A Split Luciferase Complementation Assay for the Quantification of β-Arrestin2 Recruitment to Dopamine D2-Like Receptors


Abstract
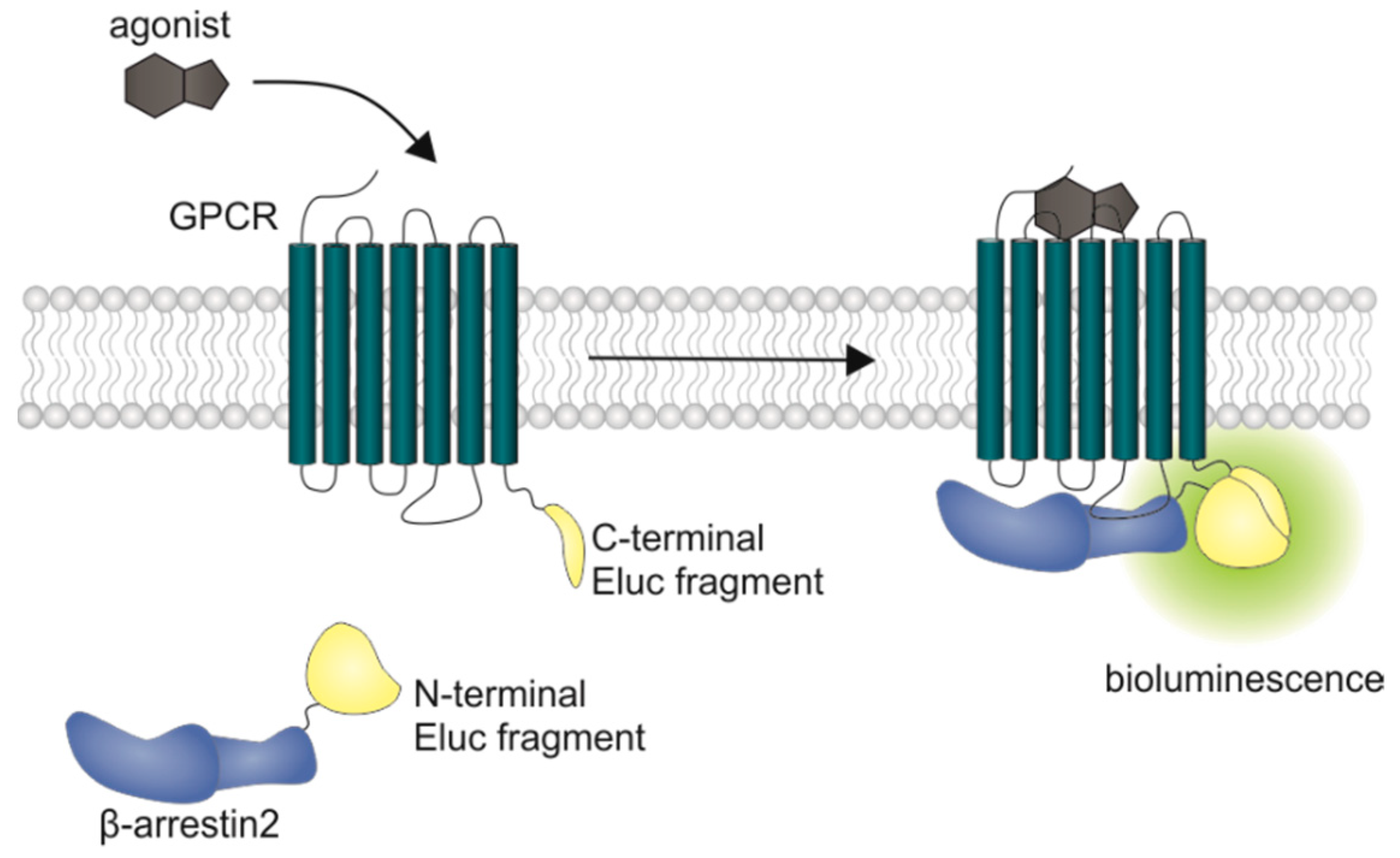
1. Introduction
2. Results and Discussion
2.1. Characterization of the Receptor Fusion Proteins
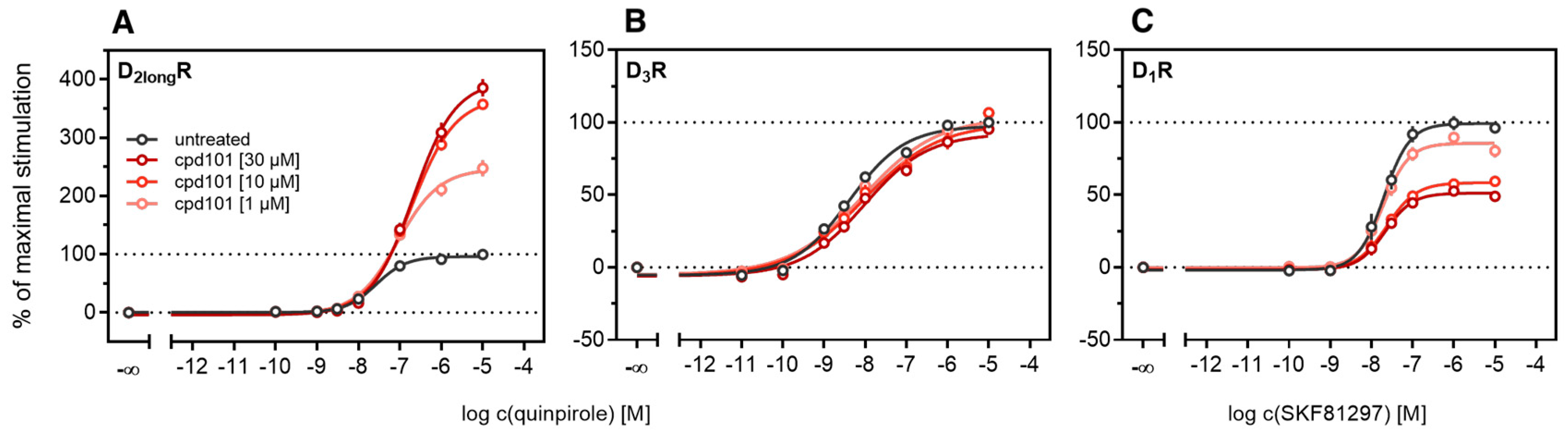
2.2. Pharmacological Characterization of Dopamine Receptor Ligands in the β-Arrestin2 Split Luciferase Complementation Assay
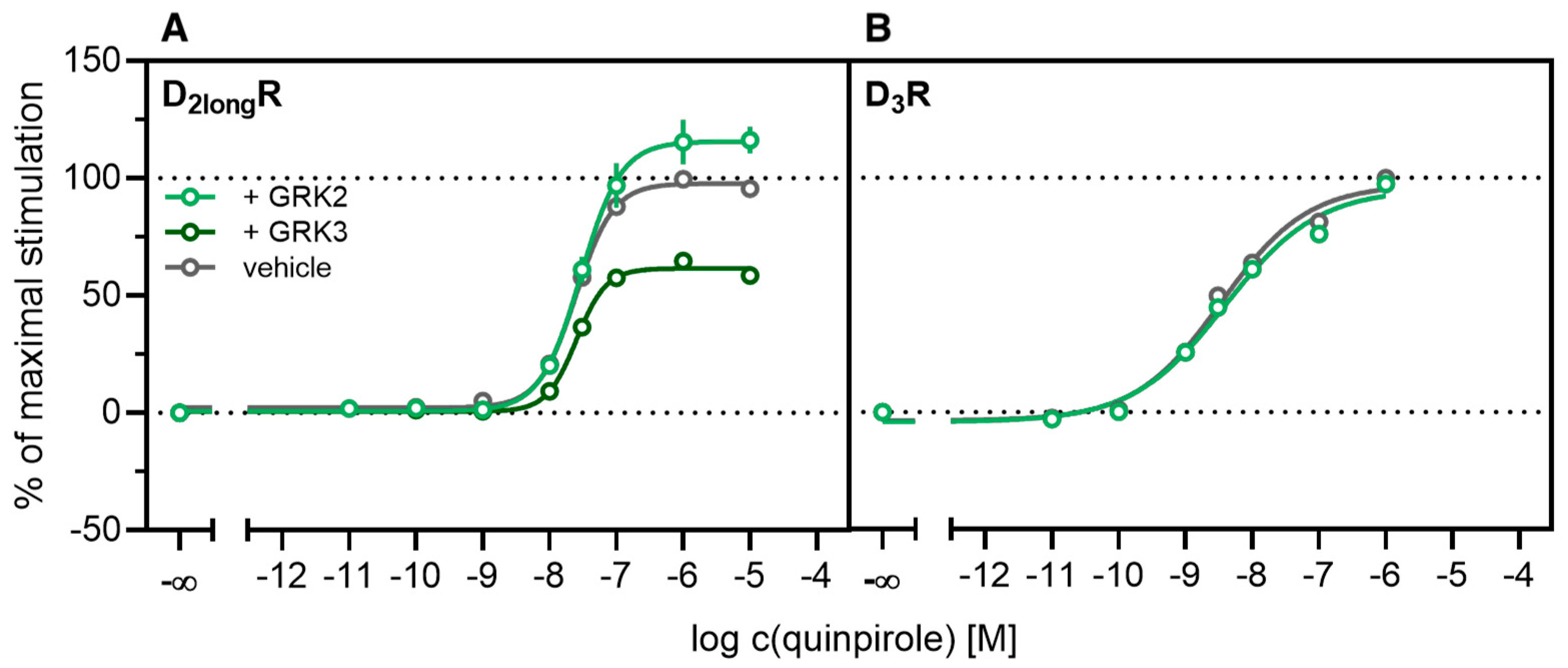
2.3. Influence of GRK2/3 on β-Arrestin2 Recruitment to the D2longR and the D3R
2.4. Influence of PKC on β-Arrestin2 Recruitment to the D3R
3. Materials and Methods
3.1. Materials
3.2. Cell Culture
3.3. Generation of Plasmids for Cells Used in the β-Arrestin2 Recruitment Assay
3.4. Generation of Plasmids for Cells Used for Homogenate Preparation
3.5. Generation of Stable Transfectants
3.6. Preparation of Cell Homogenates
3.7. Radioligand Binding Experiments with Whole Cells
3.8. Radioligand Binding Experiments with Homogenates
3.9. Quantification of β-Arrestin2 Recruitment in Live Cells
3.10. Quantification of β-Arrestin2 Recruitment by Endpoint Measurement
3.11. Statistical Analysis
3.12. Data Availability
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cools, A.R.; Van Rossum, J.M. Excitation-mediating and inhibition-mediating dopamine-receptors: A new concept towards a better understanding of electrophysiological, biochemical, pharmacological, functional and clinical data. Psychopharmacologia 1976, 45, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Dearry, A.; Gingrich, J.A.; Falardeau, P.; Fremeau, R.T.; Bates, M.D., Jr.; Caron, M.G. Molecular cloning and expression of the gene for a human D1 dopamine receptor. Nature 1990, 347, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, P.; Giros, B.; Martres, M.-P.; Bouthenet, M.-L.; Schwartz, J.-C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Van Tol, H.H.; Bunzow, J.R.; Guan, H.C.; Sunahara, R.K.; Seeman, P.; Niznik, H.B.; Civelli, O. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 1991, 350, 610–614. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Xiao, H.; Merril, C.R. The human D5 dopamine receptor (DRD5) maps on chromosome 4. Genomics 1991, 11, 777–778. [Google Scholar] [CrossRef][Green Version]
- Andersen, P.H.; Gingrich, J.A.; Bates, M.D.; Dearry, A.; Falardeau, P.; Senogles, S.E.; Caron, M.G. Dopamine receptor subtypes: Beyond the D1/D2 classification. Trends Pharmacol. Sci. 1990, 11, 231–236. [Google Scholar] [CrossRef]
- Neve, K.A.; Seamans, J.K.; Trantham-Davidson, H. Dopamine receptor signaling. J. Recept. Signal Transduct. 2004, 24, 165–205. [Google Scholar] [CrossRef]
- Kebabian, J.W. Multiple classes of dopamine receptors in mammalian central nervous system: The involvement of dopamine-sensitive adenylyl cyclase. Life Sci. 1978, 23, 479–483. [Google Scholar] [CrossRef]
