Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the Water-Deficit and Heat Stress Response Network in Durum Wheat




Abstract
1. Introduction
2. Results
2.1. Conserved and Novel miRNAs Identified in sRNA-seq
2.2. DEMs Subject to Stress Type, Genotype, and Time-Point
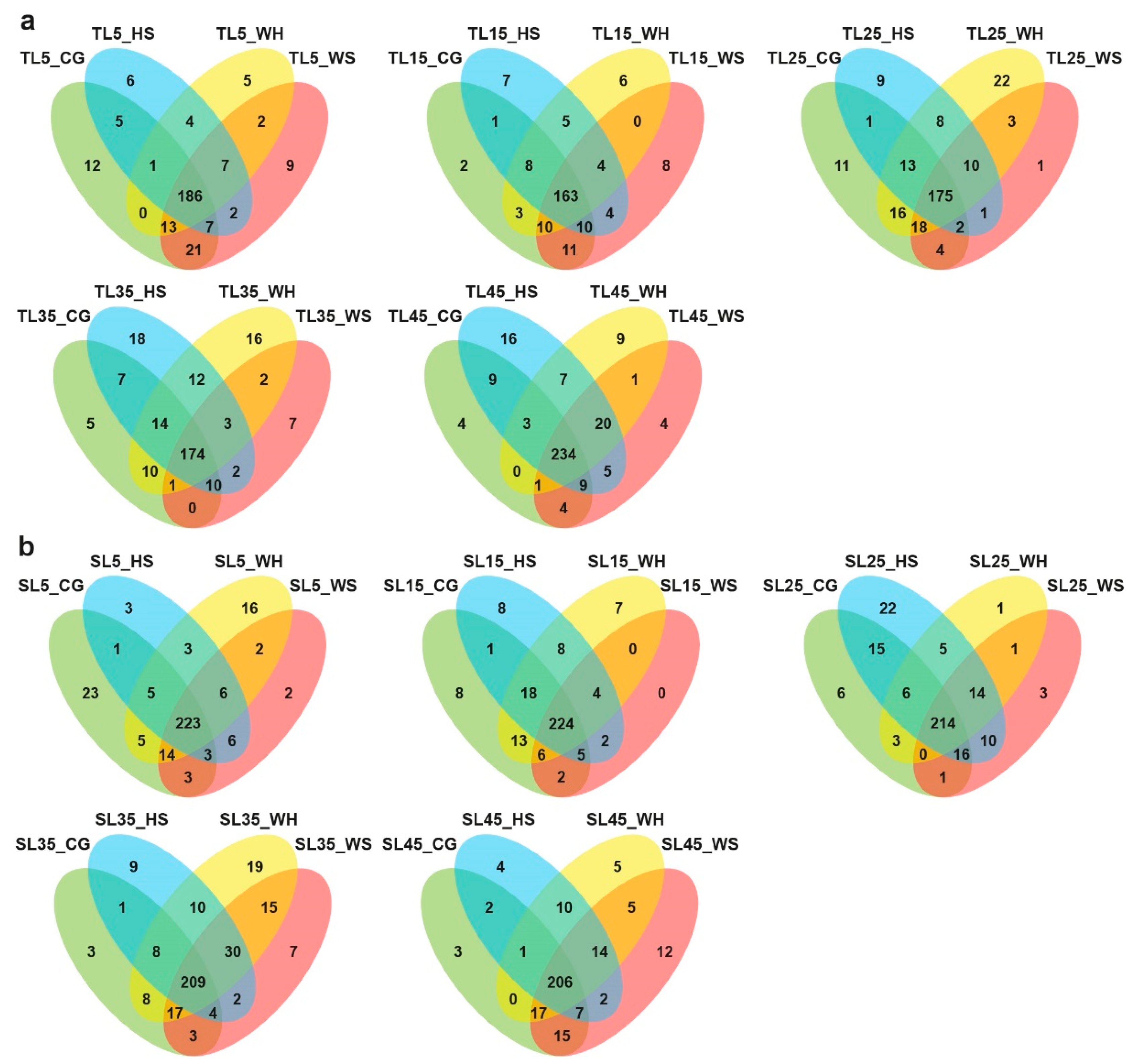
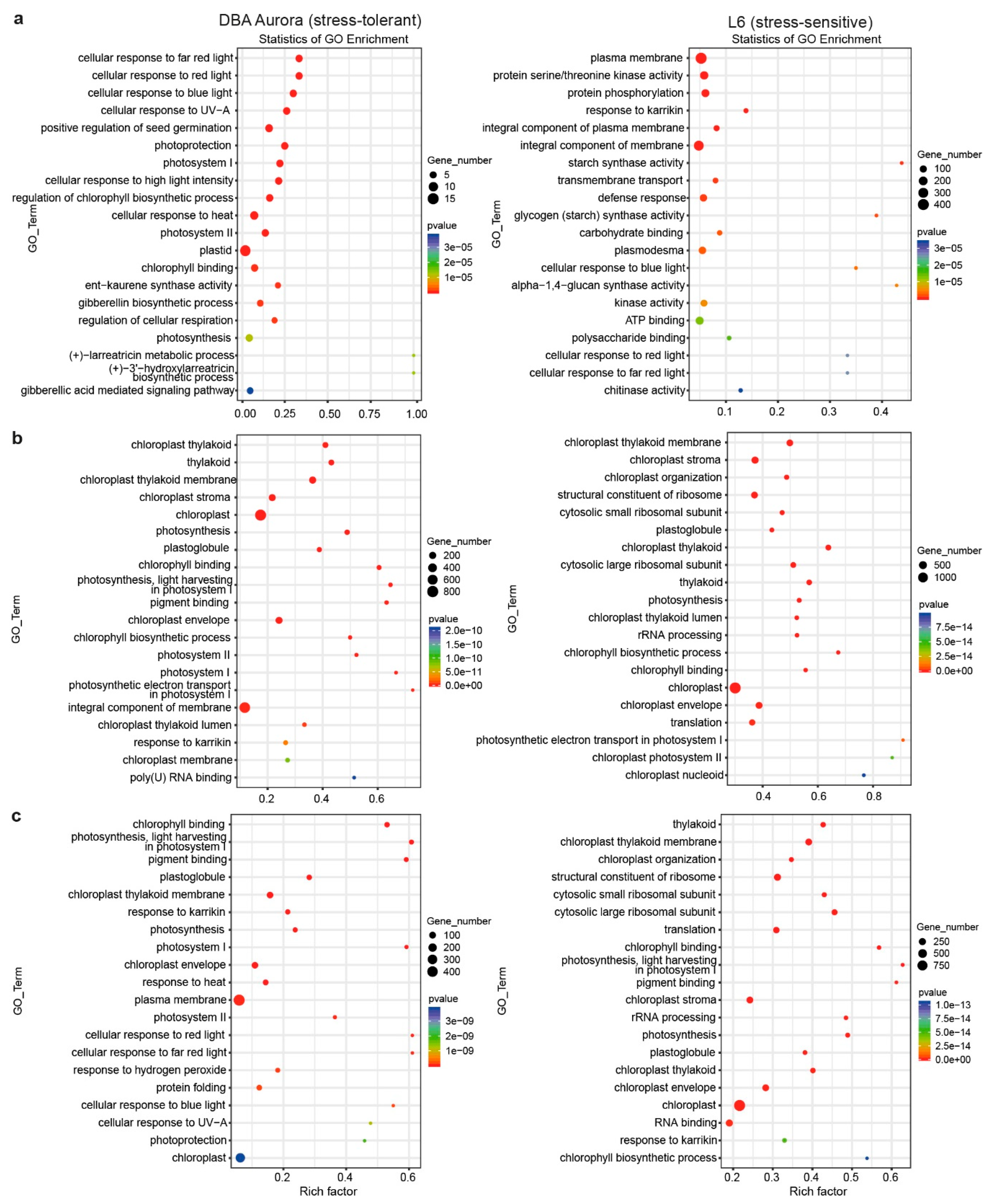
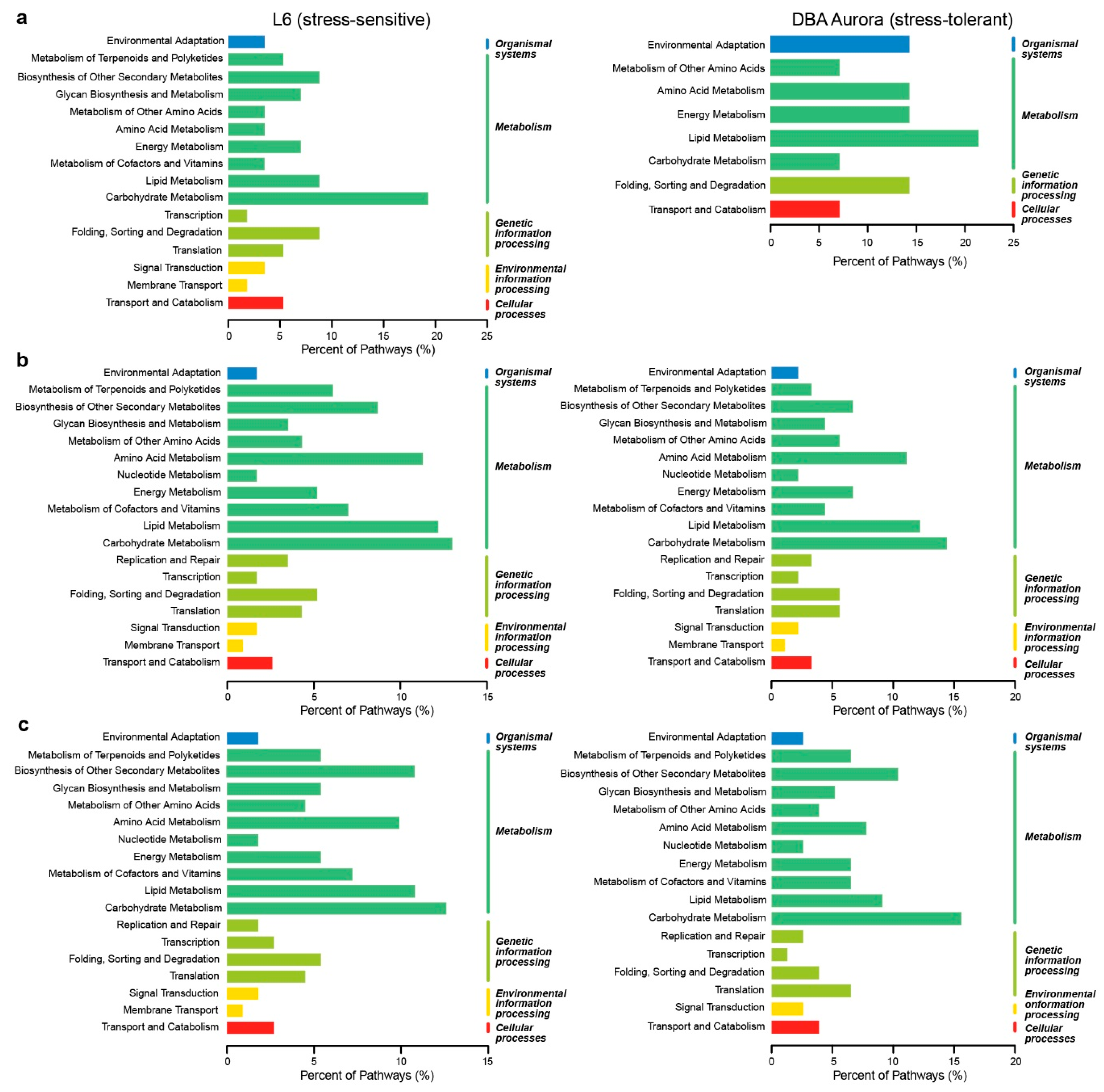
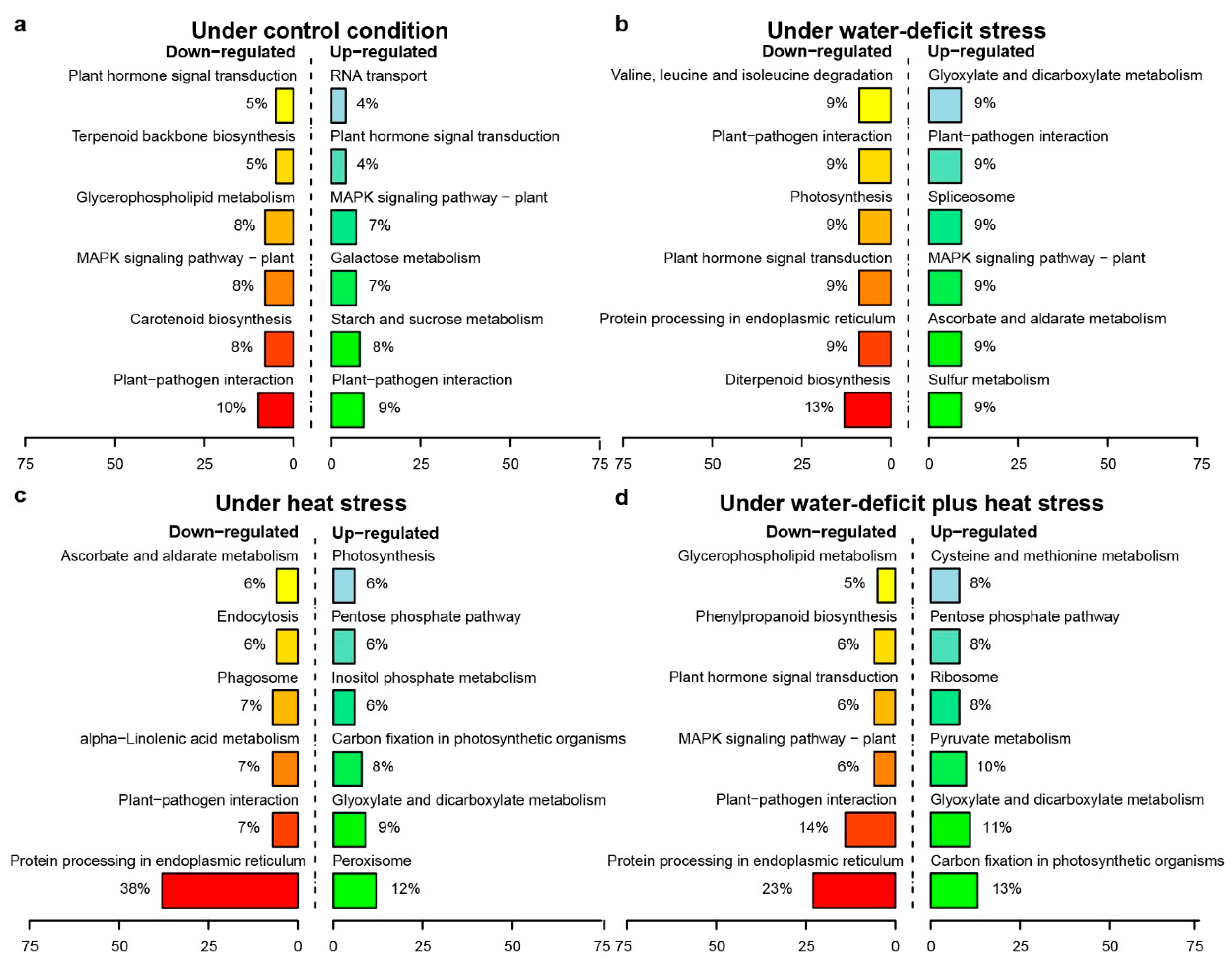
2.3. Transcriptome Sequencing and the Identification of Stress-Responsive DEGs
2.4. DEGs with Genotypic Expression
2.5. SNP (Single Nucleotide Polymorphisms) and AS (Alternative Splicing) Analysis
2.6. Degradome Signatures under Different Types of Stress
2.7. Multi-Omics Analysis: Stress-Responsive miRNA-mRNA Modules
2.8. Multi-Omics Analysis: Genotype-Dependent miRNA-mRNA Modules
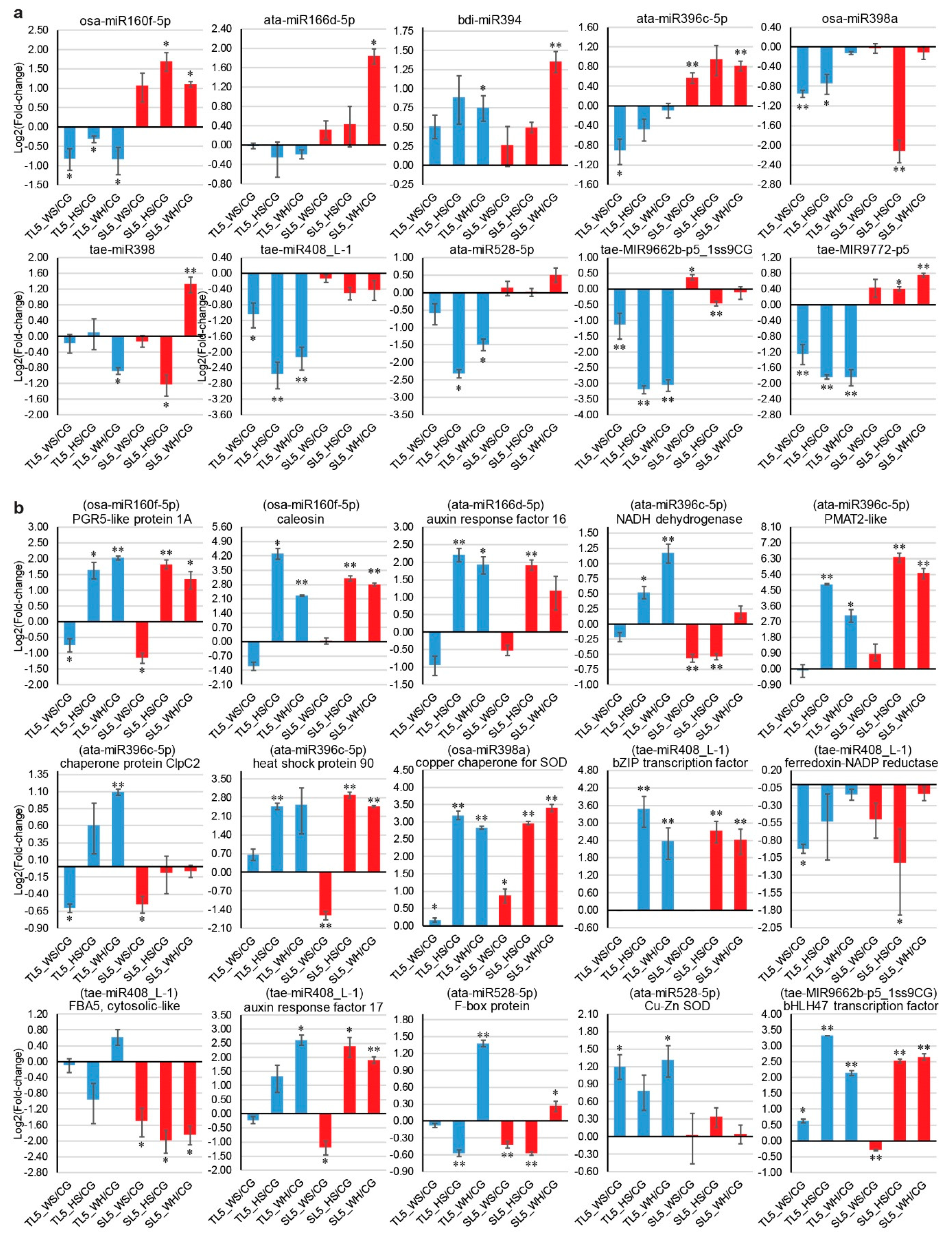
2.9. qPCR Analysis of DEMs and DEGs
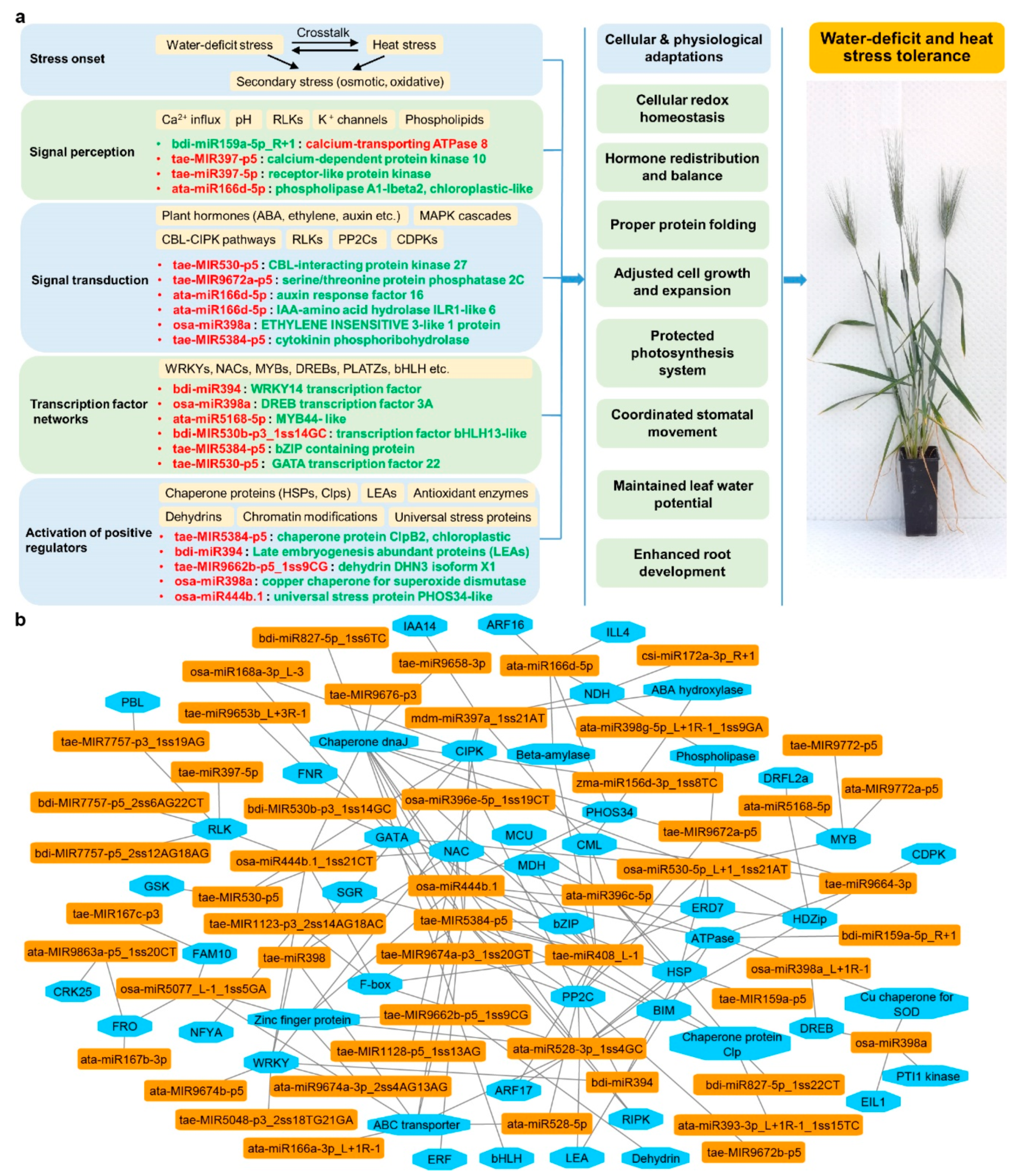
3. Discussion
3.1. miRNA Expression Specificity Provides New Insights into Their Regulatory Roles Specific to Genotype and Developmental Stage
3.2. Stress-Responsive DEMs and DEGs Reveal Synergistic Interactions between Water-Deficit Stress and Heat Stress Response
3.3. Stress-Responsive miRNA-mRNA Modules Are Valuable Attributes for Building Stress Tolerance
4. Materials and Methods
4.1. Plant Materials, Stress Treatment, and Sample Collection
4.2. Small RNA Sequencing and Data Analysis
4.3. Transcriptome Sequencing and Data Analysis
4.4. Degradome Sequencing and Data Analysis
4.5. Gene Ontology Enrichment, KEGG Pathway Analysis, and Multi-Omics Analysis
4.6. qPCR Analysis of Stress-Responsive miRNAs and Targets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barnabas, B.; Jaeger, K.; Feher, A. The effect of drought and heat stress on reproductive processes in cereals. Plant Cell Environ. 2008, 31, 11–38. [Google Scholar] [CrossRef] [PubMed]
- Sissons, M. Role of durum wheat composition on the quality of pasta and bread. Food 2008, 2, 75–90. [Google Scholar]
- Liu, H.; Bruce, D.R.; Sissons, M.; Able, A.J.; Able, J.A. Genotype-dependent changes in the phenolic content of durum under water-deficit stress. Cereal Chem. 2018, 95, 59–78. [Google Scholar] [CrossRef]
- Del Pozo, A.; Brunel-Saldias, N.; Engler, A.; Ortega-Farias, S.; Acevedo-Opazo, C.; Lobos, G.A.; Jara-Rojas, R.; Molina-Montenegro, M.A. Climate change impacts and adaptation strategies of agriculture in Mediterranean-Climate Regions (MCRs). Sustainability 2019, 11, 2769. [Google Scholar] [CrossRef]
- Moragues, M.; Del Moral, L.F.G.; Moralejo, M.; Royo, C. Yield formation strategies of durum wheat landraces with distinct pattern of dispersal within the Mediterranean basin I: Yield components. Field Crops Res. 2006, 95, 194–205. [Google Scholar] [CrossRef]
- Dong, B.; Yang, H.; Liu, H.; Qiao, Y.; Zhang, M.; Wang, Y.; Xie, Z.; Liu, M. Effects of shading stress on grain number, yield, and photosynthesis during early reproductive growth in wheat. Crop Sci. 2019, 59, 363–378. [Google Scholar] [CrossRef]
- Farooq, M.; Hussain, M.; Siddique, K.H. Drought stress in wheat during flowering and grain-filling periods. Crit. Rev. Plant Sci. 2014, 33, 331–349. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. Genotypic performance of Australian durum under single and combined water-deficit and heat stress during reproduction. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef]
- Liu, H.; Searle, I.R.; Mather, D.E.; Able, A.J.; Able, J.A. Morphological, physiological and yield responses of durum wheat to pre-anthesis water-deficit stress are genotype-dependent. Crop Pasture Sci. 2015, 66, 1024–1038. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, H.; Ji, H.; Wang, Y.; Dong, B.; Qiao, Y.; Liu, M.; Li, X. The wheat GT factor TaGT2L1D negatively regulates drought tolerance and plant development. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Dong, B.; Zheng, X.; Liu, H.; Able, J.A.; Yang, H.; Zhao, H.; Zhang, M.; Qiao, Y.; Wang, Y.; Liu, M. Effects of drought stress on pollen sterility, grain yield, abscisic acid and protective enzymes in two winter wheat cultivars. Front. Plant Sci. 2017, 8, 1008. [Google Scholar] [CrossRef] [PubMed]
