Lipophagy and Lipolysis Status in Lipid Storage and Lipid Metabolism Diseases

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Lipids in Eukaryotic Cells
2.1. Fatty Acids and Cholesterol—Essential and Toxic
2.2. Lipid Droplets—Storage of Neutral Lipids
3. Lipophagy and Lipolysis—Two Pathways that Play a Crucial Role in Lipid Metabolism
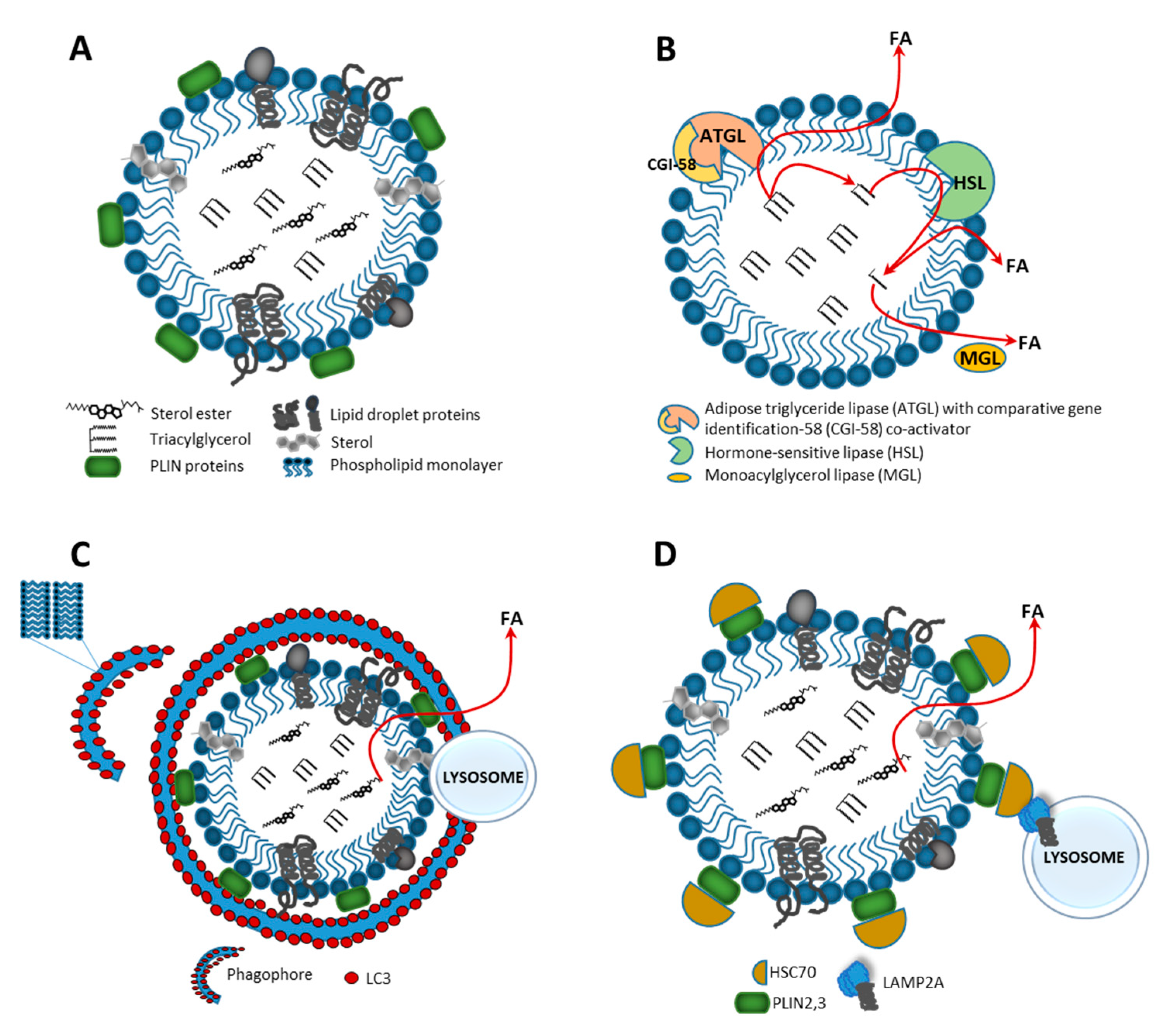
3.1. Catabolism of Lipid Droplets
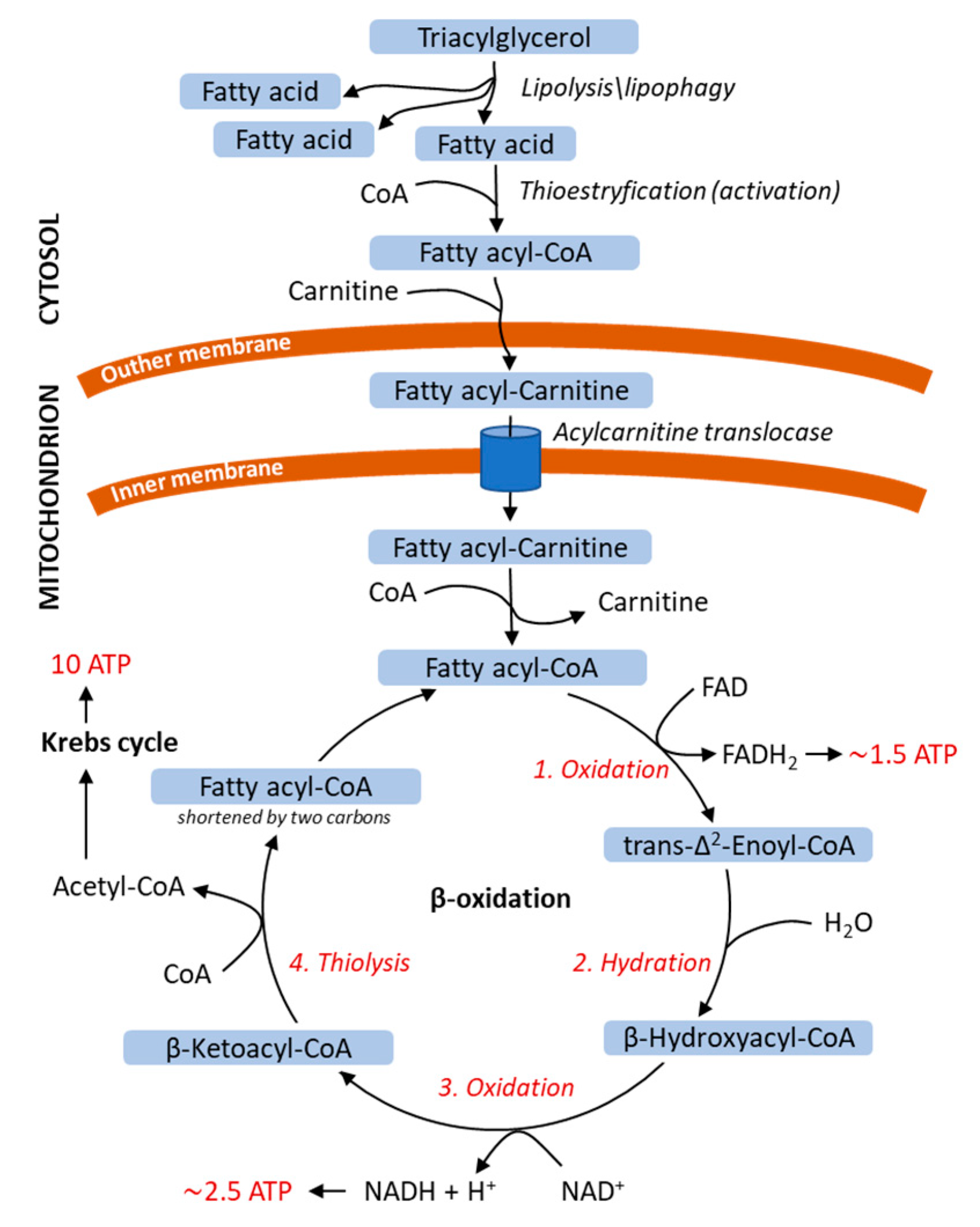
3.2. Energy Release from Fatty Acids
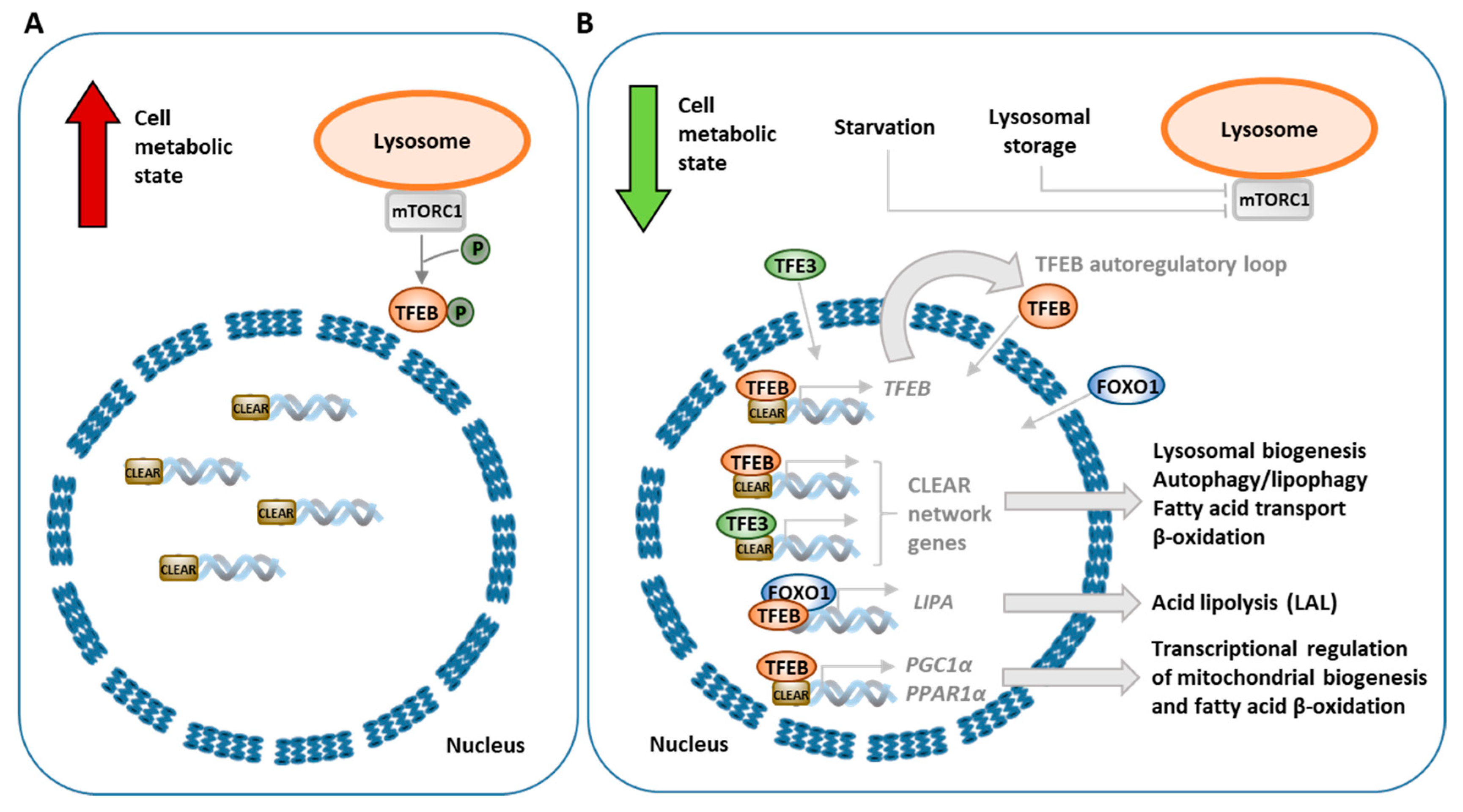
3.3. Transcriptional Regulation of Lipophagy, Lipolysis and Lipid Metabolism
4. Lipid Metabolism and Diseases
4.1. Characterization of Lipid Storage Diseases and Lipid Metabolism Diseases
4.2. Dysregulation of Autophagy or Lipolysis in Diseases
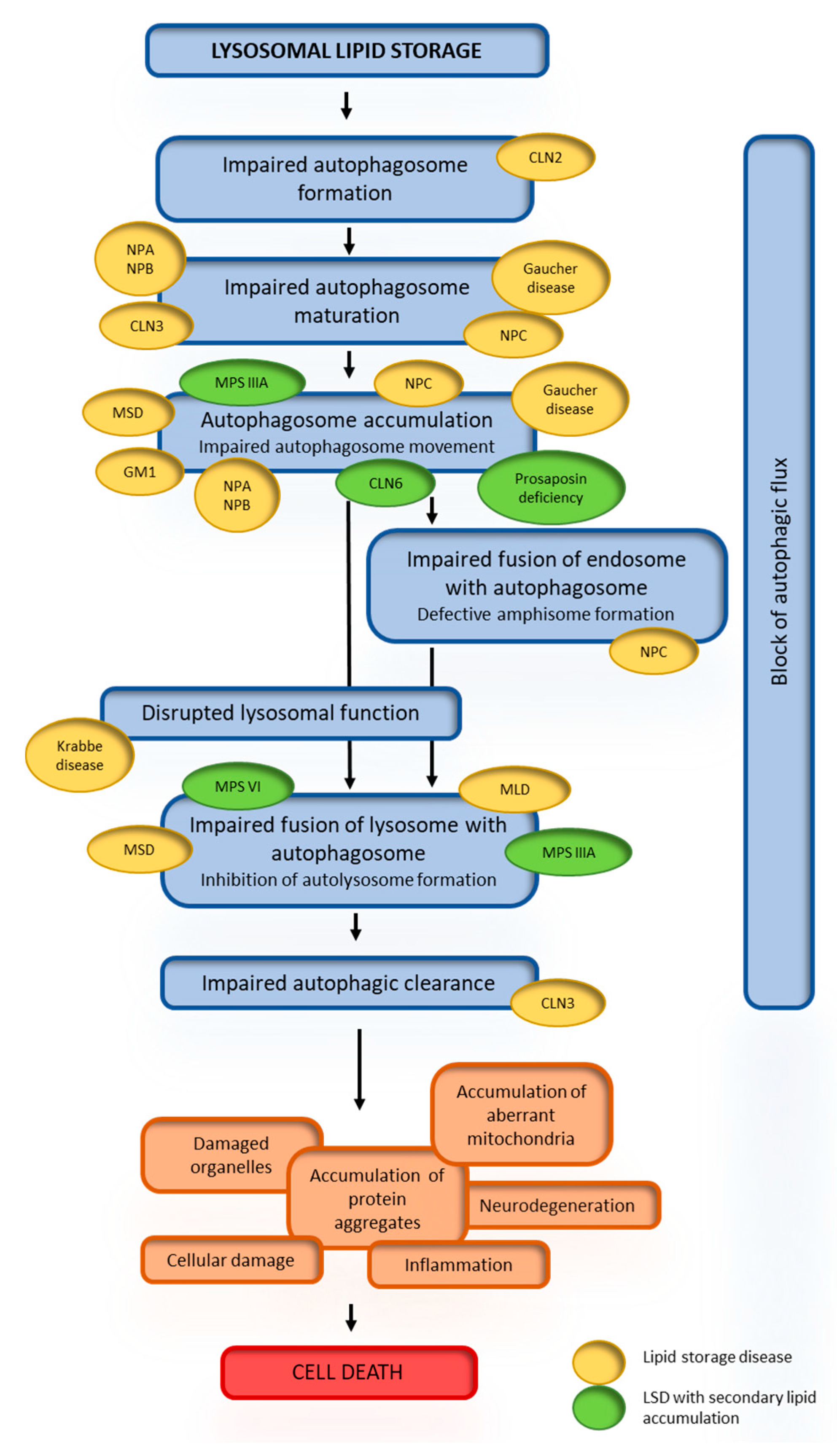
4.3. Secondary Lipid Accumulation in Lysosomal Storage Diseases
4.4. Consequences of Secondary Lipid Storage
4.5. mTOR–TFEB Signaling Pathway and Dysregulation of Autophagy in Lipid Storage Diseases
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADRP | adipophilin |
| AFSM | autofluorescent storage material |
| AMPK | AMP-activated protein kinase |
| ARF | ADP-ribosylation factor |
| ATGL | adipose triglyceride lipase |
| BMP | bis(monoacylglycero)phosphate |
| CaMKKβ | calcium/calmodulin-dependent protein kinase β |
| CAV | caveolin |
| CGI-58 | comparative gene identification-58 |
| CLEAR | coordinated lysosomal expression and regulation |
| CLN | ceroid lipofuscinosis, neuronal |
| CMA | chaperon-mediated autophagy |
| CoA | coenzyme A |
| COP | coat protein |
| CREB | cAMP response element-binding |
| CTX | cerebrotendinous xanthomatosis |
| DAG | diacylglycerols |
| ER | endoplasmic reticulum |
| ERK2 | extracellular signal-regulated kinase 2 |
| ES | sterol ester |
| FA | fatty acid |
| FAD | flavin-adenine dinucleotide |
| FGE | formylglycine-generating enzyme |
| FOXO | forkhead homeobox type O |
| FXR | farnesoid X receptor |
| Hsc70 | heat shock cognate 70 |
| HSL | hormone-sensitive lipase |
| LAL | lysosomal acid lipase |
| LAMP2A | lysosome-associated membrane protein 2A |
| LC3 | light chain 3 |
| LD | lipid droplet |
| LDL | low-density lipoprotein |
| LINCL | late infantile neuronal ceroid lipofuscinosis |
| LIR | LC3 interaction region |
| LSD | lysosomal storage disease |
| MAG | monoacylglycerol |
| MAPK | mitogen-activated protein kinase |
| MGL | monoglyceride lipase |
| MiT | microphthalmia |
| MLD | metachromatic leukodystrophy |
| MPS | mucopolysaccharidosis |
| MSD | multiple sulfatase deficiency |
| mTOR | mechanistic target of rapamycin |
| mTORC1 | mechanistic target of rapamycin complex 1 |
| NCoR1 | nuclear receptor co-repressor 1 |
| OSBP | oxysterol binding protein |
| PAT | PLIN/ADRP/TIP47 |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PGC1α | peroxisome proliferator-activated receptor gamma coactivator 1α |
| PI | phosphatidylinositol |
| PI3K | phosphatidylinositol 3-kinase |
| PI3P | phosphatidylinositol 3-phosphate |
| PKA | protein kinase A |
| PLIN | perilipin |
| PPAR1α | peroxisome proliferator activated receptor 1α |
| PPARGC1α | peroxisome proliferator-activated receptor gamma coactivator 1α |
| PS | phosphatidylserine |
| RBC | red blood cell |
| SM | sphingomyelin |
| SNARE | soluble N-ethylmaleimide-sensitive factor attachment receptor |
| TAG | triacylglycerol |
| TFE3 | transcription factor E3 |
| TFEB | transcription factor EB |
| TIP47 | tail-interacting protein of 47 kDa |
| TRPML1 | mucolipin transient receptor potential 1 |
| VLDL | very low-density lipoprotein |
References
- De Weer, P. A Century of Thinking about Cell Membranes. Annu. Rev. Physiol. 2000, 62, 919–926. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta BBA Biomembr. 2004, 1666, 62–87. [Google Scholar] [CrossRef]
- Holowka, D.; Gosse, J.A.; Hammond, A.T.; Han, X.; Sengupta, P.; Smith, N.L.; Wagenknecht-Wiesner, A.; Wu, M.; Young, R.M.; Baird, B. Lipid segregation and IgE receptor signaling: A decade of progress. Biochim. Biophys. Acta BBA Bioenerg. 2005, 1746, 252–259. [Google Scholar] [CrossRef][Green Version]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar] [CrossRef]
- Gibbons, G.F.; Islam, K.; Pease, R.J. Mobilisation of triacylglycerol stores. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2000, 1483, 37–57. [Google Scholar] [CrossRef]
- Fahy, E.; Cotter, D.; Sud, M.; Subramaniam, S. Lipid classification, structures and tools. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2011, 1811, 637–647. [Google Scholar] [CrossRef]
- Watkins, P.A. Fatty acids: Metabolism. In Encyclopedia of Human Nutrition; Caballero, B., Third, E., Eds.; Elsevier: Waltham, MA, USA, 2013; pp. 220–230. ISBN 978-0-12-384885-7. [Google Scholar]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Cortés, V.A. Physiological and pathological implications of cholesterol. Front. Biosci. 2014, 19, 416. [Google Scholar] [CrossRef]
