. Methods Mol. Biol. 2018, 1706, 17–38. [Google Scholar]
- Piñeiro-Ramil, M.; Sanjurjo-Rodríguez, C.; Castro-Viñuelas, R.; Rodríguez-Fernández, S.; Fuentes-Boquete, I.; Blanco, F.; Díaz-Prado, S. Usefulness of Mesenchymal Cell Lines for Bone and Cartilage Regeneration Research. Int. J. Mol. Sci. 2019, 20, 6286. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.; Holtzer, H. The Loss of Phenotypic Traits by Differentiated Cells. 3. The Reversible Behavior of Chondrocytes in Primary Cultures. J. Cell Biol. 1966, 28, 473–487. [Google Scholar] [CrossRef]
- Holtzer, H.; Abbott, J.; Lash, J.; Holtzer, S. The loss of phenotypic traits by differentiated cells in vitro, I. Dedifferentiation of cartilage cells. Proc. Natl. Acad. Sci. USA 1960, 46, 1533–1542. [Google Scholar] [CrossRef]
- Khodabukus, A.; Prabhu, N.; Wang, J.; Bursac, N. In Vitro Tissue-Engineered Skeletal Muscle Models for Studying Muscle Physiology and Disease. Adv. Health Mater. 2018, 7, 1701498. [Google Scholar] [CrossRef]
- Allen, D.D.; Caviedes, R.; Cardenas, A.M.; Shimahara, T.; Segura-Aguilar, J.; Caviedes, P.A. Cell Lines As In Vitro Models for Drug Screening and Toxicity Studies. Drug Dev. Ind. Pharm. 2005, 31, 757–768. [Google Scholar] [CrossRef]
- Ebert, A.D.; Yu, J.; Rose, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced Pluripotent Stem Cells from a Spinal Muscular Atrophy Patient. Nature 2008, 457, 277–280. [Google Scholar] [CrossRef]
- Piñeiro-Ramil, M.; Castro-Viñuelas, R.; Sanjurjo-Rodríguez, C.; Rodríguez-Fernández, S.; Hermida-Gómez, T.; Blanco, F.J.; Fuentes-Boquete, I.; Díaz-Prado, S. Immortalizing Mesenchymal Stromal Cells from Aged Donors While Keeping Their Essential Features. Stem Cells Int. 2020, 2020, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Mamchaoui, K.; Trollet, C.; Bigot, A.; Negroni, E.; Chaouch, S.; Wolff, A.; Kandalla, P.K.; Marie, S.; Di Santo, J.P.; Guily, J.L.S.; et al. Immortalized Pathological Human Myoblasts: Towards a Universal Tool for the Study of Neuromuscular Disorders. Skelet. Muscle 2011, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Kartsogiannis, V.; Ng, K.W. Cell Lines and Primary Cell Cultures in the Study of Bone Cell Biology. Mol. Cell. Endocrinol. 2004, 228, 79–102. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Aulner, N.; Bickle, M.; Davies, A.M.; Del Nery, E.; Ebner, D.V.; Montoya, M.C.; Ostling, P.; Pietiäinen, V.; Price, L.S.; et al. Screening Out Irrelevant Cell-Based Models of Disease. Nat. Rev. Drug Discov. 2016, 15, 751–769. [Google Scholar] [CrossRef]
- Akpancar, S.; Tatar, O.; Turgut, H.; Akyildiz, F.; Ekinci, S. The Current Perspectives of Stem Cell Therapy in Orthopedic Surgery. Arch. Trauma Res. 2016, 5, 37976. [Google Scholar] [CrossRef]
- Castro-Viñuelas, R.; Sanjurjo-Rodríguez, C.; Piñeiro-Ramil, M.; Hermida-Gómez, T.; Fuentes-Boquete, I.; De Toro-Santos, F.; Blanco-García, F.; Díaz-Prado, S. Induced Pluripotent Stem Cells for Cartilage Repair: Current Status and Future Perspectives. Eur. Cells Mater. 2018, 36, 96–109. [Google Scholar] [CrossRef]
- Diederichs, S.; Klampfleuthner, F.A.M.; Moradi, B.; Richter, W. Chondral Differentiation of Induced Pluripotent Stem Cells Without Progression into the Endochondral Pathway. Front. Cell Dev. Boil. 2019, 7, 270. [Google Scholar] [CrossRef]
- Diekman, B.O.; Christoforou, N.; Willard, V.P.; Sun, H.; Sanchez-Adams, J.; Leong, K.W.; Guilak, F. Cartilage Tissue Engineering Using Differentiated and Purified Induced Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 19172–19177. [Google Scholar] [CrossRef]
- Jevons, L.A.; Houghton, F.D.; Tare, R. Augmentation of Musculoskeletal Regeneration: Role for Pluripotent Stem Cells. Regen. Med. 2018, 13, 189–206. [Google Scholar] [CrossRef]
- Kouroupis, D.; Sanjurjo-Rodriguez, C.; Jones, E.; Correa, D. Mesenchymal Stem Cell Functionalization for Enhanced Therapeutic Applications. Tissue Eng. Part B Rev. 2019, 25, 55–77. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced Pluripotent Stem Cell Technology: A Decade of Progress. Nat. Rev. Drug Discov. 2016, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Daley, G.Q. Induced Pluripotent Stem Cells in Disease Modelling and Drug Discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef]
- Doss, M.X.; Sachinidis, A. Current Challenges of IPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef]
- Scudellari, M. How IPS Cells Changed the World. Nature 2016, 534, 310–312. [Google Scholar] [CrossRef]
- Zhao, C.; Ikeya, M. Generation and Applications of Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells. Stem Cells Int. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Dayem, A.A.; Bin Lee, S.; Kim, K.; Lim, K.M.; Jeon, T.-I.; Seok, J.; Cho, S.-G.; Cho, A.S.-G. Production of Mesenchymal Stem Cells Through Stem Cell Reprogramming. Int. J. Mol. Sci. 2019, 20, 1922. [Google Scholar] [CrossRef]
- Okur, F.; Cevher, I.; Özdemir, C.; Kocaefe, Y.C.; Uckan-Cetinkaya, D. Osteopetrotic Induced Pluripotent Stem Cells Derived from Patients with Different Disease-Associated Mutations by Non-Integrating Reprogramming Methods. Stem Cell Res. Ther. 2019, 10, 211. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, Y.; Zhao, B.; Niu, X.; Hu, B.; Li, Q.; Zhang, J.; Ding, J.; Chen, Y.; Wang, Y. Comparison of Exosomes Secreted by Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells and Synovial Membrane-Derived Mesenchymal Stem Cells for the Treatment of Osteoarthritis. Stem Cell Res. Ther. 2017, 8, 64. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, J.; Yuan, H.; Xu, Z.; Li, Q.; Niu, X.; Hu, B.; Wang, Y.; Li, X. Exosomes Secreted by Human-Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Repair Critical-Sized Bone Defects through Enhanced Angiogenesis and Osteogenesis in Osteoporotic Rats. Int. J. Boil. Sci. 2016, 12, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Csobonyeiova, M.; Polak, S.; Zamborsky, R.; Danisovic, L. IPS Cell Technologies and Their Prospect for Bone Regeneration and Disease Modeling: A Mini Review. J. Adv. Res. 2017, 8, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, B.; Hu, K.; Cao, C.; Man, Y.; Wang, P. Deriving Osteogenic Cells from Induced Pluripotent Stem Cells for Bone Tissue Engineering. Tissue Eng. Part B Rev. 2017, 23, 1–8. [Google Scholar] [CrossRef]
- Perez, J.R.; Kouroupis, D.; Li, D.J.; Best, T.M.; Kaplan, L.; Correa, D. Tissue Engineering and Cell-Based Therapies for Fractures and Bone Defects. Front. Bioeng. Biotechnol. 2018, 6, 105. [Google Scholar] [CrossRef] [PubMed]