- Živcak, M.; Repkova, J.; Olsovka, K.; Brestic, M. Osmotic adjustment in winter wheat varieties and its importance as a mechanism of drought tolerance. Cereal Res. Commun. 2009, 37, 569–572. [Google Scholar]
- Dias, A.; Barreiro, M.; Campos, P.; Ramalho, J.; Lidon, F. Wheat cellular membrane thermotolerance under heat stress. J. Agron. Crop Sci. 2010, 196, 100–108. [Google Scholar] [CrossRef]
- Farooq, M.; Bramley, H.; Palta, J.A.; Siddique, K.H. Heat stress in wheat during reproductive and grain-filling phases. Crit. Rev. Plant Sci. 2011, 30, 491–507. [Google Scholar] [CrossRef]
- Kumar, R.R.; Goswami, S.; Sharma, S.K.; Singh, K.; Gadpayle, K.A.; Singh, S.; Pathak, H.; Rai, R.D. Differential expression of heat shock protein and alteration in osmolyte accumulation under heat stress in wheat. J. Plant Biochem. Biotechnol. 2013, 22, 16–26. [Google Scholar] [CrossRef]
- Stone, P.; Nicolas, M. Wheat cultivars vary widely in their responses of grain yield and quality to short periods of post-anthesis heat stress. Funct. Plant Biol. 1994, 21, 887–900. [Google Scholar] [CrossRef]
- Blumenthal, C.; Batey, I.; Bekes, F.; Wrigley, C.; Barlow, E. Seasonal changes in wheat-grain quality associated with high temperatures during grain filling. Aust. J. Agric. Res. 1991, 42, 21–30. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. Genotypic water-deficit stress responses in durum wheat: Association between physiological traits, microRNA regulatory modules and yield components. Funct. Plant Biol. 2017, 44, 538–551. [Google Scholar] [CrossRef]
- Cosentino, S.L.; Sanzone, E.; Testa, G.; Patanè, C.; Anastasi, U.; Scordia, D. Does post-anthesis heat stress affect plant phenology, physiology, grain yield and protein content of durum wheat in a semi-arid Mediterranean environment? J. Agron. Crop Sci. 2019, 205, 309–323. [Google Scholar] [CrossRef]
- Guerra, D.; Crosatti, C.; Khoshro, H.H.; Mastrangelo, A.M.; Mica, E.; Mazzucotelli, E. Post-transcriptional and post-translational regulations of drought and heat response in plants: A spider’s web of mechanisms. Front. Plant Sci. 2015, 6, 57. [Google Scholar] [CrossRef]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, organization and origins. Funct. Integr. Genom. 2015, 15, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B. MicroRNA: A new target for improving plant tolerance to abiotic stress. J. Exp. Bot. 2015, 66, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. SMARTER de-stressed cereal breeding. Trends Plant Sci. 2016, 21, 909–925. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Mallory, A.C.; Vaucheret, H. Functions of microRNAs and related small RNAs in plants. Nat. Genet. 2006, 38, S31–S36. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Viswanathan, C.; Zhu, J.H.; Zhu, J.K. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007, 12, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Pagliarani, C.; Gambino, G. Small RNA mobility: Spread of RNA silencing effectors and its effect on developmental processes and stress adaptation in plants. Int. J. Mol. Sci. 2019, 20, 4306. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Kantar, M.; Bulut, R.; Akpinar, B.A. Stress responsive miRNAs and isomiRs in cereals. Plant Sci. 2015, 235, 1–13. [Google Scholar] [CrossRef]
- Alptekin, B.; Langridge, P.; Budak, H. Abiotic stress miRNomes in the Triticeae. Funct. Integr. Genom. 2016, 17, 145–170. [Google Scholar] [CrossRef]
- Wang, Y.; Li, K.; Chen, L.; Zou, Y.; Liu, H.; Tian, Y.; Li, D.; Wang, R.; Zhao, F.; Ferguson, B.J.; et al. microRNA167-directed regulation of the auxin response factors, GmARF8a and GmARF8b, is required for soybean nodulation and lateral root development. Plant Physiol. 2015, 168, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, F.; Cannarozzi, G.; Balan, B.; Siegrist, F.; Weichert, A.; Blösch, R.; Tadele, Z. Identification of miRNAs linked with the drought response of tef [Eragrostis tef (Zucc.) Trotter]. J. Plant Physiol. 2018, 224, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Fileccia, V.; Ingraffia, R.; Amato, G.; Giambalvo, D.; Martinelli, F. Identification of microRNAS differentially regulated by water deficit in relation to mycorrhizal treatment in wheat. Mol. Biol. Rep. 2019, 46, 5163–5174. [Google Scholar] [CrossRef]