- Schmid, K.E.; Woollett, L.A. Differential effects of polyunsaturated fatty acids on sterol synthesis rates in adult and fetal tissues of the hamster: Consequence of altered sterol balance. Am. J. Physiol. Liver Physiol. 2003, 285, G796–G803. [Google Scholar] [CrossRef]
- Yao, L.; Jenkins, K.; Horn, P.S.; Lichtenberg, M.H.; Woollett, L.A. Inability to fully suppress sterol synthesis rates with exogenous sterol in embryonic and extraembyronic fetal tissues. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2007, 1771, 1372–1379. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shen, W.-J.; Azhar, S.; Kraemer, F.B. Lipid droplets and steroidogenic cells. Exp. Cell Res. 2016, 340, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Saxena, R.; Srinivas, G.; Pande, G.; Chattopadhyay, A. Cholesterol Biosynthesis and Homeostasis in Regulation of the Cell Cycle. PLoS ONE 2013, 8, e58833. [Google Scholar] [CrossRef]
- Lasunción, M.A.; Martín-Sánchez, C.; Canfrán-Duque, A.; Busto, R. Post-lanosterol biosynthesis of cholesterol and cancer. Curr. Opin. Pharmacol. 2012, 12, 717–723. [Google Scholar] [CrossRef]
- Fernandez, C.; Lobo, M.D.V.T.; Gómez-Coronado, D.; Lasunción, M.A. Cholesterol is essential for mitosis progression and its deficiency induces polyploid cell formation. Exp. Cell Res. 2004, 300, 109–120. [Google Scholar] [CrossRef]
- Jackson, C.L. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 2019, 59, 88–96. [Google Scholar] [CrossRef]
- Welte, M.A. Expanding roles for lipid droplets. Curr. Biol. 2015, 25, R470–R481. [Google Scholar] [CrossRef]
- Zehmer, J.K.; Huang, Y.; Peng, G.; Pu, J.; Anderson, R.G.W.; Liu, P. A role for lipid droplets in inter-membrane lipid traffic. Proteomics 2009, 9, 914–921. [Google Scholar] [CrossRef]
- Penno, A.; Hackenbroich, G.; Thiele, C. Phospholipids and lipid droplets. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2013, 1831, 589–594. [Google Scholar] [CrossRef]
- Bickel, P.E.; Tansey, J.T.; Welte, M.A. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2009, 1791, 419–440. [Google Scholar] [CrossRef]
- Tansey, J.; Sztalryd, C.; Hlavin, E.; Kimmel, A.; Londos, C. The Central Role of Perilipin A in Lipid Metabolism and Adipocyte Lipolysis. IUBMB Life Int. Union Biochem. Mol. Biol. Life 2004, 56, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Proteins under new management: Lipid droplets deliver. Trends Cell Biol. 2007, 17, 363–369. [Google Scholar] [CrossRef]
- Martin, S. Caveolae, lipid droplets, and adipose tissue biology: Pathophysiological aspects. Horm. Mol. Biol. Clin. Investig. 2013, 15, 11–18. [Google Scholar] [CrossRef]
- Yang, H.; Galea, A.; Sytnyk, V.; Crossley, M. Controlling the size of lipid droplets: Lipid and protein factors. Curr. Opin. Cell Biol. 2012, 24, 509–516. [Google Scholar] [CrossRef]
- Ducharme, N.A.; Bickel, P.E. Minireview: Lipid Droplets in Lipogenesis and Lipolysis. Endocrinology 2008, 149, 942–949. [Google Scholar] [CrossRef]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS—Lipases and Lipolysis in Lipid Metabolism and Signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef]
- Osuga, J.-I.; Ishibashi, S.; Oka, T.; Yagyu, H.; Tozawa, R.; Fujimoto, A.; Shionoiri, F.; Yahagi, N.; Kraemer, F.B.; Tsutsumi, O.; et al. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 787–792. [Google Scholar] [CrossRef]
- Casado, M.E.; Pastor, Ó.; García-Seisdedos, D.; Huerta, L.; Kraemer, F.B.; Lasunción, M.A.; Martin, A.; Busto, R. Hormone-sensitive lipase deficiency disturbs lipid composition of plasma membrane microdomains from mouse testis. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2016, 1861, 1142–1150. [Google Scholar] [CrossRef]
- Haemmerle, G.; Zimmermann, R.; Hayn, M.; Theussl, C.; Waeg, G.; Wagner, E.; Sattler, W.; Magin, T.M.; Wagner, E.F.; Zechner, R. Hormone-sensitive Lipase Deficiency in Mice Causes Diglyceride Accumulation in Adipose Tissue, Muscle, and Testis. J. Biol. Chem. 2001, 277, 4806–4815. [Google Scholar] [CrossRef]
- Taschler, U.; Radner, F.P.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Köfeler, H.; et al. Monoglyceride Lipase Deficiency in Mice Impairs Lipolysis and Attenuates Diet-induced Insulin Resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef]