- Driessen, B.J.; Logie, C.; Vonk, L. Cellular Reprogramming for Clinical Cartilage Repair. Cell Boil. Toxicol. 2017, 33, 329–349. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Jing, L.; Willard, V.P.; Wu, C.-L.; Guilak, F.; Chen, J.; Setton, L.A. Differentiation of Human Induced Pluripotent Stem Cells into Nucleus Pulposus-Like Cells. Stem Cell Res. Ther. 2018, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Zhu, J.; Hua, J.; Gong, Z.; Yu, C.; Zhou, X.; Wang, J.; Huang, X.; Yu, W.; Li, L.; et al. Intradiscal Injection of Induced Pluripotent Stem Cell-Derived Nucleus Pulposus-Like Cell-Seeded Polymeric Microspheres Promotes Rat Disc Regeneration. Stem Cells Int. 2019, 2019, 6806540. [Google Scholar] [CrossRef]
- Zhu, Y.-X.; Liang, Y.; Zhu, H.; Lian, C.; Wang, L.; Wang, Y.; Gu, H.; Zhou, G.; Yu, X. The Generation and Functional Characterization of Induced Pluripotent Stem Cells from Human Intervertebral Disc Nucleus Pulposus Cells. Oncotarget 2017, 8, 42700–42711. [Google Scholar] [CrossRef]
- De Oñate, L.; Garreta, E.; Tarantino, C.; Martínez, E.; Capilla, E.; Navarro, I.; Gutiérrez, J.; Samitier, J.; Campistol, J.M.; Muñoz-Cánovas, P.; et al. Research on Skeletal Muscle Diseases Using Pluripotent Stem Cells. In Muscle Cell and Tissue; IntechOpen: London, UK, 2015. [Google Scholar]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent Stem Cells in Disease Modelling and Drug Discovery. Nat. Rev. Mol. Cell Boil. 2016, 17, 170–182. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Liu, C.-L.; Ting, C.-Y.; Chiu, Y.-T.; Cheng, Y.-C.; Nicholson, M.W.; Hsieh, P.C. Human IPSC Banking: Barriers and Opportunities. J. Biomed. Sci. 2019, 26, 87. [Google Scholar] [CrossRef]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, K.; Inamura, M.; Kawabata, K.; Sakurai, F.; Yamanishi, K.; Hayakawa, T.; Mizuguchi, H. Efficient Adipocyte and Osteoblast Differentiation from Mouse Induced Pluripotent Stem Cells by Adenoviral Transduction. Stem Cells 2009, 27, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.-L.; Tai, L.-K.; Chiou, S.-H.; Chen, Y.-J.; Lee, K.-H.; Chou, S.-J.; Chang, Y.-L.; Chang, C.-M.; Chen, S.-J.; Ku, H.-H.; et al. Resveratrol Promotes Osteogenic Differentiation and Protects Against Dexamethasone Damage in Murine Induced Pluripotent Stem Cells. Stem Cells Dev. 2010, 19, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Ardeshirylajimi, A.; Soleimani, M. Enhanced Growth and Osteogenic Differentiation of Induced Pluripotent Stem Cells by Extremely Low-Frequency Electromagnetic Field. Cell. Mol. Biol. 2015, 61, 36–41. [Google Scholar]
- Umeda, K.; Zhao, J.; Simmons, P.; Stanley, E.; Elefanty, A.; Nakayama, N. Human Chondrogenic Paraxial Mesoderm, Directed Specification and Prospective Isolation from Pluripotent Stem Cells. Sci. Rep. 2012, 2, 455. [Google Scholar] [CrossRef]
- Boreström, C.; Simonsson, S.; Enochson, L.; Bigdeli, N.; Brantsing, C.; Ellerström, C.; Hyllner, J.; Lindahl, A. Footprint-Free Human Induced Pluripotent Stem Cells from Articular Cartilage with Redifferentiation Capacity: A First Step Toward a Clinical-Grade Cell Source. Stem Cells Transl. Med. 2014, 3, 433–447. [Google Scholar] [CrossRef]
- Yokoyama, K.; Ikeya, M.; Umeda, K.; Oda, H.; Nodomi, S.; Nasu, A.; Matsumoto, Y.; Izawa, K.; Horigome, K.; Kusaka, T.; et al. Enhanced Chondrogenesis of Induced Pluripotent Stem Cells from Patients with Neonatal-Onset Multisystem Inflammatory Disease Occurs via the Caspase 1-Independent cAMP/Protein Kinase A/CREB Pathway. Arthritis Rheumatol. 2014, 67, 302–314. [Google Scholar] [CrossRef]
- Yamashita, A.; Morioka, M.; Yahara, Y.; Okada, M.; Kobayashi, T.; Kuriyama, S.; Matsuda, S.; Tsumaki, N. Generation of Scaffoldless Hyaline Cartilaginous Tissue from Human IPSCs. Stem Cell Rep. 2015, 4, 404–418. [Google Scholar] [CrossRef]
- Chal, J.; Oginuma, M.; Al Tanoury, Z.; Gobert, B.; Sumara, O.; Hick, A.; Bousson, F.; Zidouni, Y.; Mursch, C.; Moncuquet, P.; et al. Differentiation of Pluripotent Stem Cells to Muscle Fiber to Model Duchenne Muscular Dystrophy. Nat. Biotechnol. 2015, 33, 962–969. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, H.; Liu, H.; Chen, X.; Lu, P.; Zhu, T.; Yang, L.; Yin, Z.; Heng, B.C.; Zhang, Y.; et al. Well-Aligned Chitosan-Based Ultrafine Fibers Committed Teno-Lineage Differentiation of Human Induced Pluripotent Stem Cells for Achilles Tendon Regeneration. Biomaterials 2015, 53, 716–730. [Google Scholar] [CrossRef]
- Klein, S.; Dvornik, J.L.; Yarrabothula, A.R.; Schaniel, C. A Marfan Syndrome Human Induced Pluripotent Stem Cell Line with a Heterozygous FBN1 c.4082G > A Mutation, ISMMSi002-B, for Disease Modeling. Stem Cell Res. 2017, 23, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Yan, L.; Stoddard, C.; Wang, X.; Yue, Z.; Crandall, L.; Robinson, T.; Chang, Y.; Denton, K.; Li, E.; et al. Recapitulating and Correcting Marfan Syndrome in a Cellular Model. Int. J. Boil. Sci. 2017, 13, 588–603. [Google Scholar] [CrossRef] [PubMed]
- Quarto, N.P.; Leonard, B.; Li, S.; Marchand, M.; Anderson, E.; Behr, B.; Francke, U.; Pera, R.A.R.; Chiao, E.; Longaker, M.T. Skeletogenic Phenotype of Human Marfan Embryonic Stem Cells Faithfully Phenocopied by Patient-Specific Induced-Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2011, 109, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Panicker, L.M.; Srikanth, M.P.; Castro-Gomes, T.; Miller, D.; Andrews, N.W.; Feldman, R.A. Gaucher Disease IPSC-Derived Osteoblasts Have Developmental and Lysosomal Defects That Impair Bone Matrix Deposition. Hum. Mol. Genet. 2018, 27, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Sun, S.; Li, Z.; Zhang, X.; Ke, Y.; Yang, J.; Li, X. Application of CRISPR/Cas9 Technologies Combined with IPSCs in the Study and Treatment of Retinal Degenerative Diseases. Qual. Life Res. 2018, 137, 679–688. [Google Scholar] [CrossRef]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome Editing in Induced Pluripotent Stem Cells Using CRISPR/Cas9. Stem Cell Rev. Rep. 2018, 14, 323–336. [Google Scholar] [CrossRef]
- Chuang, K.; Fields, M.A.; Del Priore, L.V. Potential of Gene Editing and Induced Pluripotent Stem Cells (iPSCs) in Treatment of Retinal Diseases. Yale J. Biol. Med. 2017, 90, 635–642. [Google Scholar]
- Seah, Y.F.S.; El Farran, C.; Warrier, T.; Xu, J.; Loh, Y.-H. Induced Pluripotency and Gene Editing in Disease Modelling: Perspectives and Challenges. Int. J. Mol. Sci. 2015, 16, 28614–28634. [Google Scholar] [CrossRef]