- Pantaleo, V.; Vitali, M.; Boccacci, P.; Miozzi, L.; Cuozzo, D.; Chitarra, W.; Mannini, F.; Lovisolo, C.; Gambino, G. Novel functional microRNAs from virus-free and infected Vitis vinifera plants under water stress. Sci. Rep. 2016, 6, 20167. [Google Scholar] [CrossRef] [PubMed]
- Pagliarani, C.; Vitali, M.; Ferrero, M.; Vitulo, N.; Incarbone, M.; Lovisolo, C.; Valle, G.; Schubert, A. The accumulation of miRNAs differentially modulated by drought stress is affected by grafting in grapevine. Plant Physiol. 2017, 173, 2180–2195. [Google Scholar] [CrossRef]
- Visentin, I.; Pagliarani, C.; Deva, E.; Caracci, A.; Turečková, V.; Novák, O.; Lovisolo, C.; Schubert, A.; Cardinale, F. A novel strigolactone-miR156 module controls stomatal behaviour during drought recovery. Plant Cell Environ. 2020. [Google Scholar] [CrossRef]
- Liu, H.; Jia, S.; Shen, D.; Liu, J.; Li, J.; Zhao, H.; Han, S.; Wang, Y. Four AUXIN RESPONSE FACTOR genes downregulated by microRNA167 are associated with growth and development in Oryza sativa. Funct. Plant Biol. 2012, 39, 736–744. [Google Scholar] [CrossRef]
- Wang, J.W.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef]
- Sunkar, R.; Kapoor, A.; Zhu, J.-K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 2006, 18, 2051–2065. [Google Scholar] [CrossRef]
- Kumar, R.R.; Pathak, H.; Sharma, S.K.; Kala, Y.K.; Nirjal, M.K.; Singh, G.P.; Goswami, S.; Rai, R. Novel and conserved heat-responsive microRNAs in wheat (Triticum aestivum L.). Funct. Integr. Genom. 2014, 15, 323–348. [Google Scholar] [CrossRef]
- Giusti, L.; Mica, E.; Bertolini, E.; De Leonardis, A.M.; Faccioli, P.; Cattivelli, L.; Crosatti, C. microRNAs differentially modulated in response to heat and drought stress in durum wheat cultivars with contrasting water use efficiency. Funct. Integr. Genom. 2017, 17, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Bakhshi, B.; Fard, E.M.; Gharechahi, J.; Safarzadeh, M.; Nikpay, N.; Fotovat, R.; Azimi, M.R.; Salekdeh, G.H. The contrasting microRNA content of a drought tolerant and a drought susceptible wheat cultivar. J. Plant Physiol. 2017, 216, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Fard, E.M.; Bakhshi, B.; Keshavarznia, R.; Nikpay, N.; Shahbazi, M.; Salekdeh, G.H. Drought responsive microRNAs in two barley cultivars differing in their level of sensitivity to drought stress. Plant Physiol. Biochem. 2017, 118, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Zuluaga, D.L.; Liuzzi, V.; Curci, P.L.; Sonnante, G. MicroRNAs in durum wheat seedlings under chronic and short-term nitrogen stress. Funct. Integr. Genom. 2018, 18, 645–657. [Google Scholar] [CrossRef]
- Zuluaga, D.L.; De Paola, D.; Janni, M.; Curci, P.L.; Sonnante, G. Durum wheat miRNAs in response to nitrogen starvation at the grain filling stage. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Liu, H.; Searle, I.R.; Watson-Haigh, N.S.; Baumann, U.; Mather, D.E.; Able, A.J.; Able, J.A. Genome-wide identification of microRNAs in leaves and the developing head of four durum genotypes during water deficit stress. PLoS ONE 2015, 10, e0142799. [Google Scholar] [CrossRef]
- Akpinar, B.A.; Kantar, M.; Budak, H. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct. Integr. Genom. 2015, 15, 587–598. [Google Scholar] [CrossRef]
- De Paola, D.; Zuluaga, D.L.; Sonnante, G. The miRNAome of durum wheat: Isolation and characterisation of conserved and novel microRNAs and their target genes. BMC Genom. 2016, 17, 505. [Google Scholar] [CrossRef]