- Spano, P.F.; Govoni, S.; Trabucchi, M. Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Adv. Biochem. Psychopharmacol. 1978, 19, 155–165. [Google Scholar]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Davis, K.L.; Kahn, R.S.; Ko, G.; Davidson, M. Dopamine in schizophrenia: A review and reconceptualization. Am. J. Psychiatry 1991, 148, 1474–1486. [Google Scholar] [CrossRef] [PubMed]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef]
- Hoglinger, G.U.; Rizk, P.; Muriel, M.P.; Duyckaerts, C.; Oertel, W.H.; Caille, I.; Hirsch, E.C. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci. 2004, 7, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G. Drug addiction as dopamine-dependent associative learning disorder. Eur. J. Pharmacol. 1999, 375, 13–30. [Google Scholar] [CrossRef]
- Di Chiara, G.; Bassareo, V.; Fenu, S.; De Luca, M.A.; Spina, L.; Cadoni, C.; Acquas, E.; Carboni, E.; Valentini, V.; Lecca, D. Dopamine and drug addiction: The nucleus accumbens shell connection. Neuropharmacology 2004, 47, 227–241. [Google Scholar] [CrossRef]
- Albrecht, F.E.; Drago, J.; Felder, R.A.; Printz, M.P.; Eisner, G.M.; Robillard, J.E.; Sibley, D.R.; Westphal, H.J.; Jose, P.A. Role of the D1A dopamine receptor in the pathogenesis of genetic hypertension. J. Clin. Investig. 1996, 97, 2283–2288. [Google Scholar] [CrossRef]
- Berk, M.; Dodd, S.; Kauer-Santanna, M.; Malhi, G.S.; Bourin, M.; Kapczinski, F.; Norman, T. Dopamine dysregulation syndrome: Implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr. Scand. 2007, 116, 41–49. [Google Scholar] [CrossRef]
- Cousins, D.A.; Butts, K.; Young, A.H. The role of dopamine in bipolar disorder. Bipolar Disord. 2009, 11, 787–806. [Google Scholar] [CrossRef]
- Linke, R.; Eisensehr, I.; Wetter, T.C.; Gildehaus, F.J.; Popperl, G.; Trenkwalder, C.; Noachtar, S.; Tatsch, K. Presynaptic dopaminergic function in patients with restless legs syndrome: Are there common features with early Parkinson’s disease? Mov. Disord. 2004, 19, 1158–1162. [Google Scholar] [CrossRef]
- Stiasny-Kolster, K.; Kohnen, R.; Schollmayer, E.; Moller, J.C.; Oertel, W.H.; Rotigotine Sp 666 Study Group. Patch application of the dopamine agonist rotigotine to patients with moderate to advanced stages of restless legs syndrome: A double-blind, placebo-controlled pilot study. Mov. Disord. 2004, 19, 1432–1438. [Google Scholar] [CrossRef]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Beta-Arrestin: A protein that regulates beta-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S.; Downey, W.E., III; Colapietro, A.M.; Barak, L.S.; Menard, L.; Caron, M.G. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 1996, 271, 363–366. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed]
