Microglial Phagocytosis—Rational but Challenging Therapeutic Target in Multiple Sclerosis



Abstract
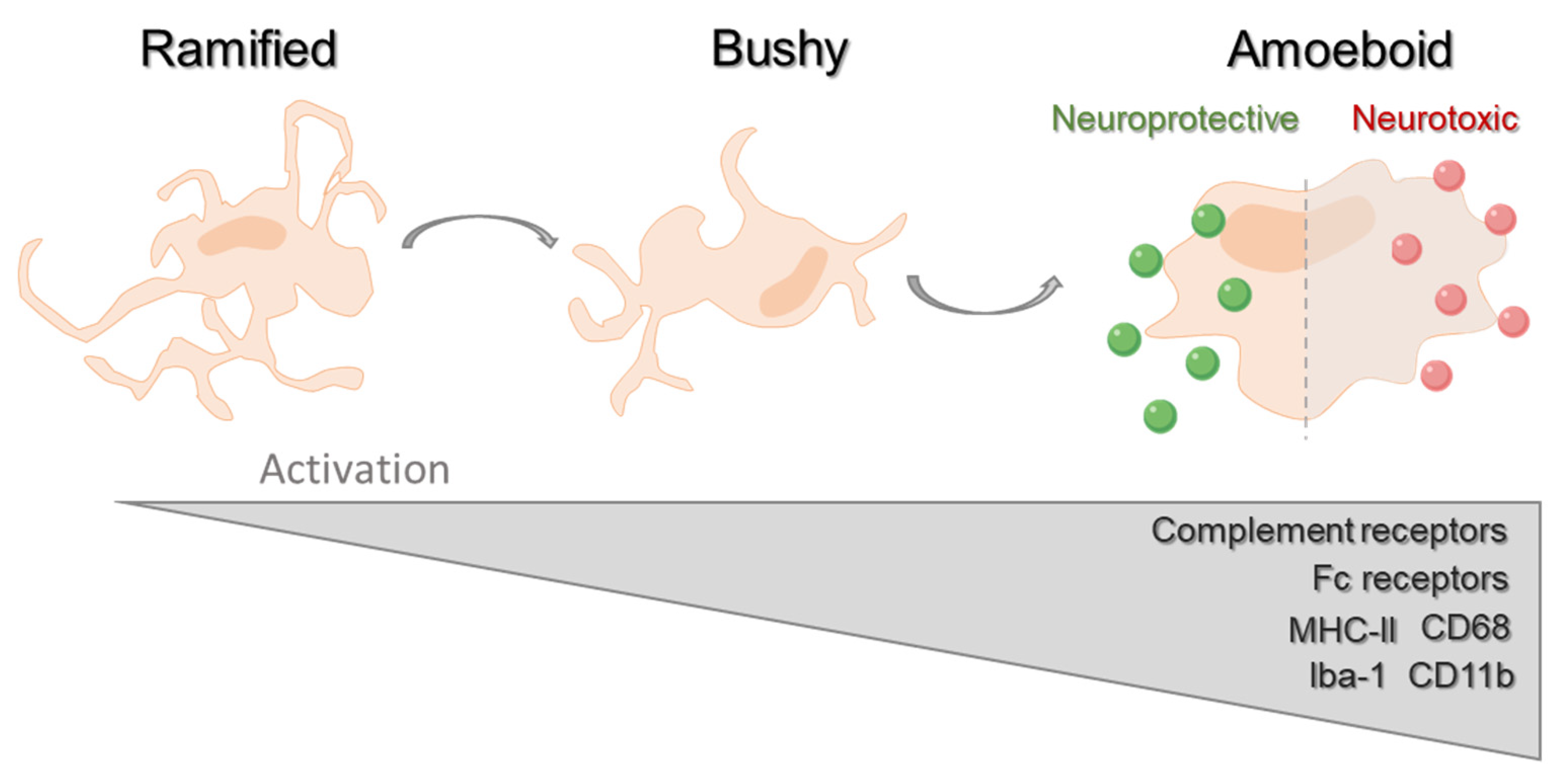
1. Introduction
2. Microglial Phagocytosis
2.1. Microglial Phagocytosis during Neurodevelopment
2.2. Microglial Phagocytosis in Multiple Sclerosis: From Targets to Therapeutic Strategies
2.2.1. Phagocytosis and Myelin Clearance
2.2.2. Phagocytosis and Microglial Inflammatory Profile
2.2.3. Phagocytosis and Cognitive Impairment
3. Conclusions
Funding
Conflicts of Interest
References
- WHO. Atlas Multiple Sclerosis resources. In The World 2008; WHO Press: Geneva, Switzerland, 2008; Volume 56. [Google Scholar]
- Bogie, J.F.; Stinissen, P.; Hendriks, J.J. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Uleng Bahrun, C.W. Immunology of multiple sclerosis. Indones. J. Clin. Pathol. Med Lab. 2018, 24, 191–195. [Google Scholar] [CrossRef][Green Version]
- Yamout, B.I.; Alroughani, R. Multiple sclerosis. Semin. Neurol. 2018, 38, 212–225. [Google Scholar] [CrossRef]
- Weil, M.T.; Mobius, W.; Winkler, A.; Ruhwedel, T.; Wrzos, C.; Romanelli, E.; Bennett, J.L.; Enz, L.; Goebels, N.; Nave, K.A.; et al. Loss of myelin basic protein function triggers myelin breakdown in models of demyelinating diseases. Cell Rep. 2016, 16, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Grigoriadis, N.; van Pesch, V.; Paradig, M.S.G. A basic overview of multiple sclerosis immunopathology. Eur. J. Neurol. 2015, 22 (Suppl. 2), 3–13. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. Animal models of multiple sclerosis: The good, the bad and the bottom line. Nat. Neurosci. 2012, 15, 1074–1077. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Hojati, Z.; Kay, M.; Dehghanian, F. Mechanism of action of interferon beta in treatment of multiple sclerosis. In Multiple Sclerosis; Academic Press: Cambridge, MA, USA, 2016; pp. 365–392. [Google Scholar] [CrossRef]
- Tselis, A.; Khan, O.; Lisak, R.P. Glatiramer acetate in the treatment of multiple sclerosis. Neuropsychiatr. Dis. Treat. 2007, 3, 259–267. [Google Scholar] [CrossRef]
- Racke, M.K.; Lovett-Racke, A.E.; Karandikar, N.J. The mechanism of action of glatiramer acetate treatment in multiple sclerosis. Neurology 2010, 74, S25–S30. [Google Scholar] [CrossRef]
- Ayzenberg, I.; Hoepner, R.; Kleiter, I. Fingolimod for multiple sclerosis and emerging indications: Appropriate patient selection, safety precautions, and special considerations. Ther. Clin. Risk Manag. 2016, 12, 261–272. [Google Scholar] [CrossRef]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Theodore Phillips, J.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef]