- Pini, J.; Rouleau, M.; Desnuelle, C.; Sacconi, S.; Bendahhou, S. Modeling Andersen’s Syndrome in Human Induced Pluripotent Stem Cells. Stem Cells Dev. 2016, 25, 151–159. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Hayashi, Y.; Schlieve, C.R.; Ikeya, M.; Kim, H.; Nguyen, T.D.; Sami, S.; Baba, S.; Barruet, E.; Nasu, A.; et al. Induced Pluripotent Stem Cells from Patients with Human Fibrodysplasia Ossificans Progressiva Show Increased Mineralization and Cartilage Formation. Orphanet J. Rare Dis. 2013, 8, 190. [Google Scholar] [CrossRef]
- Nakajima, T.; Shibata, M.; Nishio, M.; Nagata, S.; Alev, C.; Sakurai, H.; Toguchida, J.; Ikeya, M. Modeling Human Somite Development and Fibrodysplasia Ossificans Progressiva with Induced Pluripotent Stem Cells. Development 2018, 145, dev165431. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Stattin, E.-L.; Murphy, M.; Barry, F.P. Generation of Induced Pluripotent Stem Cells (ARO-IPSC1-11) from a Patient with Autosomal Recessive Osteopetrosis Harboring the c.212 + 1G > T Mutation in SNX10 Gene. Stem Cell Res. 2017, 24, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Ou, M.; Li, C.; Tang, D.; Xue, W.; Xu, Y.; Zhu, P.; Li, B.; Xie, J.; Chen, J.; Sui, W.; et al. Genotyping, Generation and Proteomic Profiling of the First Human Autosomal Dominant Osteopetrosis Type II-Specific Induced Pluripotent Stem Cells. Stem Cell Res. Ther. 2019, 10, 251. [Google Scholar] [CrossRef] [PubMed]
- Kawai, S.; Yoshitomi, H.; Sunaga, J.; Alev, C.; Nagata, S.; Nishio, M.; Hada, M.; Koyama, Y.; Uemura, M.; Sekiguchi, K.; et al. In Vitro Bone-Like Nodules Generated from Patient-Derived IPSCs Recapitulate Pathological Bone Phenotypes. Nat. Biomed. Eng. 2019, 3, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Howden, S.; Far, H.H.; Motazedian, A.; Elefanty, A.G.; Stanley, E.G.; Lamandé, S.R.; Bateman, J.F. The Use of Simultaneous Reprogramming and Gene Correction to Generate an Osteogenesis Imperfecta Patient COL1A1 c. 3936 G > T IPSC Line and an Isogenic Control IPSC Line. Stem Cell Res. 2019, 38, 101453. [Google Scholar] [CrossRef] [PubMed]
- Deyle, D.R.; Khan, I.F.; Ren, G.; Wang, P.-R.; Kho, J.; Schwarze, U.; Russell, D.W. Normal Collagen and Bone Production by Gene-Targeted Human Osteogenesis Imperfecta IPSCs. Mol. Ther. 2012, 20, 204–213. [Google Scholar] [CrossRef]
- Cui, X.; Cui, Y.; Shi, L.; Luan, J.; Zhou, X.; Han, J. A Preliminary Study on the Mechanism of Skeletal Abnormalities in Turner Syndrome Using Inducing Pluripotent Stem Cells (iPS)—Based Disease Models. Intractable Rare Dis. Res. 2019, 8, 113–119. [Google Scholar] [CrossRef]
- Lee, D.-F.; Su, J.; Kim, H.S.; Chang, B.; Papatsenko, D.; Zhao, R.; Yuan, Y.; Gingold, J.; Xia, W.; Darr, H.; et al. Modeling Familial Cancer with Induced Pluripotent Stem Cells. Cell 2015, 161, 240–254. [Google Scholar] [CrossRef]
- Zhou, R.-J.; Xu, A.; Tu, J.; Liu, M.; Gingold, J.; Zhao, R.; Lee, D.-F. Modeling Osteosarcoma Using Li-Fraumeni Syndrome Patient-Derived Induced Pluripotent Stem Cells. J. Vis. Exp. 2018, 136, e57664. [Google Scholar] [CrossRef]
- Xu, M.; Stattin, E.-L.; Shaw, G.; Heinegård, D.; Sullivan, G.J.; Wilmut, I.; Colman, A.; Önnerfjord, P.; Khabut, A.; Aspberg, A.; et al. Chondrocytes Derived from Mesenchymal Stromal Cells and Induced Pluripotent Cells of Patients with Familial Osteochondritis Dissecans Exhibit an Endoplasmic Reticulum Stress Response and Defective Matrix Assembly. Stem Cells Transl. Med. 2016, 5, 1171–1181. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Oda, H.; Ito, J.; Niwa, A.; Tanaka, T.; Hijikata, A.; Seki, R.; Nagahashi, A.; Osawa, M.; Asaka, I.; et al. Identification of a High-Frequency Somatic NLRC4 Mutation as a Cause of Autoinflammation by Pluripotent Cell-Based Phenotype Dissection. Arthritis Rheumatol. 2017, 69, 447–459. [Google Scholar] [CrossRef]
- Kimura, T.; Ozaki, T.; Fujita, K.; Yamashita, A.; Morioka, M.; Ozono, K.; Tsumaki, N. Proposal of Patient-Specific Growth Plate Cartilage Xenograft Model for FGFR3 Chondrodysplasia. Osteoarthr. Cartil. 2018, 26, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Morioka, M.; Kishi, H.; Kimura, T.; Yahara, Y.; Okada, M.; Fujita, K.; Sawai, H.; Ikegawa, S.; Tsumaki, N. Statin Treatment Rescues FGFR3 Skeletal Dysplasia Phenotypes. Nature 2014, 513, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.; Yi, H.; Diecke, S.; Kim, J.; Jung, H.; Rim, Y.A.; Jung, S.M.; Kim, M.; Kim, Y.G.; et al. Generation of Disease-Specific Induced Pluripotent Stem Cells from Patients with Rheumatoid Arthritis and Osteoarthritis. Arthr. Res. Ther. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Castro-Viñuelas, R.; Sanjurjo-Rodríguez, C.; Piñeiro-Ramil, M.; Rodríguez-Fernández, S.; Fuentes-Boquete, I.; Blanco, F.; Díaz-Prado, S. Generation of a Human Control IPS Cell Line (ESi080-A) from a Donor with No Rheumatic Diseases. Stem Cell Res. 2020, 43, 101683. [Google Scholar] [CrossRef] [PubMed]
- Castro-Viñuelas, R.; Sanjurjo-Rodríguez, C.; Piñeiro-Ramil, M.; Hermida-Gómez, T.; Rodríguez-Fernández, S.; Oreiro, N.; De Toro, J.; Fuentes, I.; Blanco, F.J.; Díaz-Prado, S. Generation and Characterization of Human Induced Pluripotent Stem Cells (iPSCs) from Hand Osteoarthritis Patient-Derived Fibroblasts. Sci. Rep. 2020, 10, 1–13. [Google Scholar]
- Kim, M.-J.; Son, M.J.; Son, M.-Y.; Seol, B.; Kim, J.; Park, J.; Kim, J.H.; Kim, Y.-H.; Park, S.A.; Lee, C.; et al. Generation of Human Induced Pluripotent Stem Cells from Osteoarthritis Patient-Derived Synovial Cells. Arthritis Rheum. 2011, 63, 3010–3021. [Google Scholar] [CrossRef]
- Tanaka, A.; Woltjen, K.; Miyake, K.; Hotta, A.; Ikeya, M.; Yamamoto, T.; Nishino, T.; Shoji, E.; Sehara-Fujisawa, A.; Manabe, Y.; et al. Efficient and Reproducible Myogenic Differentiation from Human IPS Cells: Prospects for Modeling Miyoshi Myopathy in Vitro. PLoS ONE 2013, 8, e61540. [Google Scholar] [CrossRef]
- Kokubu, Y.; Nagino, T.; Sasa, K.; Oikawa, T.; Miyake, K.; Kume, A.; Fukuda, M.; Fuse, H.; Tozawa, R.; Sakurai, H. Phenotypic Drug Screening for Dysferlinopathy Using Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Transl. Med. 2019, 8, 1017–1029. [Google Scholar] [CrossRef]
- Turan, S.; Farruggio, A.P.; Srifa, W.; Day, J.W.; Calos, M.P. Precise Correction of Disease Mutations in Induced Pluripotent Stem Cells Derived from Patients with Limb Girdle Muscular Dystrophy. Mol. Ther. 2016, 24, 685–696. [Google Scholar] [CrossRef]
- Selvaraj, S.; Dhoke, N.R.; Kiley, J.; Mateos-Aierdi, A.J.; Tungtur, S.; Mondragon-Gonzalez, R.; Killeen, G.; Oliveira, V.K.; De Munain, A.L.; Perlingeiro, R.C. Gene Correction of LGMD2A Patient-Specific IPSCs for the Development of Targeted Autologous Cell Therapy. Mol. Ther. 2019, 27, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Steele-Stallard, H.B.; Pinton, L.; Sarcar, S.; Ozdemir, T.; Maffioletti, S.M.; Zammit, P.S.; Tedesco, F.S. Modeling Skeletal Muscle Laminopathies Using Human Induced Pluripotent Stem Cells Carrying Pathogenic LMNA Mutations. Front. Physiol. 2018, 9, 1332. [Google Scholar] [CrossRef] [PubMed]