- D’Andrea, S. Lipid droplet mobilization: The different ways to loosen the purse strings. Biochimie 2016, 120, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Oral, O.; Akkoc, Y.; Bayraktar, O.; Gozuacik, D. Physiological and pathological significance of the molecular cross-talk between autophagy and apoptosis. Histol. Histopathol. 2015, 31, 479–498. [Google Scholar] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, N.; García-Macia, M.; Sahu, S.; Athonvarangkul, D.; Liebling, E.; Merlo, P.; Cecconi, F.; Schwartz, G.J.; Singh, R. Autophagy in the CNS and Periphery Coordinate Lipophagy and Lipolysis in the Brown Adipose Tissue and Liver. Cell Metab. 2016, 23, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.S.; Nilsson, T. Rab proteins implicated in lipid storage and mobilization. J. Biomed. Res. 2014, 28, 169–177. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Zimmermann, A.; Madeo, F. A molecular mechanism for lipophagy regulation in the liver. Hepatology 2015, 61, 1781–1783. [Google Scholar] [CrossRef]
- Li, Z.; Schulze, R.J.; Weller, S.G.; Krueger, E.W.; Schott, M.B.; Zhang, X.; Casey, C.A.; Liu, J.; Stöckli, J.; James, D.E.; et al. A novel Rab10-EHBP1-EHD2 complex essential for the autophagic engulfment of lipid droplets. Sci. Adv. 2016, 2, e1601470. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Triacylglycerols Are Highly Concentrated Energy Stores. In Biochemistry; W.H. Freeman: New York, NY, USA, 2002; ISBN 0-7167-3051-0. [Google Scholar]
- Adeva, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty Acid Oxidation Disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef] [PubMed]
- Janssen, U.; Stoffel, W. Disruption of Mitochondrial β-Oxidation of Unsaturated Fatty Acids in the 3,2- trans -Enoyl-CoA Isomerase-deficient Mouse. J. Biol. Chem. 2002, 277, 19579–19584. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Certain Fatty Acids Require Additional Steps for Degradation. In Biochemistry; W.H. Freeman: New York, NY, USA, 2002; ISBN 0-7167-3051-0. [Google Scholar]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Pelz, C.; Kang, H.; Di Ronza, A.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, N.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef]
- O’Rourke, E.J.; Ruvkun, G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat. Cell Biol. 2013, 15, 668–676. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.; Fu, T.; Choi, S.-E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional regulation of autophagy by an FXR–CREB axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Lettieri-Barbato, D.; Tatulli, G.; Aquilano, K.; Ciriolo, M.R. FoxO1 controls lysosomal acid lipase in adipocytes: Implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis. 2013, 4, e861. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, R.; Sergin, I.; Bhattacharya, S.; Turner, J.N.; Epelman, S.; Settembre, C.; Diwan, A.; Ballabio, A.; Razani, B. Induction of Lysosomal Biogenesis in Atherosclerotic Macrophages Can Rescue Lipid-Induced Lysosomal Dysfunction and Downstream Sequelae. Arter. Thromb. Vasc. Biol. 2014, 34, 1942–1952. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Wang, K.; He, J.; Zhang, G.; Zhang, D.; Chen, F. TFE3 Alleviates Hepatic Steatosis through Autophagy-Induced Lipophagy and PGC1α-Mediated Fatty Acid β-Oxidation. Int. J. Mol. Sci. 2016, 17, 387. [Google Scholar] [CrossRef]
- Pastore, N.; Vainshtein, A.; Klisch, T.J.; Armani, A.; Huynh, T.; Herz, N.J.; Polishchuk, E.V.; Sandri, M.; Ballabio, A. TFE 3 regulates whole-body energy metabolism in cooperation with TFEB. EMBO Mol. Med. 2017, 9, 605–621. [Google Scholar] [CrossRef]
- Saito, T.; Kuma, A.; Sugiura, Y.; Ichimura, Y.; Obata, M.; Kitamura, H.; Okuda, S.; Lee, H.-C.; Ikeda, K.; Kanegae, Y.; et al. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019, 10, 1567. [Google Scholar] [CrossRef]
- Gross, D.A.; Silver, D.L. Cytosolic lipid droplets: From mechanisms of fat storage to disease. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 304–326. [Google Scholar] [CrossRef]
- Tang, Q.-Q. Lipid metabolism and diseases. Sci. Bull. 2016, 61, 1471–1472. [Google Scholar] [CrossRef]
- Smith, C.L.; Eppig, J.T. The mammalian phenotype ontology: Enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 390–399. [Google Scholar] [CrossRef]
- Samie, M.A.; Xu, H. Lysosomal exocytosis and lipid storage disorders. J. Lipid Res. 2014, 55, 995–1009. [Google Scholar] [CrossRef]
- Schulze, H.; Sandhoff, K. Lysosomal Lipid Storage Diseases. Cold Spring Harb. Perspect. Biol. 2011, 3, a004804. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010, 584, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ren, D. Lysosomal Physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Rieger, D.; Auerbach, S.; Robinson, P.; Gropman, A.L. Neuroimaging of lipid storage disorders. Dev. Disabil. Res. Rev. 2013, 17, 269–282. [Google Scholar] [CrossRef]
- Lipid Storage Diseases Information Page. National Institute of Neurological Disorders and Stroke. Available online: https://www.ninds.nih.gov/Disorders/All-Disorders/Lipid-Storage-Diseases-Information-Page (accessed on 22 June 2020).