- Brandstadter, R.; Katz Sand, I. The use of natalizumab for multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1691–1702. [Google Scholar] [CrossRef]
- Brown, J.W.; Coles, A.J. Alemtuzumab: Evidence for its potential in relapsing-remitting multiple sclerosis. Drug Des. Dev. Ther. 2013, 7, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Mulero, P.; Midaglia, L.; Montalban, X. Ocrelizumab: A new milestone in multiple sclerosis therapy. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418773025. [Google Scholar] [CrossRef]
- Chari, D.M. Remyelination in multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 589–620. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef]
- Moyon, S.; Dubessy, A.L.; Aigrot, M.S.; Trotter, M.; Huang, J.K.; Dauphinot, L.; Potier, M.C.; Kerninon, C.; Melik Parsadaniantz, S.; Franklin, R.J.; et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 4–20. [Google Scholar] [CrossRef]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Goldman, S.A. Glia disease and repair-remyelination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020594. [Google Scholar] [CrossRef] [PubMed]
- McMurran, C.E.; Jones, C.A.; Fitzgerald, D.C.; Franklin, R.J. CNS Remyelination and the innate immune system. Front. Cell Dev. Biol. 2016, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef]
- Cardona, A.E.; Huang, D.; Sasse, M.E.; Ransohoff, R.M. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat. Protoc. 2006, 1, 1947–1951. [Google Scholar] [CrossRef]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Soulet, D.; Rivest, S. Bone-marrow-derived microglia: Myth or reality? Curr. Opin. Pharmacol. 2008, 8, 508–518. [Google Scholar] [CrossRef]
- Ling, E.; Penney, D.; Leblond, C. Use of carbon labeling to demonstrate the role of blood monocytes as precursors of the ‘Ameboid Cells’ present in the corpus callosum of postnatal rats. J. Comp. Neurol. 1980, 193, 631–657. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- von Bernhardi, R.; Heredia, F.; Salgado, N.; Munoz, P. Microglia function in the normal brain. Adv. Exp. Med. Biol. 2016, 949, 67–92. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia actively regulate the number of functional synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.H.; Warden, S.; Lenz, K.M. Sex differences in microglial phagocytosis in the neonatal hippocampus. Brain Behav. Immun. 2017, 64, 11–22. [Google Scholar] [CrossRef] [PubMed]