- Inglese, J.; Freedman, N.J.; Koch, W.J.; Lefkowitz, R.J. Structure and mechanism of the G protein-coupled receptor kinases. J. Biol. Chem. 1993, 268, 23735–23738. [Google Scholar]
- Pippig, S.; Andexinger, S.; Daniel, K.; Puzicha, M.; Caron, M.G.; Lefkowitz, R.J.; Lohse, M.J. Overexpression of beta-arrestin and beta-adrenergic receptor kinase augment desensitization of beta 2-adrenergic receptors. J. Biol. Chem. 1993, 268, 3201–3208. [Google Scholar]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Barak, L.S.; Caron, M.G. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis. J. Biol. Chem. 2001, 276, 19452–19460. [Google Scholar] [CrossRef]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Caron, M.G.; Barak, L.S. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 2000, 275, 17201–17210. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Premont, R.T.; Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 2004, 27, 107–144. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef]
- Ito, K.; Haga, T.; Lameh, J.; Sadee, W. Sequestration of dopamine D2 receptors depends on coexpression of G-protein-coupled receptor kinases 2 or 5. Eur. J. Biochem. 1999, 260, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Ito, K.; Fukuzaki, A.; Inaki, K.; Haga, T. Dynamin and rab5 regulate GRK2-dependent internalization of dopamine D2 receptors. Eur. J. Biochem. 1999, 263, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Valenzano, K.J.; Robinson, S.R.; Yao, W.D.; Barak, L.S.; Caron, M.G. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J. Biol. Chem. 2001, 276, 37409–37414. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Caron, M.G. Complementary roles of the DRY motif and C-terminus tail of GPCRS for G protein coupling and beta-arrestin interaction. Biochem. Biophys. Res. Commun. 2008, 366, 42–47. [Google Scholar] [CrossRef]
- Spooren, A.; Rondou, P.; Debowska, K.; Lintermans, B.; Vermeulen, L.; Samyn, B.; Skieterska, K.; Debyser, G.; Devreese, B.; Vanhoenacker, P.; et al. Resistance of the dopamine D4 receptor to agonist-induced internalization and degradation. Cell. Signal. 2010, 22, 600–609. [Google Scholar] [CrossRef]
- Deming, J.D.; Shin, J.A.; Lim, K.; Lee, E.J.; Van Craenenbroeck, K.; Craft, C.M. Dopamine receptor D4 internalization requires a beta-arrestin and a visual arrestin. Cell. Signal. 2015, 27, 2002–2013. [Google Scholar] [CrossRef]
- Min, C.; Zheng, M.; Zhang, X.; Caron, M.G.; Kim, K.M. Novel roles for beta-arrestins in the regulation of pharmacological sequestration to predict agonist-induced desensitization of dopamine D3 receptors. Br. J. Pharmacol. 2013, 170, 1112–1129. [Google Scholar] [CrossRef]