- Semenkovich, C.F.; Goldberg, A.C.; Goldberg, I.J. Disorders of Lipid Metabolism. In Williams Textbook of Endocrinology; Melmed, S., Polonsky, K.S., Larsen, P.R., Kronenberg, H.M., Eds.; Elsevier: Philadelphia, PA, USA, 2016; pp. 1660–1700. [Google Scholar]
- Sullivan, D.R.; Lewis, B. A classification of lipoprotein disorders: Implications for clinical management. Clin. Lipidol. 2011, 6, 327–338. [Google Scholar] [CrossRef]
- Patni, N.; Ahmad, Z.; Wilson, D.P. Genetics and Dyslipidemia, 2000th ed.; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Gabandé-Rodríguez, E.; Boya, P.; Labrador, V.; Dotti, C.G.; Ledesma, M.D. High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A. Cell Death Differ. 2014, 21, 864–875. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef]
- Pipalia, N.; Hao, M.; Mukherjee, S.; Maxfield, F.R. Sterol, Protein and Lipid Trafficking in Chinese Hamster Ovary Cells with Niemann-Pick Type C1 Defect. Traffic 2007, 8, 130–141. [Google Scholar] [CrossRef]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Milstien, S.; Spiegel, S. Niemann-Pick type C disease: The atypical sphingolipidosis. Adv. Biol. Regul. 2018, 70, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Hammond, N.; Munkacsi, A.B.; Sturley, S.L. The complexity of a monogenic neurodegenerative disease: More than two decades of therapeutic driven research into Niemann-Pick type C disease. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Bräuer, A.U.; Kuhla, A.; Holzmann, C.; Wree, A.; Witt, M. Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann-Pick Disease Type C1. Int. J. Mol. Sci. 2019, 20, 4392. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, R.; Ng, A.H.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Elrick, M.J.; Yu, T.; Chung, C.; Lieberman, A.P. Impaired proteolysis underlies autophagic dysfunction in Niemann–Pick type C disease. Hum. Mol. Genet. 2012, 21, 4876–4887. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.; Adam, D.N. A Review of Fabry Disease. Ski. Ther. Lett. 2018, 23, 4–6. [Google Scholar]
- Juchniewicz, P.; Kloska, A.; Tylki-Szymańska, A.; Jakóbkiewicz-Banecka, J.; Węgrzyn, G.; Moskot, M.; Gabig-Cimińska, M.; Piotrowska, E. Female Fabry disease patients and X-chromosome inactivation. Gene 2018, 641, 259–264. [Google Scholar] [CrossRef]
- Cairns, T.; Müntze, J.; Gernert, J.; Spingler, L.; Nordbeck, P.; Wanner, C. Hot topics in Fabry disease. Postgrad. Med. J. 2018, 94, 709–713. [Google Scholar] [CrossRef]
- Chévrier, M.; Brakch, N.; Lesueur, C.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Barbey, F.; Bekri, S.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef]
- Spassieva, S.; Bieberich, E. Lysosphingolipids and sphingolipidoses: Psychosine in Krabbe’s disease. J. Neurosci. Res. 2016, 94, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Won, J.-S.; Singh, A.K.; Singh, I. Biochemical, cell biological, pathological, and therapeutic aspects of Krabbe’s disease. J. Neurosci. Res. 2016, 94, 990–1006. [Google Scholar] [CrossRef]
- Lin, D.-S.; Ho, C.-S.; Huang, Y.-W.; Wu, T.-Y.; Lee, T.-H.; Huang, Z.-D.; Wang, T.-J.; Yang, S.-J.; Chiang, M.-F. Impairment of Proteasome and Autophagy Underlying the Pathogenesis of Leukodystrophy. Cells 2020, 9, 1124. [Google Scholar] [CrossRef] [PubMed]
- Del Grosso, A.; Angella, L.; Tonazzini, I.; Moscardini, A.; Giordano, N.; Caleo, M.; Rocchiccioli, S.; Cecchini, M. Dysregulated autophagy as a new aspect of the molecular pathogenesis of Krabbe disease. Neurobiol. Dis. 2019, 129, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Nguyen, Y.; Stirnemann, J.; Belmatoug, N. Gaucher disease: A review. Rev. Med. Interne 2019, 40, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Sgambato, J.A.; Lipinski, M.M.; Miller, D.; Zeng, X.; Feldman, R.A. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 2015, 24, 5775–5788. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, D. Molecular Pathogenesis and Therapeutic Approach of GM2 Gangliosidosis. Yakugaku Zasshi J. Pharm. Soc. Jpn. 2013, 133, 269–274. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cachón-González, M.B.; Zaccariotto, E.; Cox, T.M. Genetics and Therapies for GM2 Gangliosidosis. Curr. Gene Ther. 2018, 18, 68–89. [Google Scholar] [CrossRef]
- Vitner, E.B.; Platt, F.M.; Futerman, A.H. Common and Uncommon Pathogenic Cascades in Lysosomal Storage Diseases. J. Biol. Chem. 2010, 285, 20423–20427. [Google Scholar] [CrossRef]
- Xu, Y.-H.; Barnes, S.; Sun, Y.; Grabowski, G. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J. Lipid Res. 2010, 51, 1643–1675. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-C.; Hama, Y.; Li, Y.-T. Ineraction of GM2 Activator Protein with Glycosphingolipids. Adv. Exp. Med. Biol. 2001, 491, 351–367. [Google Scholar]
- Sandhoff, K. Neuronal sphingolipidoses: Membrane lipids and sphingolipid activator proteins regulate lysosomal sphingolipid catabolism. Biochimie 2016, 130, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Gieselmann, V.; Krägeloh-Mann, I. Metachromatic Leukodystrophy—An Update. Neuropediatrics 2010, 41, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Breiden, B.; Sandhoff, K. Lysosomal Glycosphingolipid Storage Diseases. Annu. Rev. Biochem. 2019, 88, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Berry, J.; Wenger, D.A.; Escolar, M.; Sobeih, M.; Raymond, G.; Eichler, F. Metachromatic Leukodystrophy: A Case of Triplets with the Late Infantile Variant and a Systematic Review of the Literature. J. Child Neurol. 2010, 25, 572–580. [Google Scholar] [CrossRef]
- Maegawa, G. Patil Developing therapeutic approaches for metachromatic leukodystrophy. Drug Des. Devel. Ther. 2013, 7, 729. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Corry, P.C.; Wraith, J.E.; Besley, G.T.N.; Cooper, A.; Ferrie, C.D. Juvenile Sandhoff disease—Nine new cases and a review of the literature. J. Inherit. Metab. Dis. 2004, 27, 241–249. [Google Scholar] [CrossRef]
- Kolodny, E.H. Tay–Sachs Disease. Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Cambridge, MA, USA, 2009; pp. 895–902. ISBN 9780080450469. [Google Scholar]
- Tamboli, I.Y.; Hampel, H.; Tien, N.T.; Tolksdorf, K.; Breiden, B.; Mathews, P.M.; Saftig, P.; Sandhoff, K.; Walter, J. Sphingolipid Storage Affects Autophagic Metabolism of the Amyloid Precursor Protein and Promotes Aβ Generation. J. Neurosci. 2011, 31, 1837–1849. [Google Scholar] [CrossRef]
- Keilani, S.; Lun, Y.; Stevens, A.C.; Williams, H.N.; Sjoberg, E.R.; Khanna, R.; Valenzano, K.J.; Checler, F.; Buxbaum, J.D.; Yanagisawa, K.; et al. Lysosomal Dysfunction in a Mouse Model of Sandhoff Disease Leads to Accumulation of Ganglioside-Bound Amyloid-Peptide. J. Neurosci. 2012, 32, 5223–5236. [Google Scholar] [CrossRef]
- Annunziata, I.; Bouché, V.; Lombardi, A.; Settembre, C.; Ballabio, A. Multiple sulfatase deficiency is due to hypomorphic mutations of theSUMF1 gene. Hum. Mutat. 2007, 28, 928. [Google Scholar] [CrossRef] [PubMed]
- Schlotawa, L.; Adang, L.; Radhakrishnan, K.; Ahrens-Nicklas, R.C. Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification. Int. J. Mol. Sci. 2020, 21, 3448. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.L.; De Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2007, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Scaglia, F. GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 2008, 94, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Smith, D.A.; Mooney, D.; Jung, S.S.; Walsh, D.M.; Platt, F.M. Macroautophagy Is Not Directly Involved in the Metabolism of Amyloid Precursor Protein. J. Biol. Chem. 2010, 285, 37415–37426. [Google Scholar] [CrossRef]
- Schindler, D.; Desnick, R.J. Schindler Disease: Deficient α-N-acetylgalactosaminidase Activity. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease; Rosenberg, R.N., Pascual, J.M., Eds.; Elsevier: Boston, MA, USA, 2015; pp. 431–439. ISBN 9780124105294. [Google Scholar]
- Wu, T.T.; Hoff, D.S. Fish Oil Lipid Emulsion-Associated Sea-Blue Histiocyte Syndrome in a Pediatric Patient. J. Pediatr. Pharmacol. Ther. 2015, 20, 217–221. [Google Scholar]
- Mirza, M.; Vainshtein, A.; DiRonza, A.; Chandrachud, U.; Haslett, L.J.; Palmieri, M.; Storch, S.; Groh, J.; Dobzinski, N.; Napolitano, G.; et al. The CLN3 gene and protein: What we know. Mol. Genet. Genom. Med. 2019, 7, e859. [Google Scholar] [CrossRef]
- Mukherjee, A.B.; Appu, A.P.; Sadhukhan, T.; Casey, S.; Mondal, A.; Zhang, Z.; Bagh, M.B. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 2019, 14, 4. [Google Scholar] [CrossRef]
- Cao, Y.; Espinola, J.A.; Fossale, E.; Massey, A.C.; Cuervo, A.M.; Macdonald, M.E.; Cotman, S.L. Autophagy Is Disrupted in a Knock-in Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis. J. Biol. Chem. 2006, 281, 20483–20493. [Google Scholar] [CrossRef]
- Lojewski, X.; Staropoli, J.F.; Biswas-Legrand, S.; Simas, A.M.; Haliw, L.; Selig, M.K.; Coppel, S.H.; Goss, K.A.; Petcherski, A.; Chandrachud, U.; et al. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum. Mol. Genet. 2013, 23, 2005–2022. [Google Scholar] [CrossRef]