- Fileccia, V.; Bertolini, E.; Ruisi, P.; Giambalvo, D.; Frenda, A.S.; Cannarozzi, G.; Tadele, Z.; Crosatti, C.; Martinelli, F. Identification and characterization of durum wheat microRNAs in leaf and root tissues. Funct. Integr. Genom. 2017, 17, 583–598. [Google Scholar] [CrossRef]
- Kapustová, V.; Tulpová, Z.; Toegelová, H.; Novák, P.; Macas, J.; Karafiátová, M.; Hřibová, E.; Doležel, J.; Šimková, H. The dark matter of large cereal genomes: Long tandem repeats. Int. J. Mol. Sci. 2019, 20, 2483. [Google Scholar] [CrossRef]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.A.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the breadwheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.B.; Liu, H.; Rajamani, A.; Saha, T. Transcriptome profiling reveals genetic basis of disease resistance against Corynespora cassiicola in rubber tree (Hevea brasiliensis). Cur. Plant Biol. 2019, 17, 2–16. [Google Scholar] [CrossRef]
- Benny, J.; Pisciotta, A.; Caruso, T.; Martinelli, F. Identification of key genes and its chromosome regions linked to drought responses in leaves across different crops through meta-analysis of RNA-Seq data. BMC Plant Biol. 2019, 19, 194. [Google Scholar] [CrossRef]
- Venu, R.; Sreerekha, M.; Madhav, M.S.; Nobuta, K.; Mohan, K.M.; Chen, S.; Jia, Y.; Meyers, B.C.; Wang, G.-L. Deep transcriptome sequencing reveals the expression of key functional and regulatory genes involved in the abiotic stress signaling pathways in rice. J. Plant Biol. 2013, 56, 216–231. [Google Scholar] [CrossRef]
- Li, Y.C.; Meng, F.R.; Zhang, C.Y.; Zhang, N.; Sun, M.S.; Ren, J.P.; Niu, H.B.; Wang, X.; Yin, J. Comparative analysis of water stress-responsive transcriptomes in drought-susceptible and-tolerant wheat (Triticum aestivum L.). J. Plant Biol. 2012, 55, 349–360. [Google Scholar] [CrossRef]
- Liu, Z.; Xin, M.; Qin, J.; Peng, H.; Ni, Z.; Yao, Y.; Sun, Q. Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.). BMC Plant Biol. 2015, 15, 152. [Google Scholar] [CrossRef]
- Aprile, A.; Havlickova, L.; Panna, R.; Marè, C.; Borrelli, G.M.; Marone, D.; Perrotta, C.; Rampino, P.; De Bellis, L.; Curn, V. Different stress responsive strategies to drought and heat in two durum wheat cultivars with contrasting water use efficiency. BMC Genom. 2013, 14, 1–18. [Google Scholar] [CrossRef]
- Rampino, P.; Mita, G.; Fasano, P.; Borrelli, G.M.; Aprile, A.; Dalessandro, G.; De Bellis, L.; Perrotta, C. Novel durum wheat genes up-regulated in response to a combination of heat and drought stress. Plant Physiol. Biochem. 2012, 56, 72–78. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef]
- Folkes, L.; Moxon, S.; Woolfenden, H.C.; Stocks, M.B.; Szittya, G.; Dalmay, T.; Moulton, V. PAREsnip: A tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res. 2012, 40, e103. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ma, L.; Geng, Y.; Hao, C.; Chen, X.; Zhang, X. Small RNA and degradome sequencing reveal complex roles of miRNAs and their targets in developing wheat grains. PLoS ONE 2015, 10, e0139658. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, Z.; Mo, Q.; Zou, C.; Li, W.; Xu, Y.; Xie, C. Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann. Bot 2013, 112, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Gu, L.; Li, P.; Song, X.; Wei, L.; Chen, Z.; Cao, X. Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica). Front. Biol 2010, 5, 67–90. [Google Scholar] [CrossRef]
- Ozhuner, E.; Eldem, V.; Ipek, A.; Okay, S.; Sakcali, S.; Zhang, B.; Boke, H.; Unver, T. Boron stress responsive microRNAs and their targets in barley. PLoS ONE 2013, 8, e59543. [Google Scholar] [CrossRef]
- Zhang, X.; Xie, S.; Han, J.; Zhou, Y.; Liu, C.; Zhou, Z.; Wang, F.; Cheng, Z.; Zhang, J.; Hu, Y. Integrated transcriptome, small RNA, and degradome analysis reveals the complex network regulating starch biosynthesis in maize. BMC Genom. 2019, 20, 574. [Google Scholar] [CrossRef]
- Garg, V.; Khan, A.W.; Kudapa, H.; Kale, S.M.; Chitikineni, A.; Qiwei, S.; Sharma, M.; Li, C.; Zhang, B.; Xin, L. Integrated transcriptome, small RNA and degradome sequencing approaches provide insights into Ascochyta blight resistance in chickpea. Plant Biotech. J. 2019, 17, 914–931. [Google Scholar] [CrossRef]