- Cho, E.Y.; Cho, D.I.; Park, J.H.; Kurose, H.; Caron, M.G.; Kim, K.M. Roles of protein kinase C and actin-binding protein 280 in the regulation of intracellular trafficking of dopamine D3 receptor. Mol. Endocrinol. 2007, 21, 2242–2254. [Google Scholar] [CrossRef]
- Masri, B.; Salahpour, A.; Didriksen, M.; Ghisi, V.; Beaulieu, J.M.; Gainetdinov, R.R.; Caron, M.G. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. USA 2008, 105, 13656–13661. [Google Scholar] [CrossRef]
- Littmann, T.; Buschauer, A.; Bernhardt, G. Split luciferase-based assay for simultaneous analyses of the ligand concentration- and time-dependent recruitment of beta-arrestin2. Anal. Biochem. 2019, 573, 8–16. [Google Scholar] [CrossRef]
- Song, Y.B.; Park, C.O.; Jeong, J.Y.; Huh, W.K. Monitoring G protein-coupled receptor activation using an adenovirus-based beta-arrestin bimolecular fluorescence complementation assay. Anal. Biochem. 2014, 449, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Laroche, G.; Giguere, P.M. Measurement of beta-Arrestin Recruitment at GPCRs Using the Tango Assay. Methods Mol. Biol. 2019, 1947, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Eishingdrelo, H.; Cai, J.; Weissensee, P.; Sharma, P.; Tocci, M.J.; Wright, P.S. A cell-based protein-protein interaction method using a permuted luciferase reporter. Curr. Chem. Genom. 2011, 5, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Viviani, V.R.; Arnoldi, F.G.; Neto, A.J.; Oehlmeyer, T.L.; Bechara, E.J.; Ohmiya, Y. The structural origin and biological function of pH-sensitivity in firefly luciferases. Photochem. Photobiol. Sci. 2008, 7, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Misawa, N.; Kafi, A.K.; Hattori, M.; Miura, K.; Masuda, K.; Ozawa, T. Rapid and high-sensitivity cell-based assays of protein-protein interactions using split click beetle luciferase complementation: An approach to the study of G-protein-coupled receptors. Anal. Chem. 2010, 82, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Tanaka, M.; Takakura, H.; Aoki, K.; Miura, K.; Anzai, T.; Ozawa, T. Analysis of temporal patterns of GPCR-beta-arrestin interactions using split luciferase-fragment complementation. Mol. Biosyst. 2013, 9, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Sibley, D.R.; De Lean, A.; Creese, I. Anterior pituitary dopamine receptors. Demonstration of interconvertible high and low affinity states of the D-2 dopamine receptor. J. Biol. Chem. 1982, 257, 6351–6361. [Google Scholar]
- Sibley, D.R.; Mahan, L.C.; Creese, I. Dopamine receptor binding on intact cells. Absence of a high-affinity agonist-receptor binding state. Mol. Pharmacol. 1983, 23, 295–302. [Google Scholar]
- Newman-Tancredi, A.; Cussac, D.; Audinot, V.; Nicolas, J.P.; De Ceuninck, F.; Boutin, J.A.; Millan, M.J. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. II. Agonist and antagonist properties at subtypes of dopamine D(2)-like receptor and alpha(1)/alpha(2)-adrenoceptor. J. Pharmacol. Exp. Ther. 2002, 303, 805–814. [Google Scholar] [CrossRef]