- Nita, A.A.; Mole, S.E.; Minassian, B.A. Neuronal ceroid lipofuscinoses. Epileptic Disord 2016, 18, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Donet, J.M.; Carcel-Trullols, J.; Casanova, B.; Aguado, C.; Knecht, E. Alterations in ROS Activity and Lysosomal pH Account for Distinct Patterns of Macroautophagy in LINCL and JNCL Fibroblasts. PLoS ONE 2013, 8, e55526. [Google Scholar] [CrossRef] [PubMed]
- Aguisanda, F.; Thorne, N.; Zheng, W. Targeting Wolman Disease and Cholesteryl Ester Storage Disease: Disease Pathogenesis and Therapeutic Development. Curr. Chem. Genom. Transl. Med. 2017, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pericleous, M.; Kelly, C.; Wang, T.; Livingstone, C.; Ala, A. Wolman’s disease and cholesteryl ester storage disorder: The phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol. Hepatol. 2017, 2, 670–679. [Google Scholar] [CrossRef]
- Settembre, C.; Ballabio, A. Lysosome: Regulator of lipid degradation pathways. Trends Cell Biol. 2014, 24, 743–750. [Google Scholar] [CrossRef]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 1178–1187. [Google Scholar] [CrossRef]
- Ruivo, R.; Anne, C.; Sagné, C.; Gasnier, B. Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1793, 636–649. [Google Scholar] [CrossRef]
- Jezela-Stanek, A.; Ciara, E.; Stepien, K.M. Neuropathophysiology, Genetic Profile, and Clinical Manifestation of Mucolipidosis IV—A Review and Case Series. Int. J. Mol. Sci. 2020, 21, 4564. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Long, A.A.; Elsaesser, R.; Nikolaeva, D.; Broadie, K.; Montell, C. Motor Deficit in a Drosophila Model of Mucolipidosis Type IV due to Defective Clearance of Apoptotic Cells. Cell 2008, 135, 838–851. [Google Scholar] [CrossRef]
- Venugopal, B.; Mesires, N.T.; Kennedy, J.C.; Laplante, J.M.; Dice, J.F.; Slaugenhaupt, S.A.; Curcio-Morelli, C. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J. Cell. Physiol. 2009, 219, 344–353. [Google Scholar] [CrossRef]
- Khan, A.; Sergi, C. Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder. Diagnostics 2018, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Natori, Y.; Nasui, M.; Kihara-Negishi, F. Neu1 sialidase interacts with perilipin 1 on lipid droplets and inhibits lipolysis in 3T3-L1 adipocytes. Genes Cells 2017, 22, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Davaadorj, O.; Akatsuka, H.; Yamaguchi, Y.; Okada, C.; Ito, M.; Fukunishi, N.; Sekijima, Y.; Ohnota, H.; Kawai, K.; Suzuki, T.; et al. Impaired Autophagy in Retinal Pigment Epithelial Cells Induced from iPS Cells obtained from a Patient with Sialidosis. Cell Dev. Biol. 2017, 6, 1–7. [Google Scholar] [CrossRef]
- Missaglia, S.; Coleman, R.A.; Mordente, A.; Tavian, D. Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function. Cells 2019, 8, 187. [Google Scholar] [CrossRef]
- Massa, R.; Pozzessere, S.; Rastelli, E.; Serra, L.; Terracciano, C.; Gibellini, M.; Bozzali, M.; Arca, M. Neutral lipid-storage disease with myopathy and extended phenotype with novelPNPLA2mutation. Muscle Nerve 2016, 53, 644–648. [Google Scholar] [CrossRef]
- Angelini, C.; Nascimbeni, A.C.; Cenacchi, G.; Tasca, E. Lipolysis and lipophagy in lipid storage myopathies. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1862, 1367–1373. [Google Scholar] [CrossRef]
- Yoneda, K. Inherited ichthyosis: Syndromic forms. J. Dermatol. 2016, 43, 252–263. [Google Scholar] [CrossRef]
- Mogahed, E.A.; El-Hennawy, A.; El-Sayed, R.; El-Karaksy, H. Chanarin–Dorfman syndrome: A case report and review of the literature. Arab. J. Gastroenterol. 2015, 16, 142–144. [Google Scholar] [CrossRef]
- Peng, Y.; Miao, H.; Wu, S.; Yang, W.; Zhang, Y.; Xie, G.; Xie, X.; Li, J.; Shi, C.; Ye, L.; et al. ABHD5 interacts with BECN1 to regulate autophagy and tumorigenesis of colon cancer independent of PNPLA2. Autophagy 2016, 12, 2167–2182. [Google Scholar] [CrossRef]
- Nie, S.; Chen, G.; Cao, X.; Zhang, Y. Cerebrotendinous xanthomatosis: A comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 2014, 9, 179. [Google Scholar] [CrossRef]
- Salen, G.; Steiner, R. Epidemiology, diagnosis, and treatment of cerebrotendinous xanthomatosis (CTX). J. Inherit. Metab. Dis. 2017, 40, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, E.; Mao, W.; Qiao, H.; Zhou, Y.; Yang, Q.; Liu, S.; Chan, P. Parkinsonism with Normal Dopaminergic Presynaptic Terminals in Cerebrotendinous Xanthomatosis. Mov. Disord. Clin. Pr. 2019, 7, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Liebeskind, A.; Wilson, D.P. Sitosterolemia in the Pediatric Population. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Bao, L.; Li, Y.; Deng, S.-X.; Landry, D.; Tabas, I. Sitosterol-containing Lipoproteins Trigger Free Sterol-induced Caspase-independent Death in ACAT-competent Macrophages. J. Biol. Chem. 2006, 281, 33635–33649. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.Q.; Le Trionnaire, E.; Ribeiro, I.; Carpentier, S.; Harzer, K.; Levade, T.; Ribeiro, M.G. Molecular basis of acid ceramidase deficiency in a neonatal form of Farber disease: Identification of the first large deletion in ASAH1 gene. Mol. Genet. Metab. 2013, 109, 276–281. [Google Scholar] [CrossRef]
- Wali, G.; Wali, G.M.; Sue, C.M.; Kumar, K. A Novel Homozygous Mutation in the FUCA1 Gene Highlighting Fucosidosis as a Cause of Dystonia: Case Report and Literature Review. Neuropediatrics 2019, 50, 248–252. [Google Scholar] [CrossRef]
- Koopal, C.; Marais, A.D.; Westerink, J.; Visseren, F.L. Autosomal dominant familial dysbetalipoproteinemia: A pathophysiological framework and practical approach to diagnosis and therapy. J. Clin. Lipidol. 2017, 11, 12–23. [Google Scholar] [CrossRef]
- Koopal, C.; Marais, A.D.; Visseren, F.L.J. Familial dysbetalipoproteinemia. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 133–139. [Google Scholar] [CrossRef]
- Evans, D.; Beil, F.U. Genetic factors that modify the expression of type III hyperlipidemia in probands with apolipoprotein E ε2/2 genotype. Future Lipidol. 2009, 4, 137–140. [Google Scholar] [CrossRef]
- Henneman, P.; Beer, F.V.D.S.-D.; Moghaddam, P.H.; Huijts, P.; Stalenhoef, A.F.; Kastelein, J.J.; Van Duijn, C.M.; Havekes, L.M.; Frants, R.R.; Van Dijk, K.W.; et al. The expression of type III hyperlipoproteinemia: Involvement of lipolysis genes. Eur. J. Hum. Genet. 2009, 17, 620–628. [Google Scholar] [CrossRef]
- Hendricks-Sturrup, R.; Clark-LoCascio, J.; Lu, C.Y. A Global Review on the Utility of Genetic Testing for Familial Hypercholesterolemia. J. Pers. Med. 2020, 10, 23. [Google Scholar] [CrossRef]
- Pang, J.; Sullivan, D.R.; Brett, T.; Kostner, K.M.; Hare, D.L.; Watts, G.F. Familial Hypercholesterolaemia in 2020: A Leading Tier 1 Genomic Application. Hear. Lung Circ. 2020, 29, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.M.; De La Mata, M.; Pavón, A.D.; Villanueva-Paz, M.; Povea-Cabello, S.; Cotán, D.; Álvarez-Córdoba, M.; Villalón-García, I.; Ybot-Gonzalez, P.; Salas, J.J.; et al. Intracellular cholesterol accumulation and coenzyme Q10 deficiency in Familial Hypercholesterolemia. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 3697–3713. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.H.; Miserez, A.R.; Ahmad, Z.; Andersen, R.L. Familial defective apolipoprotein B-100: A review. J. Clin. Lipidol. 2016, 10, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Forte, T.M.; Ryan, R.O. Influence of apolipoprotein A-V on the metabolic fate of triacylglycerol. Curr. Opin. Lipidol. 2013, 24, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Kwiterovich, P.O. Diagnosis and Management of Familial Dyslipoproteinemias. Curr. Cardiol. Rep. 2013, 15, 371. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; Eckel, R.H. The Chylomicronemia Syndrome Is Most Often Multifactorial: A Narrative Review of Causes and Treatment. Ann. Intern. Med. 2019, 170, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Davies, B.S.J.; Voss, C.V.; Gin, P.; Weinstein, M.M.; Tontonoz, P.; Reue, K.; Bensadoun, A.; Fong, L.G.; Beigneux, A.P. GPIHBP1, an endothelial cell transporter for lipoprotein lipase. J. Lipid Res. 2011, 52, 1869–1884. [Google Scholar] [CrossRef]
- Feingold, K.R. Triglyceride Lowering Drugs. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Burnett, J.R.; Hooper, A.J.; Hegele, R.A. Familial Lipoprotein Lipase Deficiency; Adam, M., Ardinger, H., Pagon, R., Wallace, S., Bean, L., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993; ISBN 0444810781. [Google Scholar]
- Wolska, A.; Dunbar, R.L.; Freeman, L.A.; Ueda, M.; Amar, M.J.; Sviridov, D.O.; Remaley, A.T. Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis 2017, 267, 49–60. [Google Scholar] [CrossRef]
- Wilson, C.; Oliva, C.P.; Maggi, F.; Catapano, A.L.; Calandra, S. Apolipoprotein C-II deficiency presenting as a lipid encephalopathy in infancy. Ann. Neurol. 2003, 53, 807–810. [Google Scholar] [CrossRef]