- Glazińska, P.; Kulasek, M.; Glinkowski, W.; Wojciechowski, W.; Kosiński, J. Integrated analysis of small rna, transcriptome and degradome sequencing provides new insights into floral development and abscission in yellow lupine (Lupinus luteus L.). Int. J. Mol. 2019, 20, 5122. [Google Scholar] [CrossRef]
- Li, W.; Jia, Y.; Liu, F.; Wang, F.; Fan, F.; Wang, J.; Zhu, J.; Xu, Y.; Zhong, W.; Yang, J. Integration analysis of small RNA and degradome sequencing reveals microRNAs responsive to Dickeya zeae in resistant rice. Int. J. Mol. 2019, 20, 222. [Google Scholar] [CrossRef]
- Cheng, Z.; Hou, D.; Ge, W.; Li, X.; Xie, L.; Zheng, H.; Cai, M.; Liu, J.; Gao, J. Integrated mRNA, microRNA transcriptome and degradome analyses provide insights into stamen development in moso bamboo. Plant Cell Physiol. 2020, 61, 76–87. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, P.; Chen, J.; Pennerman, K.K.; Liang, X.; Yan, H.; Zhou, S.; Feng, G.; Wang, C.; Yin, G. Combinations of small RNA, RNA, and degradome sequencing uncovers the expression pattern of microRNA-mRNA pairs adapting to drought stress in leaf and root of Dactylis glomerata L. Int. J. Mol. Sci. 2018, 19, 3114. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Sun, X.; Niu, F.; Hu, Z.; Chen, R.; Zhang, H. GmDREB1 overexpression affects the expression of microRNAs in GM wheat seeds. PLoS ONE 2017, 12, e0175924. [Google Scholar] [CrossRef] [PubMed]
- Hernández, Y.; Sanan-Mishra, N. miRNA mediated regulation of NAC transcription factors in plant development and environment stress response. Plant Gene 2017, 11, 190–198. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, X.; Wang, Y.; Ming, F. The suppression of WRKY44 by GIGANTEA-miR172 pathway is involved in drought response of Arabidopsis thaliana. PLoS ONE 2013, 8, e73541. [Google Scholar] [CrossRef]
- Tang, W.; Thompson, W.A. OsmiR528 enhances cold stress tolerance by repressing expression of stress response-related transcription factor genes in plant cells. Curr. Genom. 2019, 20, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Baranwal, V.K.; Khurana, P. Genome-wide analysis of bZIP transcription factors in wheat and functional characterization of a TabZIP under abiotic stress. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef]
- Debernardi, J.M.; Rodriguez, R.E.; Mecchia, M.A.; Palatnik, J.F. Functional specialization of the plant miR396 regulatory network through distinct microRNA-target interactions. PLoS Genet 2012, 8, e1002419. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. Water-deficit stress responsive microRNAs and their targets in four durum wheat genotypes. Funct. Integr. Genom. 2016, 17, 237–251. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, S.; Mao, T.; Qu, X.; Cao, W.; Zhang, L.; Zhang, W.; He, L.; Li, S.; Ren, S. The plant-specific actin binding protein SCAB1 stabilizes actin filaments and regulates stomatal movement in Arabidopsis. Plant Cell 2011, 23, 2314–2330. [Google Scholar] [CrossRef]
- Wang, A.; Hou, Q.; Si, L.; Huang, X.; Luo, J.; Lu, D.; Zhu, J.; Shangguan, Y.; Miao, J.; Xie, Y. The PLATZ transcription factor GL6 affects grain length and number in rice. Plant Physiol. 2019, 180, 2077–2090. [Google Scholar] [CrossRef]
- Budak, H.; Khan, Z.; Kantar, M. History and current status of wheat miRNAs using next-generation sequencing and their roles in development and stress. Brief. Funct. Genom. 2015, 14, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Ragupathy, R.; Ravichandran, S.; Mahdi, M.S.R.; Huang, D.; Reimer, E.; Domaratzki, M.; Cloutier, S. Deep sequencing of wheat sRNA transcriptome reveals distinct temporal expression pattern of miRNAs in response to heat, light and UV. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, S.; Ragupathy, R.; Edwards, T.; Domaratzki, M.; Cloutier, S. MicroRNA-guided regulation of heat stress response in wheat. BMC Genom. 2019, 20, 488. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.; Molinier, J.; Chatter, W.; Laroche, A.; Kovalchuk, I. Acute but not chronic exposure to abiotic stress results in transient reduction of expression levels of the transgene driven by the 35S promoter. New Biotechnol. 2010, 27, 70–77. [Google Scholar] [CrossRef]