- Allen, J.A.; Yost, J.M.; Setola, V.; Chen, X.; Sassano, M.F.; Chen, M.; Peterson, S.; Yadav, P.N.; Huang, X.P.; Feng, B.; et al. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. USA 2011, 108, 18488–18493. [Google Scholar] [CrossRef]
- George, S.E.; Bungay, P.J.; Naylor, L.H. Functional analysis of the D2L dopamine receptor expressed in a cAMP-responsive luciferase reporter cell line. Biochem. Pharmacol. 1998, 56, 25–30. [Google Scholar] [CrossRef]
- Klewe, I.V.; Nielsen, S.M.; Tarpo, L.; Urizar, E.; Dipace, C.; Javitch, J.A.; Gether, U.; Egebjerg, J.; Christensen, K.V. Recruitment of beta-arrestin2 to the dopamine D2 receptor: Insights into anti-psychotic and anti-parkinsonian drug receptor signaling. Neuropharmacology 2008, 54, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.; Kiss, D.J.; Keseru, G.M.; Stark, H. Binding kinetics of cariprazine and aripiprazole at the dopamine D3 receptor. Sci. Rep. 2018, 8, 12509. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Reith, M.E.A.; Chen, L.Y.; Kortagere, S. Biased signaling agonist of dopamine D3 receptor induces receptor internalization independent of beta-arrestin recruitment. Pharmacol. Res. 2019, 143, 48–57. [Google Scholar] [CrossRef]
- Free, R.B.; Chun, L.S.; Moritz, A.E.; Miller, B.N.; Doyle, T.B.; Conroy, J.L.; Padron, A.; Meade, J.A.; Xiao, J.; Hu, X.; et al. Discovery and characterization of a G protein-biased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Mol. Pharmacol. 2014, 86, 96–105. [Google Scholar] [CrossRef]
- Sautel, F.; Griffon, N.; Levesque, D.; Pilon, C.; Schwartz, J.C.; Sokoloff, P. A functional test identifies dopamine agonists selective for D3 versus D2 receptors. Neuroreport 1995, 6, 329–332. [Google Scholar] [CrossRef]
- McDonald, W.M.; Sibley, D.R.; Kilpatrick, B.F.; Caron, M.G. Dopaminergic inhibition of adenylate cyclase correlates with high affinity agonist binding to anterior pituitary D2 dopamine receptors. Mol. Cell. Endocrinol. 1984, 36, 201–209. [Google Scholar] [CrossRef]
- Sokoloff, P.; Andrieux, M.; Besancon, R.; Pilon, C.; Martres, M.P.; Giros, B.; Schwartz, J.C. Pharmacology of human dopamine D3 receptor expressed in a mammalian cell line: Comparison with D2 receptor. Eur. J. Pharmacol. 1992, 225, 331–337. [Google Scholar] [CrossRef]
- Gao, Y.; Peterson, S.; Masri, B.; Hougland, M.T.; Adham, N.; Gyertyan, I.; Kiss, B.; Caron, M.G.; El-Mallakh, R.S. Cariprazine exerts antimanic properties and interferes with dopamine D2 receptor beta-arrestin interactions. Pharmacol. Res. Perspect. 2015, 3, e00073. [Google Scholar] [CrossRef]