- Abujarour, R.; Bennett, M.; Valamehr, B.; Lee, T.T.; Robinson, M.; Robbins, D.; Le, T.; Lai, K.; Flynn, P. Myogenic Differentiation of Muscular Dystrophy-Specific Induced Pluripotent Stem Cells for Use in Drug Discovery. Stem Cells Transl. Med. 2014, 3, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Lim, H.; Estrellas, K.; Mula, J.; Cohen, T.V.; Zhang, Y.; Donnelly, C.J.; Richard, J.-P.; Kim, Y.J.; Kim, H.; et al. Concordant But Varied Phenotypes Among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived Using a Human IPSC-Based Model. Cell Rep. 2016, 15, 2301–2312. [Google Scholar] [CrossRef]
- Ferrari, G.; Muntoni, F.; Tedesco, F.S. Generation of Two Genomic-Integration-Free DMD IPSC Lines with Mutations Affecting All Dystrophin Isoforms and Potentially Amenable to Exon-Skipping. Stem Cell Res. 2020, 43, 101688. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.-H.; Bassel-Duby, R.; Olson, E.N. Functional Correction of Dystrophin Actin Binding Domain Mutations by Genome Editing. JCI Insight 2017, 2, e95918. [Google Scholar] [CrossRef]
- Modragón-González, R.; Perlingeiro, R.C.R. Recapitulating Muscle Disease Phenotypes with Myotonic Dystrophy 1 Induced Pluripotent Stem Cells: A Tool for Disease Modeling and Drug Discovery. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-Mediated Editing of Trinucleotide Repeat Expansion in Myotonic Dystrophy Patient-Derived IPS and Myogenic Cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef]
- Ho, J.C.Y.; Zhou, T.; Lai, W.-H.; Huang, Y.; Chan, Y.-C.; Li, X.; Wong, N.L.Y.; Li, Y.; Au, K.-W.; Guo, D.; et al. Generation of Induced Pluripotent Stem Cell Lines from 3 Distinct Laminopathies Bearing Heterogeneous Mutations in Lamin A/C. Aging 2011, 3, 380–390. [Google Scholar] [CrossRef]
- Siu, C.-W.; Lee, Y.-K.; Ho, J.C.-Y.; Lai, W.-H.; Chan, Y.-C.; Ng, K.-M.; Wong, L.-Y.; Au, K.-W.; Lau, Y.-M.; Zhang, J.; et al. Modeling of Lamin A/C Mutation Premature Cardiac Aging Using Patient-Specific Induced Pluripotent Stem Cells. Aging 2012, 4, 803–822. [Google Scholar] [CrossRef]
- Yasuno, T.; Osafune, K.; Sakurai, H.; Asaka, I.; Tanaka, A.; Yamaguchi, S.; Yamada, K.; Hitomi, H.; Arai, S.; Kurose, Y.; et al. Functional Analysis of IPSC-Derived Myocytes from a Patient with Carnitine Palmitoyltransferase II Deficiency. Biochem. Biophys. Res. Commun. 2014, 448, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, K.J.; Nalbandian, A.; Weiss, L.N.; Chang, I.; Yu, H.; Khatib, B.; Tan, B.; Scarfone, V.; Kimonis, V. Myogenic Differentiation of VCP Disease-Induced Pluripotent Stem Cells: A Novel Platform for Drug Discovery. PLoS ONE 2017, 12, e0176919. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Arber, C.E.; Bartolome, F.; De Vicente, M.; Preza, E.; Carro, E.; Houlden, H.; Gandhi, S.; Wray, S.; Abramov, A.Y. Mutations in Valosin-Containing Protein (VCP) Decrease ADP/ATP Translocation across the Mitochondrial Membrane and Impair Energy Metabolism in Human Neurons. J. Boil. Chem. 2017, 292, 8907–8917. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Awaya, T.; Jonouchi, T.; Kimura, R.; Kimura, S.; Era, T.; Heike, T.; Sakurai, H. A Skeletal Muscle Model of Infantile-Onset Pompe Disease with Patient-Specific IPS Cells. Sci. Rep. 2017, 7, 13473. [Google Scholar] [CrossRef]
- Higuchi, T.; Kawagoe, S.; Otsud, M.; Shimad, Y.; Kobayashi, H.; Hirayama, R.; Eto, K.; Ida, H.; Ohashi, T.; Nakauchi, H.; et al. The Generation of Induced Pluripotent Stem Cells (iPSCs) from Patients with Infantile and Late-Onset Types of Pompe Disease and the Effects of Treatment with Acid-α-Glucosidase in Pompe’s IPSCs. Mol. Genet. Metab. 2014, 112, 44–48. [Google Scholar] [CrossRef]
- Raval, K.K.; Tao, R.; White, B.E.; De Lange, W.J.; Koonce, C.H.; Yu, J.; Kishnani, P.S.; Thomson, J.A.; Mosher, D.F.; Ralphe, J.C.; et al. Pompe Disease Results in a Golgi-Based Glycosylation Deficit in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. J. Boil. Chem. 2014, 290, 3121–3136. [Google Scholar] [CrossRef]
- Huang, H.-P.; Chen, H.F.; Chuang, C.-Y.; Stone, L.; Li, L.-T.; Ho, H.-N.; Hwu, W.; Chien, C.; Chiang, S.-C.; Kuo, H.-C. Human Pompe Disease-Induced Pluripotent Stem Cells for Pathogenesis Modeling, Drug Testing and Disease Marker Identification. Hum. Mol. Genet. 2011, 20, 4851–4864. [Google Scholar] [CrossRef]
- Mikami, Y.; Matsumoto, T.; Kano, K.; Toriumi, T.; Somei, M.; Honda, M.J.; Komiyama, K. Current Status of Drug Therapies for Osteoporosis and the Search for Stem Cells Adapted for Bone Regenerative Medicine. Anat. Sci. Int. 2013, 89, 1–10. [Google Scholar] [CrossRef]
- Nakajima, T.; Ikeya, M. Insights into the Biology of Fibrodysplasia Ossificans Progressiva Using Patient-Derived Induced Pluripotent Stem Cells. Regen. Ther. 2019, 11, 25–30. [Google Scholar] [CrossRef]
- Kuroda, Y.; Yuasa, S.; Watanabe, Y.; Ito, S.; Egashira, T.; Seki, T.; Hattori, T.; Ohno, S.; Kodaira, M.; Suzuki, T.; et al. Flecainide Ameliorates Arrhythmogenicity through NCX Flux in Andersen-Tawil Syndrome-IPS Cell-Derived Cardiomyocytes. Biochem. Biophys. Rep. 2017, 9, 245–256. [Google Scholar] [CrossRef]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Sgambato, J.A.; Lipinski, M.M.; Miller, D.; Zeng, X.; Feldman, R.A. Altered TFEB-Mediated Lysosomal Biogenesis in Gaucher Disease IPSC-Derived Neuronal Cells. Hum. Mol. Genet. 2015, 24, 5775–5788. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Florer, J.; Mayhew, C.N.; Jia, Z.; Zhao, Z.; Xu, K.; Ran, H.; Liou, B.; Zhang, W.; Setchell, K.D.R.; et al. Properties of Neurons Derived from Induced Pluripotent Stem Cells of Gaucher Disease Type 2 Patient Fibroblasts: Potential Role in Neuropathology. PLoS ONE 2015, 10, e0118771. [Google Scholar] [CrossRef] [PubMed]
- Kodaka, Y.; Rabu, G.; Asakura, A. Skeletal Muscle Cell Induction from Pluripotent Stem Cells. Stem Cells Int. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C.R. Human ES- and IPS-Derived Myogenic Progenitors Restore DYSTROPHIN and Improve Contractility Upon Transplantation in Dystrophic Mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef]
- Xia, G.; Terada, N.; Ashizawa, T. Human IPSC Models to Study Orphan Diseases: Muscular Dystrophies. Curr. Stem Cell Rep. 2018, 4, 299–309. [Google Scholar] [CrossRef]