- VanRyzin, J.W.; Marquardt, A.E.; Argue, K.J.; Vecchiarelli, H.A.; Ashton, S.E.; Arambula, S.E.; Hill, M.N.; McCarthy, M.M. Microglial phagocytosis of newborn cells is induced by endocannabinoids and sculpts sex differences in juvenile rat social play. Neuron 2019, 102, 435–449.e6. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, R.S.; Wing, M.G.; Compston, A. Nonactivated microglia promote oligodendrocyte precursor survival and maturation through the transcription factor NF-kappa B. Eur. J. Neurosci. 2001, 13, 959–967. [Google Scholar] [CrossRef]
- Pasquini, L.A.; Millet, V.; Hoyos, H.C.; Giannoni, J.P.; Croci, D.O.; Marder, M.; Liu, F.T.; Rabinovich, G.A.; Pasquini, J.M. Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death Differ. 2011, 18, 1746–1756. [Google Scholar] [CrossRef]
- Hoyos, H.C.; Rinaldi, M.; Mendez-Huergo, S.P.; Marder, M.; Rabinovich, G.A.; Pasquini, J.M.; Pasquini, L.A. Galectin-3 controls the response of microglial cells to limit cuprizone-induced demyelination. Neurobiol. Dis. 2014, 62, 441–455. [Google Scholar] [CrossRef]
- Orr, A.G.; Orr, A.L.; Li, X.J.; Gross, R.E.; Traynelis, S.F. Adenosine A(2A) receptor mediates microglial process retraction. Nat. Neurosci. 2009, 12, 872–878. [Google Scholar] [CrossRef]
- Stence, N.; Waite, M.; Dailey, M.E. Dynamics of microglial activation: A confocal time-lapse analysis in hippocampal slices. Glia 2001, 33, 256–266. [Google Scholar] [CrossRef]
- Lynch, M.A. The multifaceted profile of activated microglia. Mol. Neurobiol. 2009, 40, 139–156. [Google Scholar] [CrossRef]
- Hendrickx, D.A.E.; van Eden, C.G.; Schuurman, K.G.; Hamann, J.; Huitinga, I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J. Neuroimmunol. 2017, 309, 12–22. [Google Scholar] [CrossRef]
- Gomes-Leal, W. Microglial physiopathology: How to explain the dual role of microglia after acute neural disorders? Brain Behav. 2012, 2, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Jung, S.; Priller, J. Microglia biology: One century of evolving concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Haider, L.; Fischer, M.T.; Frischer, J.M.; Bauer, J.; Hoftberger, R.; Botond, G.; Esterbauer, H.; Binder, C.J.; Witztum, J.L.; Lassmann, H. Oxidative damage in multiple sclerosis lesions. Brain J. Neurol. 2011, 134, 1914–1924. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, J.X.; Zhang, G.X. Macrophages: A double-edged sword in experimental autoimmune encephalomyelitis. Immunol. Lett. 2014, 160, 17–22. [Google Scholar] [CrossRef]
- Abourbeh, G.; Theze, B.; Maroy, R.; Dubois, A.; Brulon, V.; Fontyn, Y.; Dolle, F.; Tavitian, B.; Boisgard, R. Imaging microglial/macrophage activation in spinal cords of experimental autoimmune encephalomyelitis rats by positron emission tomography using the mitochondrial 18 kDa translocator protein radioligand [(1)(8)F]DPA-714. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 5728–5736. [Google Scholar] [CrossRef]