- Wang, X.; Vignjevic, M.; Liu, F.; Jacobsen, S.; Jiang, D.; Wollenweber, B. Drought priming at vegetative growth stages improves tolerance to drought and heat stresses occurring during grain filling in spring wheat. Plant Growth Regul. 2015, 75, 677–687. [Google Scholar] [CrossRef]
- Sussmilch, F.C.; Atallah, N.M.; Brodribb, T.J.; Banks, J.A.; McAdam, S.A. Abscisic acid (ABA) and key proteins in its perception and signaling pathways are ancient, but their roles have changed through time. Plant Signal. Behav. 2017, 12, e1365210. [Google Scholar] [CrossRef]
- Singh, A.; Jha, S.K.; Bagri, J.; Pandey, G.K. ABA inducible rice protein phosphatase 2C confers ABA insensitivity and abiotic stress tolerance in Arabidopsis. PLoS ONE 2015, 10, e0125168. [Google Scholar] [CrossRef]
- Malefo, M.; Mathibela, E.; Crampton, B.; Makgopa, M. Investigating the role of Bowman-Birk serine protease inhibitor in Arabidopsis plants under drought stress. Plant Physiol. Biochem. 2020, 149, 286–293. [Google Scholar] [CrossRef]
- Shah, N.; Paulsen, G. Interaction of drought and high temperature on photosynthesis and grain-filling of wheat. Plant Soil 2003, 257, 219–226. [Google Scholar] [CrossRef]
- Ashoub, A.; Baeumlisberger, M.; Neupaertl, M.; Karas, M.; Brüggemann, W. Characterization of common and distinctive adjustments of wild barley leaf proteome under drought acclimation, heat stress and their combination. Plant Mol. Biol. 2015, 87, 459–471. [Google Scholar] [CrossRef]
- Liu, M.; Yu, H.; Zhao, G.; Huang, Q.; Lu, Y.; Ouyang, B. Profiling of drought-responsive microRNA and mRNA in tomato using high-throughput sequencing. BMC Genom. 2017, 18, 481. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, Q.; Chen, K.; Zhao, S.; Zhang, C.; Pan, R.; Cai, T.; Deng, Y.; Wang, X.; Chen, Y. Integrated microRNA and transcriptome profiling reveals a miRNA-mediated regulatory network of embryo abortion under calcium deficiency in peanut (Arachis hypogaea L.). BMC Genom. 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.N. Physiology of PSI cyclic electron transport in higher plants. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, H.; Ballester, A.; Muñoz, R.; Quiles, M.J. Chlororespiration and tolerance to drought, heat and high illumination. J. Plant Physiol. 2010, 167, 732–738. [Google Scholar] [CrossRef]
- Paredes, M.; Quiles, M.J. Stimulation of chlororespiration by drought under heat and high illumination in Rosa meillandina. J. Plant Physiol. 2013, 170, 165–171. [Google Scholar] [CrossRef]
- Muñoz, R.; Quiles, M.J. Water deficit and heat affect the tolerance to high illumination in hibiscus plants. Int. J. Mol. Sci. 2013, 14, 5432–5444. [Google Scholar] [CrossRef]
- Flores-Pérez, Ú.; Bédard, J.; Tanabe, N.; Lymperopoulos, P.; Clarke, A.K.; Jarvis, P. Functional analysis of the Hsp93/ClpC chaperone at the chloroplast envelope. Plant Physiol. 2016, 170, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, H.; Junaid, A.; Manjhi, J.; Singh, G.P.; Gaikwad, K.; Padaria, J.C. Heat stress transcripts, differential expression, and profiling of heat stress tolerant gene TaHsp90 in Indian wheat (Triticum aestivum L.) cv C306. PLoS ONE 2018, 13, e0198293. [Google Scholar] [CrossRef]
- Xu, J.; Xue, C.; Xue, D.; Zhao, J.; Gai, J.; Guo, N.; Xing, H. Overexpression of GmHsp90s, a heat shock protein 90 (Hsp90) gene family cloning from soybean, decrease damage of abiotic stresses in Arabidopsis thaliana. PLoS ONE 2013, 8, e69810. [Google Scholar] [CrossRef]