- Hagan, M.; Ashraf, M.; Kim, I.-M.; Weintraub, N.L.; Tang, Y. Effective Regeneration of Dystrophic Muscle Using Autologous IPSC-Derived Progenitors with CRISPR-Cas9 Mediated Precise Correction. Med. Hypotheses 2018, 110, 97–100. [Google Scholar] [CrossRef]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.B.; Mannhardt, I.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-Dimensional Human IPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef]
- Weiler, T.; Bashir, R.; Anderson, L.V.B.; Davison, K.; Moss, J.A.; Britton, S.; Nylen, E.; Keers, S.; Vafiadaki, E.; Greenberg, C.R.; et al. Identical Mutation in Patients with Limb Girdle Muscular Dystrophy Type 2B Or Miyoshi Myopathy Suggests a Role for Modifier Gene(s). Hum. Mol. Genet. 1999, 8, 871–877. [Google Scholar] [CrossRef]
- Huang, H.-P.; Chiang, W.; Stone, L.; Kang, C.-K.; Chuang, C.-Y.; Kuo, H.-C. Using Human Pompe Disease-Induced Pluripotent Stem Cell-Derived Neural Cells to Identify Compounds with Therapeutic Potential. Hum. Mol. Genet. 2019, 28, 3880–3894. [Google Scholar] [CrossRef]
- Mitzelfelt, K.A.; Limphong, P.; Choi, M.J.; Kondrat, F.D.L.; Lai, S.; Kolander, K.D.; Kwok, W.-M.; Dai, Q.; Grzybowski, M.N.; Zhang, H.; et al. The Human 343delT HSPB5 Chaperone Associated with Early-Onset Skeletal Myopathy Causes Defects in Protein Solubility. J. Boil. Chem. 2016, 291, 14939–14953. [Google Scholar] [CrossRef]
- Loebel, C.; Burdick, J.A. Engineering Stem and Stromal Cell Therapies for Musculoskeletal Tissue Repair. Cell Stem Cell 2018, 22, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Andersson, G.B.J. Stem Cell Therapy for Intervertebral Disc Regeneration: Obstacles and Solutions. Nat. Rev. Rheumatol. 2015, 11, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Schol, J. Cell Therapy for Intervertebral Disc Repair: Clinical Perspective. J. Orthop. Transl. 2017, 9, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, R.J.; Jones, E.; Sanjurjo-Rodríguez, C.; Lotfy, A.; Ganguly, P.; Churchman, S.M.; Castana, P.; Tan, H.B.; McGonagle, D.; Papadimitriou, E.; et al. Regulation of Angiogenesis Discriminates Tissue Resident MSCs from Effective and Defective Osteogenic Environments. J. Clin. Med. 2020, 9, 1628. [Google Scholar] [CrossRef]
- Piñeiro-Ramil, M.; Castro-Viñuelas, R.; Sanjurjo-Rodríguez, C.; Hermida-Gómez, T.; Fuentes-Boquete, I.; De Toro-Santos, F.J.; Blanco-García, F.J.; Díaz-Prado, S.M. Cell Therapy and Tissue Engineering for Cartilage Repair. In Cartilage Repair and Regeneration; IntechOpen: London, UK, 2017. [Google Scholar]
- Monaco, M.L.; Merckx, G.; Ratajczak, J.; Gervois, P.; Hilkens, P.; Clegg, P.; Bronckaers, A.; Vandeweerd, J.-M.; Lambrichts, I. Stem Cells for Cartilage Repair: Preclinical Studies and Insights in Translational Animal Models and Outcome Measures. Stem Cells Int. 2018, 2018, 1–22. [Google Scholar] [CrossRef]
- Rama, P.; Matuska, S.; Paganoni, G.; Spinelli, A.; De Luca, M.; Pellegrini, G. Limbal Stem-Cell Therapy and Long-Term Corneal Regeneration. N. Engl. J. Med. 2010, 363, 147–155. [Google Scholar] [CrossRef]
- Duarte, R.F.; Labopin, M.; Bader, P.; Basak, G.W.; Bonini, C.; Chabannon, C.; Corbacioglu, S.; Dreger, P.; Dufour, C.; Gennery, A.R.; et al. Indications for Haematopoietic Stem Cell Transplantation for Haematological Diseases, Solid Tumours and Immune Disorders: Current Practice in Europe, 2019. Bone Marrow Transplant. 2019, 54, 1525–1552. [Google Scholar] [CrossRef]
- Ong, C.S.; Yesantharao, P.; Huang, C.-Y.; Mattson, G.; Boktor, J.; Fukunishi, T.; Zhang, H.; Hibino, N. 3D Bioprinting Using Stem Cells. Pediatr. Res. 2017, 83, 223–231. [Google Scholar] [CrossRef]
- Kamei, N.; Adachi, N.; Ochi, M. Magnetic Cell Delivery for the Regeneration of Musculoskeletal and Neural Tissues. Regen. Ther. 2018, 9, 116–119. [Google Scholar] [CrossRef]
- De Peppo, G.M.; Marcos-Campos, I.; Kahler, D.J.; Alsalman, D.; Shang, L.; Vunjak-Novakovic, G.; Marolt, D. Engineering Bone Tissue Substitutes from Human Induced Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 8680–8685. [Google Scholar] [CrossRef]
- Hynes, K.; Menicanin, D.; Mrozik, K.M.; Gronthos, S.; Bartold, P.M. Generation of Functional Mesenchymal Stem Cells from Different Induced Pluripotent Stem Cell Lines. Stem Cells Dev. 2013, 23, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.D.; Kuznetsov, S.A.; Cherman, N.; Park, K.; Chen, K.G.; McClendon, B.N.; Hamilton, R.S.; McKay, R.D.; Chenoweth, J.G.; Mallon, B.S.; et al. Directed Differentiation of Human Induced Pluripotent Stem Cells Toward Bone and Cartilage: In Vitro Versus In Vivo Assays. Stem Cells Transl. Med. 2014, 3, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-X.; Wu, X.; Liang, Y.; Gu, H.; Song, K.; Zou, X.; Zhou, G.Q. Repair of Cartilage Defects in Osteoarthritis Rats with Induced Pluripotent Stem Cell Derived Chondrocytes. BMC Biotechnol. 2016, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Uto, S.; Nishizawa, S.; Hikita, A.; Takato, T.; Hoshi, K. Application of Induced Pluripotent Stem Cells for Cartilage Regeneration in CLAWN Miniature Pig Osteochondral Replacement Model. Regen. Ther. 2018, 9, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Nejadnik, H.; Diecke, S.; Lenkov, O.D.; Chapelin, F.; Donig, J.; Tong, X.-M.; Derugin, N.; Chan, R.C.F.; Gaur, A.; Yang, F.; et al. Improved Approach for Chondrogenic Differentiation of Human Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2015, 11, 242–253. [Google Scholar] [CrossRef]
- Xu, X.; Shi, D.; Liu, Y.; Yao, Y.; Dai, J.; Xu, Z.; Chen, D.; Teng, H.; Jiang, Q. In Vivo Repair of Full-Thickness Cartilage Defect with Human IPSC-Derived Mesenchymal Progenitor Cells in a Rabbit Model. Exp. Ther. Med. 2017, 14, 239–245. [Google Scholar] [CrossRef]
- Ko, J.-Y.; Kim, K.-I.; Park, S.; Im, G.-I. In Vitro Chondrogenesis and in Vivo Repair of Osteochondral Defect with Human Induced Pluripotent Stem Cells. Biomaterials 2014, 35, 3571–3581. [Google Scholar] [CrossRef]
- Saito, A.; Ooki, A.; Nakamura, T.; Onodera, S.; Hayashi, K.; Hasegawa, D.; Okudaira, T.; Watanabe, K.; Kato, H.; Onda, T.; et al. Targeted Reversion of Induced Pluripotent Stem Cells from Patients with Human Cleidocranial Dysplasia Improves Bone Regeneration in a Rat Calvarial Bone Defect Model. Stem Cell Res. Ther. 2018, 9, 12. [Google Scholar] [CrossRef]
- Jungbluth, P.; Spitzhorn, L.-S.; Grassmann, J.; Tanner, S.; Latz, D.; Rahman, S.; Bohndorf, M.; Wruck, W.; Sager, M.; Grotheer, V.; et al. Human IPSC-Derived IMSCs Improve Bone Regeneration in Mini-Pigs. Bone Res. 2019, 7, 1–11. [Google Scholar] [CrossRef]