- Desnick, R.J.; Guntinas-Lichius, O.; Padberg, G.W.; Schonfeld, G.; Lin, X.; Averna, M.; Yue, P.; Schnog, J.-J.B.; Gerdes, V.E.A.; Cutillas, P.R.; et al. Familial Lipoprotein Lipase Deficiency. In Encyclopedia of Molecular Mechanisms of Disease; Springer: Berlin/Heidelberg, Germany, 2009; p. 635. ISBN 0444810781. [Google Scholar]
- Kobayashi, J.; Miyashita, K.; Nakajima, K.; Mabuchi, H. Hepatic Lipase: A Comprehensive View of its Role on Plasma Lipid and Lipoprotein Metabolism. J. Atheroscler. Thromb. 2015, 22, 1001–1011. [Google Scholar] [CrossRef]
- Warden, B.A.; Fazio, S.; Shapiro, M.D. Familial Hypercholesterolemia: Genes and Beyond. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Fellin, R.; Arca, M.; Zuliani, G.; Calandra, S.; Bertolini, S. The history of Autosomal Recessive Hypercholesterolemia (ARH). From clinical observations to gene identification. Gene 2015, 555, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Foody, J.M.; Vishwanath, R. Familial hypercholesterolemia/autosomal dominant hypercholesterolemia: Molecular defects, the LDL-C continuum, and gradients of phenotypic severity. J. Clin. Lipidol. 2016, 10, 970–986. [Google Scholar] [CrossRef]
- Sun, H.; Krauss, R.M.; Chang, J.T.; Teng, B.-B. PCSK9 deficiency reduces atherosclerosis, apolipoprotein B secretion, and endothelial dysfunction. J. Lipid Res. 2018, 59, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1793, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, R.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2016, 1861, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef]
- Patterson, M.C.; Vanier, M.T.; Suzuki, K.K.K.; Morris, J.A.; Carstea, E.; Neufeld, E.B.; Blanchette-Mackie, E.J.; Pentchev, P.G.; Blanchette-Mackie, J.E.; Pentchev, P.G. Niemann-Pick Disease Type C: A Lipid Trafficking Disorder. In The Online Metabolic & Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill: New York, NY, USA, 2004; pp. 1–44. ISBN 9780071459969. [Google Scholar]
- Walkley, S.U.; Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1793, 726–736. [Google Scholar] [CrossRef]
- Akgoc, Z.; Sena-Esteves, M.; Martin, U.R.; Han, X.; D’Azzo, A.; Seyfried, T.N. Bis (monoacylglycero) phosphate: A secondary storage lipid in the gangliosidoses. J. Lipid Res. 2015, 56, 1006–1013. [Google Scholar] [CrossRef]
- Walkley, S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 2004, 15, 433–444. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Wasserstein, M.P. Types A and B Niemann-Pick disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 237–247. [Google Scholar] [CrossRef]
- Hulkova, H.; Cervenková, M.; Ledvinova, J.; Tochácková, M.; Hrebícek, M.; Poupětová, H.; Befekadu, A.; Berná, L.; Paton, B.; Harzer, K.; et al. A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum. Mol. Genet. 2001, 10, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Campos, D.; Monaga, M. Mucopolysaccharidosis type I: Current knowledge on its pathophysiological mechanisms. Metab. Brain Dis. 2012, 27, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of Mucopolysaccharidoses, an Update. Int. J. Mol. Sci. 2020, 21, 2515. [Google Scholar] [CrossRef] [PubMed]
- Tessitore, A.; Pirozzi, M.; Auricchio, A. Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics 2009, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, I.; D’Azzo, A. Galactosialidosis: Historic aspects and overview of investigated and emerging treatment options. Expert Opin. Orphan Drugs 2016, 5, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Seyrantepe, V.; Poupětová, H.; Froissart, R.; Pshezhetsky, A.V. Molecular pathology of NEU1 gene in sialidosis. Hum. Mutat. 2003, 22, 343–352. [Google Scholar] [CrossRef]
- Kyttälä, A.; Lahtinen, U.; Braulke, T.; Hofmann, S.L. Functional biology of the neuronal ceroid lipofuscinoses (NCL) proteins. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2006, 1762, 920–933. [Google Scholar] [CrossRef]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Price, S.M.; Verot, L.; Shen, M.M.; Tint, G.S.; Vanier, M.T.; Walkley, S.U.; Lobel, P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. USA 2004, 101, 5886–5891. [Google Scholar] [CrossRef]
- Käkelä, R.; Somerharju, P.; Tyynelä, J. Analysis of phospholipid molecular species in brains from patients with infantile and juvenile neuronal-ceroid lipofuscinosis using liquid chromatography-electrospray ionization mass spectrometry. J. Neurochem. 2003, 84, 1051–1065. [Google Scholar] [CrossRef]
- Jabs, S.; Quitsch, A.; Kkel, R.; Koch, B.; Tyynel, J.; Brade, H.; Glatzel, M.; Walkley, S.; Saftig, P.; Vanier, M.T.; et al. Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J. Neurochem. 2008, 106, 1415–1425. [Google Scholar] [CrossRef]
- Micsenyi, M.C.; Dobrenis, K.; Stephney, G.; Pickel, J.; Vanier, M.T.; Slaugenhaupt, S.A.; Walkley, S.U. Neuropathology of the Mcoln1−/− Knockout Mouse Model of Mucolipidosis Type IV. J. Neuropathol. Exp. Neurol. 2009, 68, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Lysosomal Storage Causes Cellular Dysfunction in Mucolipidosis II Skin Fibroblasts. J. Biol. Chem. 2011, 286, 35283–35290. [Google Scholar] [CrossRef] [PubMed]
- Sobo, K.; Le Blanc, I.; Luyet, P.-P.; Fivaz, M.; Ferguson, C.; Parton, R.G.; Gruenberg, J.; Van Der Goot, F.G. Late Endosomal Cholesterol Accumulation Leads to Impaired Intra-Endosomal Trafficking. PLoS ONE 2007, 2, e851. [Google Scholar] [CrossRef]
- Gondré-Lewis, M.C.; McGlynn, R.; Walkley, S.U. Cholesterol accumulation in NPC1-deficient neurons is ganglioside dependent. Curr. Biol. 2003, 13, 1324–1329. [Google Scholar] [CrossRef]
- Anheuser, S.; Breiden, B.; Sandhoff, K. Ganglioside GM2 catabolism is inhibited by storage compounds of mucopolysaccharidoses and by cationic amphiphilic drugs. Mol. Genet. Metab. 2019, 128, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Anheuser, S.; Breiden, B.; Sandhoff, K. Membrane lipids and their degradation compounds control GM2 catabolism at intralysosomal luminal vesicles. J. Lipid Res. 2019, 60, 1099–1111. [Google Scholar] [CrossRef]
- Oninla, V.O.; Breiden, B.; Babalola, J.O.; Sandhoff, K. Acid sphingomyelinase activity is regulated by membrane lipids and facilitates cholesterol transfer by NPC2. J. Lipid Res. 2014, 55, 2606–2619. [Google Scholar] [CrossRef]
- Brown, R.A.; Voit, A.; Srikanth, M.P.; Thayer, J.A.; Kingsbury, T.J.; Jacobson, M.A.; Lipinski, M.M.; Feldman, R.A.; Awad, O. mTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. Dis. Model. Mech. 2019, 12, dmm038596. [Google Scholar] [CrossRef]
- Ricoult, S.J.H.; Manning, B.D. The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 2013, 14, 242–251. [Google Scholar] [CrossRef]
- Liebau, M.C.; Braun, F.; Höpker, K.; Weitbrecht, C.; Bartels, V.; Müller, R.-U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated Autophagy Contributes to Podocyte Damage in Fabry’s Disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Hossain, M.A.; Miyajima, T.; Nagao, K.; Miyashita, T.; Eto, Y. Dysregulated DNA methylation of GLA gene was associated with dysfunction of autophagy. Mol. Genet. Metab. 2019, 126, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.P.; Tse, T.E.; O’Quinn, D.B.; Percival, S.M.; Jaimes, E.A.; Warnock, D.G.; Shacka, J.J. Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of alpha-galactosidase A-deficient mice. Acta Neuropathol. Commun. 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 2019, 14, e0210617. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and Chemical Correction of Cholesterol Accumulation and Impaired Autophagy in Hepatic and Neural Cells Derived from Niemann-Pick Type C Patient-Specific iPS Cells. Stem Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; Van Der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; Van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9–Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef]
- Lim, C.-Y.; Davis, O.B.; Shin, H.R.; Zhang, J.; Berdan, C.A.; Jiang, X.; Counihan, J.L.; Ory, D.S.; Nomura, D.K.; Zoncu, R. ER-lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann-Pick type C. Nat. Cell Biol. 2019, 21, 1206–1218. [Google Scholar] [CrossRef]
- Takamura, A.; Higaki, K.; Kajimaki, K.; Otsuka, S.; Ninomiya, H.; Matsuda, J.; Ohno, K.; Suzuki, Y.; Nanba, E. Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem. Biophys. Res. Commun. 2008, 367, 616–622. [Google Scholar] [CrossRef]
- Onyenwoke, R.U.; Sexton, J.Z.; Yan, F.; Díaz, M.C.H.; Forsberg, L.J.; Major, M.B.; Brenman, J.E. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem. J. 2015, 470, 331–342. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Rosato, A.S.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Rosato, A.S.; Montefusco, S.; Soldati, C.; Di Paola, S.; Capuozzo, A.; Monfregola, J.; Polishchuk, E.; Amabile, A.; Grimm, C.; Lombardo, A.L.; et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Leinonen, H.; Keksa-Goldsteine, V.; Ragauskas, S.; Kohlmann, P.; Singh, Y.; Savchenko, E.; Puranen, J.; Malm, T.; Kalesnykas, G.; Koistinaho, J.; et al. Retinal Degeneration in a Mouse Model of CLN5 Disease Is Associated with Compromised Autophagy. Sci. Rep. 2017, 7, 1597. [Google Scholar] [CrossRef] [PubMed]