- Li, Q.; Wang, W.; Wang, W.; Zhang, G.; Liu, Y.; Wang, Y.; Wang, W. Wheat F-box protein gene TaFBA1 is involved in plant tolerance to heat stress. Front. Plant Sci. 2018, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-M.; Kong, X.-Z.; Kang, H.-H.; Sun, X.-D.; Wang, W. The involvement of wheat F-box protein gene TaFBA1 in the oxidative stress tolerance of plants. PLoS ONE 2015, 10, e0122117. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Sun, X.; Yin, S.; Kong, X.; Zhou, S.; Xu, Y.; Luo, Y.; Wang, W. The role of the F-box gene TaFBA1 from wheat (Triticum aestivum L.) in drought tolerance. Plant Physiol. Biochem. 2014, 84, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Cohu, C.M.; Abdel-Ghany, S.E.; Reynolds, K.A.G.; Onofrio, A.M.; Bodecker, J.R.; Kimbrel, J.A.; Niyogi, K.K.; Pilon, M. Copper delivery by the copper chaperone for chloroplast and cytosolic copper/zinc-superoxide dismutases: Regulation and unexpected phenotypes in an Arabidopsis mutant. Mol. Plant 2009, 2, 1336–1350. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-C.; Lee, W.-C.; Guo, W.-Y.; Pan, S.-M.; Chen, L.-J.; Li, H.-m.; Jinn, T.-L. A copper chaperone for superoxide dismutase that confers three types of copper/zinc superoxide dismutase activity in Arabidopsis. Plant Physiol. 2005, 139, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Bartel, D.P.; Bartel, B. MicroRNA-directed regulation of Arabidopsis Auxin Response Factor17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef]
- Sharma, L.; Dalal, M.; Verma, R.K.; Kumar, S.V.; Yadav, S.K.; Pushkar, S.; Kushwaha, S.R.; Bhowmik, A.; Chinnusamy, V. Auxin protects spikelet fertility and grain yield under drought and heat stresses in rice. Environ. Exp. Bot. 2018, 150, 9–24. [Google Scholar] [CrossRef]
- Wang, B.; Xue, J.-S.; Yu, Y.-H.; Liu, S.-Q.; Zhang, J.-X.; Yao, X.-Z.; Liu, Z.-X.; Xu, X.-F.; Yang, Z.-N. Fine regulation of ARF17 for anther development and pollen formation. BMC Plant Biol. 2017, 17, 243. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. Transgenerational effects of water-deficit and heat stress on germination and seedling vigour—new insights from durum wheat microRNAs. Plants 2020, 9, 189. [Google Scholar] [CrossRef]
- Li, X.; Shahid, M.Q.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative small RNA analysis of pollen development in autotetraploid and diploid rice. Int. J. Mol. Sci. 2016, 17, 499. [Google Scholar] [CrossRef]
- Zhong, M.; Huang, F.; Luo, R.; Lv, Y.; Ali, U.; Sheng, Z.; Tang, S.; Wei, X.; Hu, P. The effect of cadmium on the microRNAome, degradome and transcriptome of rice seedlings. Plant Growth Regul. 2020, 90, 15–27. [Google Scholar] [CrossRef]
- Sun, W.; Xu, X.H.; Wu, X.; Wang, Y.; Lu, X.; Sun, H.; Xie, X. Genome-wide identification of microRNAs and their targets in wild type and phyB mutant provides a key link between microRNAs and the phyB-mediated light signaling pathway in rice. Front. Plant Sci. 2015, 6, 372. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Snyder, J.A.; Park, Y.B.; Li, Y.-F.; Sunkar, R.; Axtell, M.J. Sliced microRNA targets and precise loop-first processing of MIR319 hairpins revealed by analysis of the Physcomitrella patens degradome. RNA 2009, 15, 2112–2121. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviations | Description |
|---|---|
| CG | Control group |
| WS | Pre-anthesis water-deficit group |
| HS | Post-anthesis heat stress group |
| WH | Pre-anthesis water-deficit combined with post-anthesis heat stress group |
| TL | Libraries made from leaf tissue of the stress-tolerant genotype DBA Aurora |
| SL | Libraries made from leaf tissue of the stress-sensitive genotype L6 |
| 5, 15, 25, 35, 45 | Time-points of sampling (DPA, days post-anthesis). |
| miRNA | Target Accession | Target Annotation | miRNA log2 (FC) | Target log2 (FC) | Stress Type | Pairing Type | TFs |
|---|---|---|---|---|---|---|---|
| osa-miR398a | TRITD_1Bv1G135100-4 | DREB 1 transcription factor 3A | −1.23 | 2.95 | WS | Newly discovered | DREB |
| −2.67 | 1.75 | HS | Newly discovered | ||||
| −1.34 | 1.75 | WH | Newly discovered | ||||
| mdm-miR397a_1ss21AT | TRITD_2Bv1G034790 | NAC 1 transcription factor (plastid) | −inf 2 | 3.74 | HS | Newly discovered | NAC |
| osa-miR398a_L+1R-1 | TRITD_3Av1G235010-2 | NAC transcription factor 6A | −2.05 | 4.13 | HS | Newly discovered | |