- Goudenege, S.; Lebel, C.; Huot, N.B.; Dufour, C.; Fujii, I.; Gekas, J.; Rousseau, J.; Tremblay, J.P. Myoblasts Derived from Normal HESCs and Dystrophic HiPSCs Efficiently Fuse with Existing Muscle Fibers Following Transplantation. Mol. Ther. 2012, 20, 2153–2167. [Google Scholar] [CrossRef]
- Darabi, R.; Pan, W.; Bosnakovski, D.; Baik, J.; Kyba, M.; Perlingeiro, R.C.R. Functional Myogenic Engraftment from Mouse IPS Cells. Stem Cell Rev. Rep. 2011, 7, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Kouroupis, D.; Kyrkou, A.; Triantafyllidi, E.; Katsimpoulas, M.; Chalepakis, G.; Goussia, A.; Georgoulis, A.; Murphy, C.; Fotsis, T. Generation of Stem Cell-Based Bioartificial Anterior Cruciate Ligament (ACL) Grafts for Effective ACL Rupture Repair. Stem Cell Res. 2016, 17, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Xing, R.; Jiang, L.; Li, Z.; Liu, P.; Wang, H.; Xi-Lei, L.; Dong, J. Thermosensitive Hydrogels Loaded with human-induced Pluripotent Stem Cells Overexpressing Growth Differentiation factor-5 Ameliorate Intervertebral Disc Degeneration in Rats. J. Biomed. Mater. Res. Part B Appl. Biomater. 2020, 108, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Sheyn, D.; Ben-David, S.; Tawackoli, W.; Zhou, Z.; Salehi, K.; Bez, M.; De Mel, S.; Chan, V.; Roth, J.; Avalos, P.; et al. Human IPSCs Can Be Differentiated into Notochordal Cells That Reduce Intervertebral Disc Degeneration in a Porcine Model. Theranostics 2019, 9, 7506–7524. [Google Scholar] [CrossRef]
- Jeon, O.H.; Panicker, L.M.; Lu, Q.; Chae, J.J.; Feldman, R.A.; Elisseeff, J.H. Human IPSC-Derived Osteoblasts and Osteoclasts Together Promote Bone Regeneration in 3D Biomaterials. Sci. Rep. 2016, 6, 26761. [Google Scholar] [CrossRef]
- Wrighton, K.H. Stem Cells: The Different Flavours of IPS Cells. Nat. Rev. Genet. 2017, 18, 394. [Google Scholar] [CrossRef]
- Boland, M.J.; Nazor, K.L.; Loring, J.F. Epigenetic Regulation of Pluripotency and Differentiation. Circ. Res. 2014, 115, 311–324. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic Memory in Induced Pluripotent Stem Cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef]
- Banito, A.; Gil, J. Induced Pluripotent Stem Cells and Senescence: Learning the Biology to Improve the Technology. EMBO Rep. 2010, 11, 353–359. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Veiga, E.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence During Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Tidball, A.M.; Dang, L.T.; Glenn, T.W.; Kilbane, E.G.; Klarr, D.J.; Margolis, J.L.; Uhler, M.D.; Parent, J.M. Rapid Generation of Human Genetic Loss-of-Function IPSC Lines by Simultaneous Reprogramming and Gene Editing. Stem Cell Rep. 2017, 9, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Glicksman, M.A. Induced Pluripotent Stem Cells: The Most Versatile Source for Stem Cell Therapy. Clin. Ther. 2018, 40, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Suzuki, K.; Kim, N.Y.; Liu, G.-H.; Belmonte, J.C.I. A Cut above the Rest: Targeted Genome Editing Technologies in Human Pluripotent Stem Cells. J. Boil. Chem. 2013, 289, 4594–4599. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.E.; Melton, D. Turning Straw into Gold: Directing Cell Fate for Regenerative Medicine. Nat. Rev. Genet. 2011, 12, 243–252. [Google Scholar] [CrossRef]
- Smith, A.S.; Macadangdang, J.; Leung, W.; Laflamme, M.; Kim, D.-H. Human IPSC-Derived Cardiomyocytes and Tissue Engineering Strategies for Disease Modeling and Drug Screening. Biotechnol. Adv. 2016, 35, 77–94. [Google Scholar] [CrossRef]
- Barreca, M.M.; Cancemi, P.; Geraci, F. Mesenchymal and Induced Pluripotent Stem Cells-Derived Extracellular Vesicles: The New Frontier for Regenerative Medicine? Cells 2020, 9, 1163. [Google Scholar] [CrossRef]
- Boyd, A.S.; Rodrigues, N.P.; Lui, K.O.; Fu, X.; Xu, Y. Concise Review: Immune Recognition of Induced Pluripotent Stem Cells. Stem Cells 2012, 30, 797–803. [Google Scholar] [CrossRef]
- Alvarez-Palomo, B.; Vives, J.; Casaroli-Marano, R.P.; Gomez, S.G.; Rodriguez Gómez, L.; Edel, M.J.; Querol Giner, S. Adapting Cord Blood Collection and Banking Standard Operating Procedures for HLA-Homozygous Induced Pluripotent Stem Cells Production and Banking for Clinical Application. J. Clin. Med. 2019, 8, 476. [Google Scholar] [CrossRef]
- Simonson, O.E.; Domogatskaya, A.; Volchkov, P.; Rodin, S. The Safety of Human Pluripotent Stem Cells in Clinical Treatment. Ann. Med. 2015, 47, 370–380. [Google Scholar] [CrossRef]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic Derivatives of Induced Pluripotent Stem Cells Evade Immune Rejection in Fully Immunocompetent Allogeneic Recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Uto, S.; Nishizawa, S.; Takasawa, Y.; Asawa, Y.; Fujihara, Y.; Takato, T.; Hoshi, K. Bone and Cartilage Repair by Transplantation of Induced Pluripotent Stem Cells in Murine Joint Defect Model. Biomed. Res. 2013, 34, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Taylor, S.E.B.; Smeriglio, P.; Lai, J.; Maloney, W.J.; Yang, F.; Bhutani, N. Early Induction of a Prechondrogenic Population Allows Efficient Generation of Stable Chondrocytes from Human Induced Pluripotent Stem Cells. FASEB J. 2015, 29, 3399–3410. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.J.; Ulbright, T.M.; Pera, M.F.; Looijenga, L.H.J. Lessons from Human Teratomas to Guide Development of Safe Stem Cell Therapies. Nat. Biotechnol. 2012, 30, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, S.; Wakitani, S.; Shimamoto, A.; Kamei, N.; Sawa, M.; Adachi, N.; Ochi, M. Magnetic Targeted Delivery of Induced Pluripotent Stem Cells Promotes Articular Cartilage Repair. Stem Cells Int. 2017, 2017, 9514719. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Xiao, K.; Xiang, S.; Li, Z.; Weng, X. Emerging Role of Exosomes in the Joint Diseases. Cell. Physiol. Biochem. 2018, 47, 2008–2017. [Google Scholar] [CrossRef]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef]
- Zhang, S.; Chuah, S.J.; Lai, R.C.; Hui, J.H.P.; Lim, S.K.; Toh, W.S. MSC Exosomes Mediate Cartilage Repair by Enhancing Proliferation, Attenuating Apoptosis and Modulating Immune Reactivity. Biomaterials 2018, 156, 16–27. [Google Scholar] [CrossRef]
- Liu, X.; Li, Q.; Niu, X.; Hu, B.; Chen, S.; Song, W.; Ding, J.; Zhang, C.; Wang, Y. Exosomes Secreted from Human-Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Prevent Osteonecrosis of the Femoral Head by Promoting Angiogenesis. Int. J. Boil. Sci. 2017, 13, 232–244. [Google Scholar] [CrossRef]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying Extracellular Vesicles Based Therapeutics in Clinical Trials—An ISEV Position Paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef]