- Vanhauwe, J.F.; Fraeyman, N.; Francken, B.J.; Luyten, W.H.; Leysen, J.E. Comparison of the ligand binding and signaling properties of human dopamine D(2) and D(3) receptors in Chinese hamster ovary cells. J. Pharmacol. Exp. Ther. 1999, 290, 908–916. [Google Scholar]
- Freedman, S.B.; Patel, S.; Marwood, R.; Emms, F.; Seabrook, G.R.; Knowles, M.R.; McAllister, G. Expression and pharmacological characterization of the human D3 dopamine receptor. J. Pharmacol. Exp. Ther. 1994, 268, 417–426. [Google Scholar] [PubMed]
- Tadori, Y.; Forbes, R.A.; McQuade, R.D.; Kikuchi, T. Characterization of aripiprazole partial agonist activity at human dopamine D3 receptors. Eur. J. Pharmacol. 2008, 597, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, X.; Tocker, A.M.; Huang, P.; Reith, M.E.; Liu-Chen, L.Y.; Smith, A.B., III; Kortagere, S. Functional Characterization of a Novel Series of Biased Signaling Dopamine D3 Receptor Agonists. ACS Chem. Neurosci. 2017, 8, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Todd, R.D.; Heller, A.; O’Malley, K.L. Pharmacological and functional characterization of D2, D3 and D4 dopamine receptors in fibroblast and dopaminergic cell lines. J. Pharmacol. Exp. Ther. 1994, 268, 495–502. [Google Scholar] [PubMed]
- Slot, L.A.B.; Palmier, C.; Tardif, S.; Cussac, D. Action of novel antipsychotics at human dopamine D3 receptors coupled to G protein and ERK1/2 activation. Neuropharmacology 2007, 53, 232–241. [Google Scholar] [CrossRef]
- Vile, J.M.; D’Souza, U.M.; Strange, P.G. [3H]nemonapride and [3H]spiperone label equivalent numbers of D2 and D3 dopamine receptors in a range of tissues and under different conditions. J. Neurochem. 1995, 64, 940–943. [Google Scholar] [CrossRef]
- Namkung, Y.; Dipace, C.; Urizar, E.; Javitch, J.A.; Sibley, D.R. G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J. Biol. Chem. 2009, 284, 34103–34115. [Google Scholar] [CrossRef]
- Thal, D.M.; Yeow, R.Y.; Schoenau, C.; Huber, J.; Tesmer, J.J. Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol. Pharmacol. 2011, 80, 294–303. [Google Scholar] [CrossRef]
- Lamey, M.; Thompson, M.; Varghese, G.; Chi, H.; Sawzdargo, M.; George, S.R.; O’Dowd, B.F. Distinct residues in the carboxyl tail mediate agonist-induced desensitization and internalization of the human dopamine D1 receptor. J. Biol. Chem. 2002, 277, 9415–9421. [Google Scholar] [CrossRef]
- Pack, T.F.; Orlen, M.I.; Ray, C.; Peterson, S.M.; Caron, M.G. The dopamine D2 receptor can directly recruit and activate GRK2 without G protein activation. J. Biol. Chem. 2018, 293, 6161–6171. [Google Scholar] [CrossRef]