- Chandrachud, U.; Walker, M.W.; Simas, A.M.; Heetveld, S.; Petcherski, A.; Klein, M.; Oh, H.; Wolf, P.; Zhao, W.-N.; Norton, S.; et al. Unbiased Cell-based Screening in a Neuronal Cell Model of Batten Disease Highlights an Interaction between Ca2+ Homeostasis, Autophagy, and CLN3 Protein Function. J. Biol. Chem. 2015, 290, 14361–14380. [Google Scholar] [CrossRef] [PubMed]
- Colussi, D.J.; Jacobson, M.A. Patient-Derived Phenotypic High-Throughput Assay to Identify Small Molecules Restoring Lysosomal Function in Tay–Sachs Disease. SLAS Discov. Adv. Sci. Drug Discov. 2019, 24, 295–303. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Gene Deficient Enzyme/Protein | Accumulated Products | Symptoms | Perturbations in Autophagy/Lipophagy/Lipolysis | Reference |
|---|---|---|---|---|---|
| Lysosomal storage diseases | |||||
| Lipid storage diseases | |||||
| Sphingolipidoses | |||||
| Niemann–Pick disease types A and B | SMPD1 sphingomyelinase | Sphingomyelin in brain and red blood cells (RBCs) | Hepatosplenomegaly, psychomotor regression, clumsiness and difficulty walking, dystonia, sleep disturbances, difficulty swallowing and eating, recurrent pneumonia, thrombocytopenia, a cherry-red spot inside the eye, frequent respiratory infections, slow mineralization of bone | Impaired autolysosomal clearance; formation of late endosome/lysosome (LE/LY)-like storage organelles (LSOs) and the misdirection of lipids to the LSOs; defect in autophagosome maturation; accumulation of autophagosomes | [74,75,76,77] |
| Niemann–Pick disease type C | NPC1 or NPC2 intracellular cholesterol transporters located within lysosomal and endosomal membranes (NPC1) or inside lysosomes (NPC2) | Free cholesterol, sphingomyelin and glycosphingolipid storage in lysosomes or late endosomes | Hepatosplenomegaly, problems with speech and swallowing, dementia, seizures, ataxia, vertical supranuclear gaze palsy, dystonia, severe liver disease, interstitial lung disease | Defective amphisome formation; impaired maturation of autophagosomes; accumulation of autophagosomes and autolysosomes | [78,79,80,81,82] |
| Fabry disease | GLA α-galactosidase A | Glycolipids, particularly ceramide trihexoside, in brain, heart and kidney | Episodes of pain (particularly acroparesthesias), angiokeratomas, hypohidrosis, corneal opacity or corneal verticillate, problems with the gastrointestinal system, tinnitus, hearing loss, kidney damage, heart attack, stroke | Impairment of the autophagic pathway | [83,84,85,86] |
| Krabbe disease (globoid cell leukodystrophy) | GALC galactocerebrosidase | Glycolipids, particularly galactocerebroside, in oligodendrocytes | Irritability, muscle weakness, feeding difficulties, stiff posture, delayed mental and physical development, spasticity, hypertonia, blindness, hyperreflexia, deafness, neurodegeneration (leading to death) | Impairment of autophagy; lysosomal dysfunction; partial blocking and saturation of the autophagy flux | [87,88,89,90] |
| Gaucher disease | GBA glucocerebrosidase | Glucocerebrosides in RBCs, liver and spleen | Hepatosplenomegaly, pancytopenia, Erlenmeyer flask deformity, anemia, lung disease, bone abnormalities such as bone pain, fractures, arthritis | Impaired autophagosome maturation; accumulation of autophagosomes; autophagy block | [91,92,93] |
| Tay–Sachs disease | HEXA β-hexosaminidase A | GM2 gangliosides in neurons | Neurodegeneration, seizures, vision and hearing loss, cherry-red spot, muscle weakness, ataxia, intellectual disability, paralysis, early death | Altered lipid trafficking; impaired autophagy | [94,95,96,97] |
| Tay–Sachs Disease, AB Variant (AB-variant GM2) | GM2A GM2 ganglioside activator | GM2 ganglioside in neurons in the brain and spinal cord | Psychomotor deterioration, seizures, vision and hearing loss, intellectual disability, paralysis, cherry-red spot, early death | Impaired autophagy | [95,98,99] |
| Metachromatic leukodystrophy (MLD) | ASA or PSAP arylsulfatase A or prosaposin | Sulfatide compounds in neural tissue | Demyelination in central and peripheral nervous systems (peripheral neuropathy, mental retardation, motor dysfunction, ataxia, hyporeflexia), seizures, incontinence, paralysis, inability to speak, blindness, hearing loss | Affected trafficking due to altered chain length of the lipids; defective autophagosome–lysosome fusion, impaired autophagy | [100,101,102,103] |
| Sandhoff disease | HEXB β-hexosaminidase A and β-hexosaminidase B | GM2 ganglioside in neurons of the brain and spinal cord | Progressive nervous system deterioration, muscle weakness, ataxia, speech problems, mental retardation, blindness, seizures, spasticity, macrocephaly, cherry-red spots in the eyes, frequent respiratory infections, doll-like facial appearance, hepatosplenomegaly | Disruption of autophagy, aberrant lysosomal–autophagic turnover | [104,105,106,107] |
| Multiple sulfatase deficiency | SUMF1 formylglycine-generating enzyme (FGE) | Sulfatides, sulfated glycosaminoglycans, sphingolipids and steroid sulfates in tissues | Leukodystrophy, movement problems, seizures, developmental delay, slow growth, ichthyosis, hypertrichosis, skeletal abnormalities (scoliosis, joint stiffness, dysostosis multiplex), hypotonia, coarse facial features, mild deafness, hepatomegaly, progressive neurologic deterioration, hydrocephalus | Accumulation of autophagosomes, defective autophagosome–lysosome fusion | [108,109,110] |
| GM1 gangliosidosis | GLB1 β-galactosidase | GM1 ganglioside in tissues and organs, particularly in the brain | Hepatosplenomegaly, skeletal abnormalities, seizures, profound intellectual disability, cherry-red spot, gingival hypertrophy, cardiomyopathy, dysostosis multiplex, coarsened facial features | Accumulation of autophagosomes, impaired lysosomal flux | [101,111,112] |
| Schindler disease | NAGA α-N-acetylgalactosaminidase | Glycosphingolipids, glycoproteins and oligosaccharides with terminal or preterminal N-acetylgalactosaminyl residues in the lysosomes of most tissues | Developmental regression, blindness, seizures, loss of awareness of surroundings, unresponsive, cognitive impairment, sensorineural hearing loss, weakness and loss of sensation, angiokeratomas | No data | [113] |
| Sea-blue histiocytosis (inherited lipemic splenomegaly) | APOE apolipoprotein E | Cholesterol, triglycerides and beta-very-low-density lipoproteins (beta-VLDLs) in the blood; glycosphingolipids, particularly sphingomyelins in the histocytes | Hypertriglyceridemia, splenomegaly, liver function abnormalities, heart disease, sea-blue histiocytes in many organs (bone marrow, liver and spleen) | No data | [114] |
| Neuronal ceroid lipofuscinosis | |||||
| Batten disease (juvenile neuronal ceroid lipofuscinosis, CLN3 disease) | CLN3 battenin, hydrophobic transmembrane protein involved in lysosomal function | Lysosomal autofluorescent storage material (AFSM) in the cells of the brain, central nervous system, and retina in the eye | Progressive blindness, seizures, mental and cognitive decline, dementia, speech and motor skills problems, premature death | Disruption of autophagy, vacuole maturation and impaired mitophagy; impaired autophagic clearance, defective autophagosome maturation | [115,116,117,118] |
| Jansky–Bielschowsky disease (late infantile neuronal ceroid lipofuscinosis, LINCL, CLN2 disease) | TPP1 tripeptidyl-peptidase 1 | Lipopigments in neurons, primarily in the cerebral and cerebellar cortices | Epilepsy, ataxia, myoclonus, vision loss, speech and motor skills problems (e.g., sitting and walking), developmental regression, intellectual disability, behavioral problems | Reduction in autophagic flux, inhibition of autophagosome formation, reduction in autophagosomes and autophagic degradation | [119,120] |
| Lysosomal and lipase deficiency | |||||
| Lysosomal acid lipase deficiency (Wolman disease, cholesteryl ester storage disease) | LIPA lysosomal acid lipase | Cholesteryl esters, triglycerides, and other lipids within lysosomes of most tissues | Hepatosplenomegaly, ascites, calcified adrenal glands, vomiting, diarrhea with steatorrhea, progressive psychomotor degradation, anemia, cachexia, low muscle tone, jaundice, vomiting, developmental delay, anemia, poor absorption of nutrients from food | Impairment of the lipophagic pathway | [121,122,123,124] |
| Mucolipidosis | |||||
| Mucolipidosis IV | MCOLN1 (TRPML1) mucolipin-1 | Sphingolipids, phospholipids, mucopolysaccharides and glycoproteins in cells of almost all tissues, including liver, spleen and in fibroblasts | Intellectual disability, psychomotor retardation, hypotonia, retinal degeneration, strabismus, photophobia, myopia, amblyopia or blindness, iron-deficiency anemia, achlorhydria with elevated blood gastrin levels | Impairment of autophagy and lipolysis; accumulation of lysosomes, autophagosomes and autophagy substrates | [125,126,127,128] |