- Baglío, S.R.; Pegtel, D.M.; Baldini, N. Mesenchymal Stem Cell Secreted Vesicles Provide Novel Opportunities in (stem) Cell-Free Therapy. Front. Physiol. 2012, 3, 359. [Google Scholar] [CrossRef]
- Zhang, H.-C.; Liu, X.-B.; Yang, Y.; Guo, Z.-K.; Huang, S.; Bi, X.-Y.; Wang, H.-X.; Xie, L.-X.; Wang, Y.-Q.; Cao, X.-F.; et al. Microvesicles Derived from Human Umbilical Cord Mesenchymal Stem Cells Stimulated by Hypoxia Promote Angiogenesis Both In Vitro and In Vivo. Stem Cells Dev. 2012, 21, 3289–3297. [Google Scholar] [CrossRef] [PubMed]
- Keshtkar, S.; Azarpira, N.; Ghahremani, M.H. Mesenchymal Stem Cell-Derived Extracellular Vesicles: Novel Frontiers in Regenerative Medicine. Stem Cell Res. Ther. 2018, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, L.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal Experimental Requirements for Definition of Extracellular Vesicles and Their Functions: A Position Statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Disease | Suggested Associated Genes | Cell Line Name | Differentiate D Cell Type | Disease-Related Findings | Reference |
|---|---|---|---|---|---|
| Marfan’s syndrome (MFS) | FBN1 | ISMMSi002-B | - | - | Klein et al. [63] |
| MFS-iPSCs | Osteoblasts and vascular smooth muscle cells | Reduced osteogenesis in derivative MSCs, reduced ratio of contracting cells and altered calcium signals in derivative SMCs. | Park et al. [64] | ||
| MFSiPS | Osteoblasts and chondrocytes | Impaired osteogenic differentiation. | Quarto et al. [65] | ||
| Andersen’s syndrome (AS) | KCNJ2, KCNJ5 | AS-iPS | Osteoblasts | Osteogenic differentiation markers were lower in differentiated AS-iPS compared with control iPSC cells. | Pini et al. [71] |
| Fibrodysplasia ossificans progressiva (FOP) | ACVR1 | FOP iPS | Osteoblasts and chondrocytes | FOP-iPS cells showed a trend towards increased mineralization and enhanced chondrogenesis in vitro. | Matsumoto et al. [72] |
| Dermatome, myotome, sclerotome and syndetome cells | OP-iPSC-MSCs showed enhanced chondrogenesis but FOP-iPSC-derived sclerotome did not. | Nakajima et al. [73] | |||
| Osteopetrosis | TCIRG1, SNX10, CLCN7, CAII, PLEKHM1, RANKL | BSG-OST14-MSC, ZCD-OST10-MSC, ANC-OST3-MSC | - | - | Okur et al. [40] |
| ARO-iPSC1-11 | - | - | Xu et al. [74] | ||
| ADO2-iPSCs | - | - | Ou et al. [75] | ||
| Osteogenesis imperfecta (OI) | COL1A1 | OI-iPSCs | Osteoblasts and osteocytes | Osteogenic marker expression was similar between OI-iPSC-derived cells and healthy ones. However, amounts of calcium deposition of OI-iPSC-derived cells were lower than those of WT-iPSC-derived cells. Also, intracellular Type I collagen was much larger in OI-cells than in healthy cells. | Kawai et al. [76] |
| MCRIi018-A, MCRIi018-B | - | - | Howden et al. [77] | ||
| iPSC-OI-FV, iPSC-OI-LV, iPSCe-LV | Osteoblasts | - | Deyle et al. [78] | ||
| Turner syndrome (TS) | Monosomy X | TS-iPSC | Osteoblasts and osteoclasts | No differences between osteoblasts from TS-iPSCs and healthy iPSCs but TS-iPSCs showed increased osteoclastogenesis. | Cui et al. [79] |
| Gaucher disease (GD) | GBA1 | GD hiPSC | Osteoblasts | GD iPSC-osteoblasts showed reduced osteogenic differentiation and lysosomal abnormalities that interfered with bone matrix deposition. | Panicker et al. [66] |
| Li-Fraumeni syndrome (LFS) | TP53, H19 | LFS1-A-D, LFS2-A-D, LFS3-A-C | Osteoblasts through MSCs | p53 signaling is active in healthy iPSC-OBs but impaired in LFS. Also, slower and lower osteogenic differentiation in LFS iPSC-MSC with no mineral precipitations observed. | Lee et al. [80] |
| LFS iPSCs | Osteoblasts through MSCs | - | Zhou et al. [81] | ||
| Familial osteochondritis dissecans (FOCD) | ACAN | FOCD-NS1-iPSC-2/30, FOCD-NS2-iPSC-9/13 | Chondrocytes | Chondrocytes from FOCD-NS iPSCs showed glycosaminoglycan (GAG) in the matrix but lack aggrecan, which had a pronounced intracellular localization. | Xu et al. [82] |
| NOMID arthropathy | NLRP3 | NLPR3-iPSCs | Chondrocytes | Chondrocyte-iPSCs produced larger chondrocyte masses than healthy iPSCs owing to GAG overproduction. | Yokoyama et al. [59] |
| NLRC4 | - | - | Kawasaki et al. [83] | ||
| FGFR3-chondrodysplasia | FGFR3 | TD1-shFGFR3 | Chondrocytes | Small hypertrophic chondrocytes. | Kimura et al. [84] |
| TD1-714-3, TD1-10749-1, TD1-315H-2 | Chondrocytes | Chondrocyte-iPSCs showed low levels of GAGs in the extracellular matrix (ECM), decreased proliferation and increased apoptosis. | Yamashita et al. [85] | ||
| Rheumatoid arthritis | Multifactorial | RA iPSCs | Osteoblasts | - | Lee et al. [86] |
| Osteoarthritis | Multifactorial | OA iPSCs | Osteoblasts | - | Lee et al. [86] |
| N1-FiPS4F#7 | - | - | Castro-Viñuelas et al. [87] | ||
| MOA1-FiPS4F#7, MOA2-FiPS4F#17 | Chondrocytes | OA iPSC-chondrocytes showed lower levels of collagens and proteoglycans in the ECM than healthy iPSCs. | Castro-Viñuelas et al. [88] | ||
| hSC52/hSC65 | Chondrocytes | - | Kim et al. [89] | ||
| Myoshi myopathy (MM) | DYSFERLIN | MyoD-hiPSCs | Myocytes | Defective membrane repair in MyoD-MM hiPSC-derived myotubes. | Tanaka et al. [90] |
| MM iPS | Myocytes | Dysferlin protein was lower in MM- than in control-iPSC-myocytes. | Kokubu et al. [91] | ||
| Limb-girdle muscular dystrophies (LGMD): 1B, 2B, 2A, 2D | Several genes implicated (depending on type) | JF010i-DYSFHZ1 | Muscle progenitors | Low levels of dysferlin protein and absence of alpha-sarcoglycan protein were observed in the differentiated LGMD2B- and LGMD2D- iPSCs. | Turan et al. [92] |
| 9015, 0826 and 0989 | Myotubes | LGMD2A-iPSC-myotubes lacked expression of the Calpain 3 protein. | Selvaraj et al. [93] | ||
| R249W | Myogenic cells | Higher proportion of abnormal myotube nuclei compared to control. iPSC myotube nuclei showed morphological defects in nuclear contour ratio. | Steele-Stallard et al. [94] | ||
| Duchenne muscular dystrophy (DMD) | Dystrophin | GM05112-M5.1 | Myoblasts | Ectopic expression of MyoD in DMD iPSCs led to increased expression levels of Dystrophin but transcripts were truncated. | Abujarour et al. [95] |
| DMD-hiPSC-GM05169 | Myoblasts | DMD-iPSC-myoblasts showed aberrant expression of inflammation genes and collagens, BMP/TGFβ signaling and reduced myotube formation compared with control iPSCs. | Choi et al. [96] | ||
| UCLi011-A, UCLi012-A | - | - | Ferrari et al. [97] | ||
| ΔEx8-9 iDMD iPSCs | - | - | Kyrychenko et al. [98] | ||
| Myotonic dystrophy 1 | DMPK | DM1-1 iPS, DM1-2 iPS | Myogenic lineage | iPSC-myogenic progenitors and -myotubes showed intranuclear RNA foci and sequestration of Muscleblind-like protein 1. | Mondragón-González et al. [99] |