- Namkung, Y.; Dipace, C.; Javitch, J.A.; Sibley, D.R. G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J. Biol. Chem. 2009, 284, 15038–15051. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Gainetdinov, R.R.; Gurevich, V.V. G protein-coupled receptor kinases as regulators of dopamine receptor functions. Pharmacol. Res. 2016, 111, 1–16. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, N.; Zheng, M.; Kim, K.M. Clathrin-mediated endocytosis is responsible for the lysosomal degradation of dopamine D3 receptor. Biochem. Biophys. Res. Commun. 2016, 476, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.A.; Fredericks, Z.L.; Stone, W.C.; Premont, R.T.; Stoffel, R.H.; Koch, W.J.; Lefkowitz, R.J. Phosphatidylinositol 4,5-bisphosphate (PIP2)-enhanced G protein-coupled receptor kinase (GRK) activity: Location, structure, and regulation of the PIP2 binding site distinguishes the GRK subfamilies. J. Biol. Chem. 1996, 271, 24907–24913. [Google Scholar] [CrossRef] [PubMed]
- Lieb, S.; Littmann, T.; Plank, N.; Felixberger, J.; Tanaka, M.; Schafer, T.; Krief, S.; Elz, S.; Friedland, K.; Bernhardt, G.; et al. Label-free versus conventional cellular assays: Functional investigations on the human histamine H1 receptor. Pharmacol. Res. 2016, 114, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Bartole, E.; Gratz, L.; Littmann, T.; Wifling, D.; Seibel, U.; Buschauer, A.; Bernhardt, G. UR-DEBa242: A Py-5-Labeled Fluorescent Multipurpose Probe for Investigations on the Histamine H3 and H4 Receptors. J. Med. Chem. 2020, 63, 5297–5311. [Google Scholar] [CrossRef]
- Hübner, H.; Haubmann, C.; Utz, W.; Gmeiner, P. Conjugated Enynes as Nonaromatic Catechol Bioisosteres: Synthesis, Binding Experiments, and Computational Studies of Novel Dopamine Receptor Agonists Recognizing Preferentially the D3 Subtype. J. Med. Chem. 2000, 43, 756–762. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D2longR | D3R | D4.4R | ||||
|---|---|---|---|---|---|---|
| ELucC Fusion Protein | wt | ELucC Fusion Protein | wt | ELucC Fusion Protein | wt | |
| pKd | 10.56 ± 0.04 | 10.84 ± 0.05 | 10.31 ± 0.03 | 10.59 ± 0.01 | 9.40 ± 0.09 | 10.11 ± 0.04 |
| cpd | D2longR | D3R | D4.4R | ||||
|---|---|---|---|---|---|---|---|
| ELucC Fusion Protein | wt | ELucC Fusion Protein | wt | ELucC Fusion Protein | wt | ||
| aripiprazole | 9.25 ± 0.16 | 8.32 ± 0.02 | 8.9 ± 0.24 | 7.85 ± 0.08 | 7.64 ± 0.15 | 7.85 ± 0.08 | |
| quinpirole | 7.29 ± 0.07 | hi | 7.90 ± 0.10 | 7.63 ± 0.05 | 8.00 ± 0.08 | 6.58 ± 0.01 | 8.00 ± 0.08 |
| lo | 6.11 ± 0.02 | ||||||
| haloperidol | 9.45 ± 0.05 | 9.58 ± 0.13 | 8.27 ± 0.08 | 8.93 ± 0.02 | 8.27 ± 0.02 | 8.93 ± 0.02 | |
| nemonapride | 9.95 ± 0.13 | 9.76 ± 0.08 | 9.76 ± 0.06 | 9.33 ± 0.02 | 9.69 ± 0.08 | 9.33 ± 0.02 | |
| Receptor | cpd | β-Arrestin2 Recruitment | N | Radioligand Displacement | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| pEC50 | Emax [%] | pKb | pKi | |||||
| D2longR | R-(−)-apomorphine | 7.77 ± 0.04 | 87 ± 3 | 4 | 7.48 ± 0.14 | 7.66 [49] | ||