| −0.92 | 2.58 | WH | Newly discovered | ||||
| osa-miR444b.1 | TRITD_2Av1G026820 | NAC transcription factor (plastid) | −1.77 | 3.40 | HS | Newly discovered | |
| −1.50 | 1.94 | WH | Newly discovered | ||||
| TRITD_5Av1G170450-2 | NAC domain-containing protein 41 | −1.50 | 2.90 | WH | Newly discovered | ||
| TRITD_5Bv1G161590 | NAC domain-containing protein 41 | −1.50 | 3.59 | WH | Newly discovered | ||
| tae-MIR1128-p5_1ss13AG | TRITD_4Bv1G108860 | NAC domain-containing protein 92-like | −inf | 4.16 | HS | Newly discovered | |
| tae-MIR5384-p5 | TRITD_5Bv1G161590 | NAC domain-containing protein 41 | −1.98 | 3.59 | WH | Newly discovered | |
| ata-miR166d-5p | TRITD_7Av1G051530-5 | auxin response factor 16 | −2.37 | 1.60 | WH | Newly discovered | ARF 1 |
| osa-miR167a-5p_R+1 | TRITD_6Av1G046710 | auxin response factor 6-like | −1.00 | 1.59 | HS | Conserved | |
| tae-miR408_L-1 | TRITD_7Bv1G194180-3 | auxin response factor 17 | −3.62 | 2.21 | WH | Newly discovered | |
| ata-miR528-3p_1ss4GC | TRITD_7Bv1G194180-3 | auxin response factor 17 | −inf | 1.81 | HS | Newly discovered | |
| −2.60 | 2.21 | WH | Newly discovered | ||||
| TRITD_7Av1G245930-4 | auxin response factor 17 | −2.60 | 1.79 | WH | Newly discovered | ||
| bdi-miR394 | TRITD_1Bv1G030900-5 | WRKY14 1 transcription factor | −0.76 | 1.76 | WH | Newly discovered | WRKY |
| osa-miR396e-5p_1ss19CT | TRITD_5Bv1G146000-2 | probable WRKY transcription factor 2 | −2.32 | 2.04 | WH | Newly discovered | |
| tae-miR398 | TRITD_7Av1G028020 | putative WRKY transcription factor 72 | −inf | 1.74 | HS | Newly discovered | |
| osa-miR444b.1_1ss21CT | TRITD_3Av1G213210 | putative WRKY transcription factor 33 | −1.32 | 2.58 | HS | Newly discovered | |
| tae-MIR5048-p3_2ss18TG21GA | TRITD_3Av1G074690-4 | WRKY transcription factor, partial | −0.66 | 1.63 | HS | Newly discovered | |
| ata-MIR9674b-p5 | TRITD_3Av1G213210 | putative WRKY transcription factor 33 | −3.53 | 2.58 | HS | Newly discovered | |
| TRITD_3Bv1G195790 | WRKY27 transcription factor | −3.53 | 1.88 | HS | Newly discovered | ||
| osa-miR530-5p_L+1_1ss21AT | TRITD_5Av1G046770 | MYB 1 transcription factor 79 | −inf | 4.00 | HS | Newly discovered | MYB |
| ata-miR5168-5p | TRITD_5Av1G118710 | transcription factor MYB44-like | −inf | 4.57 | HS | Newly discovered | |
| −inf | 2.67 | WH | Newly discovered | ||||
| tae-MIR9772-p5 | TRITD_5Av1G207760-4 | MYB-related protein | −0.66 | 2.95 | HS | Newly discovered | |
| tae-miR408_L-1 | TRITD_6Av1G199570-2 | bZIP 1 transcription factor | −3.55 | 3.69 | HS | Newly discovered | bZIP |
| tae-MIR5384-p5 | TRITD_3Bv1G212830 | bZIP domain containing protein | −1.93 | 2.98 | HS | Newly discovered | |
| −1.98 | 1.97 | WH | Newly discovered | ||||
| bdi-MIR530b-p3_1ss14GC | TRITD_3Av1G067970 | bHLH13-like 1 | −inf | 2.14 | HS | Newly discovered | bHLH |
| tae-MIR9662b-p5_1ss9CG | TRITD_2Av1G047140 | bHLH47 | −1.66 | 2.42 | HS | Newly discovered |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Able, A.J.; Able, J.A. Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the Water-Deficit and Heat Stress Response Network in Durum Wheat. Int. J. Mol. Sci. 2020, 21, 6017. https://doi.org/10.3390/ijms21176017
Liu H, Able AJ, Able JA. Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the Water-Deficit and Heat Stress Response Network in Durum Wheat. International Journal of Molecular Sciences. 2020; 21(17):6017. https://doi.org/10.3390/ijms21176017
Chicago/Turabian StyleLiu, Haipei, Amanda J. Able, and Jason A. Able. 2020. "Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the Water-Deficit and Heat Stress Response Network in Durum Wheat" International Journal of Molecular Sciences 21, no. 17: 6017. https://doi.org/10.3390/ijms21176017
APA StyleLiu, H., Able, A. J., & Able, J. A. (2020). Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the Water-Deficit and Heat Stress Response Network in Durum Wheat. International Journal of Molecular Sciences, 21(17), 6017. https://doi.org/10.3390/ijms21176017