| DM1-iPSCs | Myogenic lineage | Ribonuclear foci in the undifferentiated and myo-iPSCs but not in healthy ones. | Dastidar et al. [100] | ||
| Emery-Dreifuss/LMNA-related congenital muscular dystrophy/dilated cardiomyopathy | LMNA | K32del, L35P | Myogenic cells | LMNA mutant iPSC-myogenic cell produced a higher proportion of abnormal myotube nuclei compared with control. | Steele-Stallard et al. [94] |
| DCM | - | - | Ho et al. [101] | ||
| R225X | - | - | Siu et al. [102] | ||
| Carnitine palmitotransferase II deficiency | CPT2 | CPTIID-iPSC | Myocytes | iPSC-myocytes accumulated more palmitoylcarnitine than control ones. | Yasuno et al. [103] |
| Valosin-containing protein disease | VCP | VCP-iPSC | Myogenic progenitor cells | Accumulation of autophagy markers in differentiated myogenic-iPSCs. | Llewelyn et al. [104] |
| R155C, R191Q | - | - | Ludtmann et al. [105] | ||
| Pompe disease | GAA | Pom2 iPSC | Myocytes | iPSC- myocytes showed lysosomal glycogen accumulation and impaired mTORC1 activity. | Yoshida et al. [106] |
| GM20124, GM11661 | - | - | Higuchi et al. [107] | ||
| GM20089, GM20123, GM04912 | - | - | Raval et al. [108] | ||
| PomD-iPSCs | - | - | Huang et al. [109] | ||
| Degenerative disc disease | Multifactorial | DDD NP-derived iPSCs | Nucleus pulposus cells | - | Zhu et al. [49] |
| Disease or Damaged Tissue | Cell Type Used | Animal | Application | Outcome | References |
|---|---|---|---|---|---|
| MIA-induced OA rat model | iPSCs and chondro-differentiated iPSCs | Rats | Injected as cell suspension | Histologically, gradual engraftment, improvement of subchondral integrity and articular cartilage matrix production. Better outcome using chondro-differentiated cells. | Zhu et al. [136] |
| Osteochondral defect model | Mesoderm-differentiated iPSCs | Minipigs | Cells embedded in collagen hydrogel and seeded on bTCP/PLLA scaffolds | Histologically, cartilage formation partially observed at the transplantation sites. | Uto et al. [137] |
| Osteochondral defect model | iPSC-MSCs and chondro-derived iPSCs | Rats | Pellets on PEG and CS methacrylate scaffolds | hiPSC-derived MSC implants had started to produce a chondrogenic matrix but chondro-derived cells showed stronger GAG and collagen type II staining. | Nejadnik et al. [138] |
| Osteochondral defect model | iPSC-MSCs | Rabbits | Cells seeded on Matrigel-coated PLGA scaffolds | Better histological quality of in vivo cartilage defect repair in the experimental group compared with controls. | Xu et al. [139] |
| Osteochondral defect model | Chondrogenic-iPSCs constructs or micromass | Rats | Cell pellets or alginate-hiPSCs constructs | Histologically, hiPSCs showed significantly better quality of cartilage repair than control defects. | Ko et al. [140] |
| Osteochondral defect model | Cartilaginous tissues derived from iPSCs | Rats and minipigs | Transplantation of hiPSC-derived cartilaginous particles | Histologically, cartilage-like particles were observed to be integrated into native tissue. | Yamashita et al. [60] |
| Bone defect model | Osteoblasts-differentiated iPSCs | Rats | Cells embedded on PuraMatrix | Good osteogenic properties both in vivo and in vitro. Bone volume and bone mineral content were significantly higher and more newly formed bone than in iPSC-RUNX2 mutated controls. | Saito et al. [141] |
| Critical size-defect bone model | iPSC-MSCs | Minipigs | Cells seeded on calcium phosphate granules | New bone formation with good osseous consolidation in the central and cortical defect zones but less successful than in the autograft group. | Jungbluth et al. [142] |
| Dystrophy-induced mice model | Myogenic like cells derived from iPSCs | Mice | Injection | MB1-MyoD-hiPSCs were highly fused with host muscle fibers compared with MB1- MyoD-ESCs and control myoblasts. | Goudenege et al. [143] |
| Dystrophy-induced mice model | Myogenic like progenitor cells derived from iPSCs | Mice | Injection | Pax7-induced iPS-derived myogenic progenitors resulted in extensive engraftment and improved contractility of muscles. | Darabi et al. [144] |
| Anterior cruciate ligament injury model | Ligament and osteogenic derivation of iPSCs | Swine | Leeds-Keio constructs | New ACL-like tissue showed morphological and biochemical characteristics resembling those of normal ACL. | Kouroupis et al. [145] |
| Degenerative disc disease | iPSC-nucleus pulposus cells | Rat | Injection of cells embedded on gelatin microspheres | Histology and imaging results indicated partial restoration of iPSC-nucleus pulposus cells and their ECM and disc height and water content increased. | Xia et al. [48] |
| Degenerative disc disease | GDF5+ iPSC | Rat | Injection of cells embedded on a thermosensitive hydrogel | Disc height and histology improved after treatment with GDF5+hiPSCs on hydrogel. | Hu et al. [146] |
| Degenerative disc disease | iPSC-notochordal cells | Pig | Injection of cells resuspended in Geltrex | Good notochordal cell phenotype in vitro and in vivo. Proper functionality of notochordal cells protecting from degeneration and changes in pH level. | Sheyn et al. [147] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanjurjo-Rodríguez, C.; Castro-Viñuelas, R.; Piñeiro-Ramil, M.; Rodríguez-Fernández, S.; Fuentes-Boquete, I.; Blanco, F.J.; Díaz-Prado, S. Versatility of Induced Pluripotent Stem Cells (iPSCs) for Improving the Knowledge on Musculoskeletal Diseases. Int. J. Mol. Sci. 2020, 21, 6124. https://doi.org/10.3390/ijms21176124
Sanjurjo-Rodríguez C, Castro-Viñuelas R, Piñeiro-Ramil M, Rodríguez-Fernández S, Fuentes-Boquete I, Blanco FJ, Díaz-Prado S. Versatility of Induced Pluripotent Stem Cells (iPSCs) for Improving the Knowledge on Musculoskeletal Diseases. International Journal of Molecular Sciences. 2020; 21(17):6124. https://doi.org/10.3390/ijms21176124
Chicago/Turabian StyleSanjurjo-Rodríguez, Clara, Rocío Castro-Viñuelas, María Piñeiro-Ramil, Silvia Rodríguez-Fernández, Isaac Fuentes-Boquete, Francisco J. Blanco, and Silvia Díaz-Prado. 2020. "Versatility of Induced Pluripotent Stem Cells (iPSCs) for Improving the Knowledge on Musculoskeletal Diseases" International Journal of Molecular Sciences 21, no. 17: 6124. https://doi.org/10.3390/ijms21176124
APA StyleSanjurjo-Rodríguez, C., Castro-Viñuelas, R., Piñeiro-Ramil, M., Rodríguez-Fernández, S., Fuentes-Boquete, I., Blanco, F. J., & Díaz-Prado, S. (2020). Versatility of Induced Pluripotent Stem Cells (iPSCs) for Improving the Knowledge on Musculoskeletal Diseases. International Journal of Molecular Sciences, 21(17), 6124. https://doi.org/10.3390/ijms21176124