| aripiprazole | 6.65 ± 0.15 | 8 ± 2 | 3 | 8.32 ± 0.02 | 6.84 [50] | |||
| dopamine | 7.24 ± 0.04 | 104 ± 3 | 3 | hi | 7.99 ± 0.16 | 7.05 [55] | ||
| lo | 6.30 ± 0.07 | |||||||
| pramipexole | 8.19 ± 0.05 | 86 ± 4 | 4 | hi | 7.59 ± 0.12 | 8.51 [56] | ||
| lo | 6.00 ± 0.03 | |||||||
| quinpirole | 7.55 ± 0.07 | 100 | 5 | hi | 7.90 ± 0.10 | 7.11 [51] | ||
| lo | 6.11 ± 0.02 | |||||||
| (+)-butaclamol | 8.29 ± 0.10 | 3 | 9.14 ± 0.06 | 8.04 [57] | ||||
| domperidone | 9.13 ± 0.09 | 3 | 9.47 ± 0.07 | 8.87 [58] | ||||
| haloperidol | 8.90 ± 0.05 | 3 | 9.58 ± 0.13 | 8.89 [59] | ||||
| nemonapride | 8.90 ± 0.05 | 3 | 9.76 ± 0.08 | 9.32 [60] | ||||
| S-(−)-sulpiride | 8.86 ± 0.10 | 3 | 7.51 ± 0.09 | 8.22 [61] | ||||
| D3R | R-(−)-apomorphine | 7.43 ± 0.17 | 91 ± 5 | 3 | 8.40 ± 0.03 | 7.93 [49] | ||
| aripiprazole | 7.44 ± 0.05 | 26 ± 1 | 3 | 8.26 ± 0.02 | 7.00 [62] | |||
| dopamine | 7.66 ± 0.14 | 105 ± 8 | 3 | hi | 8.78 ± 0.09 | 7.95 [63] | ||
| lo | 7.23 ± 0.09 | |||||||
| pramipexole | 9.09 ± 0.06 | 99 ± 4 | 4 | 9.18 ± 0.06 | 8.65 [49] | |||
| quinpirole | 8.75 ± 0.07 | 100 | 6 | 8.34 ± 0.07 | 9.07 [56] | |||
| (+)-butaclamol | 7.16 ± 0.17 | −27 ± 9 | 7.35 ± 0.08 | 3/3 | 8.59 ± 0.02 | 7.95 [64] | ||
| domperidone | 8.02 ± 0.14 | −26 ± 4 | 8.06 ± 0.09 | 3/3 | 8.96 ± 0.11 | 8.12 [58] | ||
| haloperidol | 8.29 ± 0.29 | −27 ± 5 | 8.68 ± 0.12 | 3/3 | 8.95 ± 0.03 | 8.70 [65] | ||
| nemonapride | 8.43 ± 0.13 | −25 ± 4 | 9.07 ± 0.12 | 3/3 | 9.99 ± 0.06 | 9.77 [66] | ||
| S-(−)-sulpiride | 8.33 ± 0.10 | −26 ± 8 | 8.23 ± 0.07 | 3/4 | 7.20 ± 0.03 | 7.70 [58] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forster, L.; Grätz, L.; Mönnich, D.; Bernhardt, G.; Pockes, S. A Split Luciferase Complementation Assay for the Quantification of β-Arrestin2 Recruitment to Dopamine D2-Like Receptors. Int. J. Mol. Sci. 2020, 21, 6103. https://doi.org/10.3390/ijms21176103
Forster L, Grätz L, Mönnich D, Bernhardt G, Pockes S. A Split Luciferase Complementation Assay for the Quantification of β-Arrestin2 Recruitment to Dopamine D2-Like Receptors. International Journal of Molecular Sciences. 2020; 21(17):6103. https://doi.org/10.3390/ijms21176103
Chicago/Turabian StyleForster, Lisa, Lukas Grätz, Denise Mönnich, Günther Bernhardt, and Steffen Pockes. 2020. "A Split Luciferase Complementation Assay for the Quantification of β-Arrestin2 Recruitment to Dopamine D2-Like Receptors" International Journal of Molecular Sciences 21, no. 17: 6103. https://doi.org/10.3390/ijms21176103
APA StyleForster, L., Grätz, L., Mönnich, D., Bernhardt, G., & Pockes, S. (2020). A Split Luciferase Complementation Assay for the Quantification of β-Arrestin2 Recruitment to Dopamine D2-Like Receptors. International Journal of Molecular Sciences, 21(17), 6103. https://doi.org/10.3390/ijms21176103