| Sialidosis (mucolipidosis I) | NEU1 neuraminidase 1 | Sialic acid–containing compounds (sialyloligosaccharides and sialolipids) in lysosomes in bodily tissues | Type I: progressive neurological impairment without bone or joint abnormalities; type II: mental retardation, severe hepatosplenomegaly, coarse facial features, dysostosis multiplex, seizures, myoclonus, ataxia, aminoaciduria, corneal opacity, macular cherry-red spot, skeletal abnormalities | Impairment of lipolysis and autophagy | [129,130,131] |
| Neutral lipid storage disease | |||||
| Neutral lipid storage disease with myopathy | PNPLA2 adipose triglyceride lipase (ATGL) | Triglycerides in muscle and other tissues | Myopathy, fatty liver, cardiomyopathy, pancreatitis, hypothyroidism, type 2 diabetes | Impairment of lipolysis | [132,133,134] |
| Chanarin–Dorfman syndrome (neutral lipid storage disease type I, neutral lipid storage disease with ichthyosis) | ABHD5 abhydrolase domain containing 5 (activator of ATGL) | Triglycerides in organs and tissues, including skin, liver, muscles, intestine, eyes and ears | Ichthyosis, hepatomegaly, cataracts, ataxia, hearing loss, short stature, myopathy, nystagmus, mild intellectual disability | Impaired long-chain fatty acid oxidation; impaired BECN1-induced autophagic flux | [135,136,137] |
| Xanthomatosis | |||||
| Cerebrotendinous xanthomatosis (CTX) | CYP27A1 sterol 27-hydroxylase | Cholestanol and bile alcohols in the blood | Neonatal cholestasis, childhood-onset cataract, tendon and brain xanthomata, neurologic dysfunction (dementia, psychiatric disturbances, pyramidal and/or cerebellar signs, seizures and neuropathy), liver dysfunction, intellectual impairment, neuropsychiatric symptoms (hallucinations, aggression and depression) | Induced autophagy | [138,139,140] |
| Plant sterol storage disease | |||||
| Sitosterolemia | ABCG5 or ABCG8 sterolin | Plant sterols, such as sitosterol, and LDL in the blood | Atherosclerosis, increased chance of a heart attack, stroke or sudden death, xanthomas, joint stiffness and pain, hemolytic anemia, macrothrombocytopenia | Accumulation of autophagic vacuoles | [141,142] |
| Farber lipogranulomatosis | |||||
| Farber disease (Farber lipogranulomatosis) | ASAH1 acid ceramidase | Lipids in cells and tissues throughout the body, particularly around the joints. | Lipogranulomas, swollen and painful joint deformity, subcutaneous nodules, hoarseness, difficulty breathing, hepatosplenomegaly, developmental delay, vomiting | Impairment of autophagic flux | [143] |
| Fucosidosis | |||||
| Fucosidosis | FUCA1 alpha-L-fucosidase | Fucose containing glyco-lipids and polysaccharides in the brain, liver, spleen, skin, heart, pancreas and kidneys | Intellectual disability, dementia, delayed development of motor skills, impaired growth, dysostosis multiplex, seizures, spasticity, angiokeratomas, coarse facial features, recurrent respiratory infections, visceromegaly | Induction of the autophagic cell death | [144] |
| Lipid metabolism diseases | |||||
| Familial hyperlipidemia | |||||
| Hyperlipoproteinemia | |||||
| Familial dysbetalipoproteinemia (hyperlipoproteinemia type III) | APOE apolipoprotein E | Chylomicrons and VLDL remnants in plasma | Palmar and tuberoeruptive xanthomas, coronary heart disease, peripheral vascular disease | Decreased lipolysis | [145,146,147,148] |
| Familial hypercholesterolemia (hyperlipoproteinemia type IIa) | LDLR LDL receptor | LDL in plasma | Tendon xanthomas, coronary heart disease, increased chance of a heart attack, stroke or sudden death | Impairment of autophagic flux; altered autophagy flux by persistent mitophagy | [149,150,151] |
| Familial defective apoB-100 (hyperlipoproteinemia type IIa) | APOB apolipoprotein B-100 | LDL in plasma | Tendon xanthomas, coronary heart disease, increased chance of a heart attack, stroke or sudden death | Impairment of autophagic flux; altered autophagy flux by persistent mitophagy | [151,152] |
| Familial chylomicronemia syndrome | |||||
| ApoA-V deficiency | APOA5 apolipoprotein A-V | Chylomicrons and VLDL in blood | Eruptive xanthomas, hepatosplenomegaly, pancreatitis | Impairment of lipolysis | [153,154] |
| GPIHBP1 deficiency | GPIHBP1 glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 | Chylomicrons in plasma | Eruptive xanthomas, pancreatitis | Impairment of lipolysis | [155,156] |
| Lipoprotein lipase deficiency (hyperlipoproteinemia type I) | LPL lipoprotein lipase | Chylomicrons in plasma | Eruptive xanthomas, abdominal pain, lipemia retinalis, hepatosplenomegaly, pancreatitis | Impairment of lipolysis | [157,158] |
| Familial apolipoprotein C-II deficiency (hyperlipoproteinemia type I) | APOC2 apolipoprotein C-II (LPL cofactor) | Chylomicrons in plasma | Eruptive xanthomas, abdominal pain, lipemia retinalis, hepatosplenomegaly, pancreatitis | Impairment of lipolysis | [159,160,161] |
| Familial hepatic lipase deficiency | LIPC hepatic lipase | VLDL remnants and IDLs in plasma | Pancreatitis, coronary heart disease, increased chance of a heart attack, stroke or sudden death | Impairment of lipolysis | [162] |
| Familial hypercholesterolemia | |||||
| Autosomal recessive hypercholesterolemia | LDLRAP1 (ARH) low-density lipoprotein receptor adaptor protein 1 | LDL in plasma | Tendon xanthomas, coronary heart disease, increased chance of a heart attack, stroke or sudden death | Induced autophagy | [73,163,164] |
| Autosomal dominant hypercholesterolemia | PCSK9 proprotein convertase subtilisin/kexin type 9 | LDL in plasma | Tendon xanthomas, coronary heart disease, increased chance of a heart attack, stroke or sudden death | Increased autophagic flux | [73,165,166] |
| Secondary Storage Lipid | Disease | Compartment | Cellular Disturbance | Reference |
|---|---|---|---|---|
| Phospholipids | ||||
| Sphingomyelin | Sphingolipidoses: Niemann–Pick type C | Lysosomes | Altered membrane lipids trafficking | [170,171] |
| Bis(monoacylglycero)phosphate (BMP) | Sphingolipidoses: Niemann–Pick type C, Fabry disease, Gaucher disease, GM1 gangliosidosis, GM2 gangliosidosis Mucopolysaccharidoses: Hurler syndrome, Hunter syndrome Neuronal ceroid lipofuscinoses: NCL 10 | Endosomes, lysosomes | Altered membrane lipids trafficking, lamellar bodies formation | [171,172] |
| Glycosphingolipids | ||||
| Gangliosides—GM1, GM2, GM3, GD1a, GD2, GD3 | Sphingolipidoses: Niemann–Pick type A, B and C, Gaucher disease, prosaposin deficiency Mucopolysaccharidoses: Hurler syndrome, Hunter syndrome, Sanfilippo syndrome, Maroteaux–Lamy syndrome, Sly syndrome Glycoproteinoses: Galactosialidosis, α-mannosidosis, sialidosis Mucolipidoses: mucolipidosis II/III, mucolipidosis IV Neuronal ceroid lipofuscinoses: NCL 3, NCL 6, NCL 10 | Late endosomes, lysosomes, cytoplasmic vesicles | Alteration of lysosomal pH, autophagy dysregulation, rupture of H+/Ca2+ homeostasis, altered vesicle trafficking, dysregulation of signaling pathways, accumulation of polyubiquitinated proteins, reduced capacity of immune cells to produce cytokines and antibodies, neurodegeneration (gliosis, demyelination of white matter, astrocyte and microglial activation) | [173,174,175,176,177,178,179,180,181] |
| Cholesterol | ||||
| Cholesterol | Sphingolipidoses: Niemann–Pick type A and B Mucopolysaccharidoses: Hurler syndrome, Hunter syndrome, Sanfilippo syndrome, Maroteaux–Lamy syndrome Glycoproteinoses: α-mannosidosis | Late endosomes, lysosomes, cytoplasmic vesicles | Impaired vesicle trafficking, abnormal sequestration of materials, foam cells in cerebral blood vessels and liver | [171,174,176,177,178] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kloska, A.; Węsierska, M.; Malinowska, M.; Gabig-Cimińska, M.; Jakóbkiewicz-Banecka, J. Lipophagy and Lipolysis Status in Lipid Storage and Lipid Metabolism Diseases. Int. J. Mol. Sci. 2020, 21, 6113. https://doi.org/10.3390/ijms21176113
Kloska A, Węsierska M, Malinowska M, Gabig-Cimińska M, Jakóbkiewicz-Banecka J. Lipophagy and Lipolysis Status in Lipid Storage and Lipid Metabolism Diseases. International Journal of Molecular Sciences. 2020; 21(17):6113. https://doi.org/10.3390/ijms21176113
Chicago/Turabian StyleKloska, Anna, Magdalena Węsierska, Marcelina Malinowska, Magdalena Gabig-Cimińska, and Joanna Jakóbkiewicz-Banecka. 2020. "Lipophagy and Lipolysis Status in Lipid Storage and Lipid Metabolism Diseases" International Journal of Molecular Sciences 21, no. 17: 6113. https://doi.org/10.3390/ijms21176113
APA StyleKloska, A., Węsierska, M., Malinowska, M., Gabig-Cimińska, M., & Jakóbkiewicz-Banecka, J. (2020). Lipophagy and Lipolysis Status in Lipid Storage and Lipid Metabolism Diseases. International Journal of Molecular Sciences, 21(17), 6113. https://doi.org/10.3390/ijms21176113