- Zhang, B.; Wei, Y.Z.; Wang, G.Q.; Li, D.D.; Shi, J.S.; Zhang, F. Targeting MAPK pathways by naringenin modulates microglia M1/M2 polarization in lipopolysaccharide-stimulated cultures. Front. Cell. Neurosci. 2018, 12, 531. [Google Scholar] [CrossRef]
- Wang, J.; Qi, Y.; Niu, X.; Tang, H.; Meydani, S.N.; Wu, D. Dietary naringenin supplementation attenuates experimental autoimmune encephalomyelitis by modulating autoimmune inflammatory responses in mice. J. Nutr. Biochem. 2018, 54, 130–139. [Google Scholar] [CrossRef]
- Djedovic, N.; Stanisavljevic, S.; Jevtic, B.; Momcilovic, M.; Lavrnja, I.; Miljkovic, D. Anti-encephalitogenic effects of ethyl pyruvate are reflected in the central nervous system and the gut. Biomed. Pharmacother. 2017, 96, 78–85. [Google Scholar] [CrossRef]
- Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Kuznetsova, I.S.; Pomytkin, I.; Lyundup, A.; Strekalova, T.; Barteneva, N.S.; Ponomarev, E.D. Cyclic AMP pathway suppress autoimmune neuroinflammation by inhibiting functions of Encephalitogenic CD4 T cells and enhancing M2 macrophage polarization at the site of inflammation. Front. Immunol. 2018, 9, 50. [Google Scholar] [CrossRef]
- Fan, C.; Long, R.; You, Y.; Wang, J.; Yang, X.; Huang, S.; Sheng, Y.; Peng, X.; Liu, H.; Wang, Z.; et al. A novel PADRE-Kv1.3 vaccine effectively induces therapeutic antibodies and ameliorates experimental autoimmune encephalomyelitis in rats. Clin. Immunol. 2018, 193, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Yu, Z.; Fang, X.; Liu, M.; Pu, Y.; Shao, Q.; Wang, D.; Zhao, X.; Huang, A.; Xiang, Z.; et al. LncRNA GAS5 inhibits microglial M2 polarization and exacerbates demyelination. EMBO Rep. 2017, 18, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Wang, J.; Yang, B.; Weng, Q.; He, Q. Targeting microglia and macrophages: A potential treatment strategy for multiple sclerosis. Front. Pharmacol. 2019, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.L. Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol. Neurobiol. 2017, 54, 7567–7584. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.R.; Franklin, R.J. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain J. Neurol. 2009, 132, 288–295. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Prefontaine, P.; Plante, M.M.; Sanchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.E.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the brain: Homeostasis and disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef]
- Tremblay, M.E.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Francesca, P.; Laura, M.; Maria, S.; Patrizia, P.; Maurizio, G.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Dissing-Olesen, L.; LeDue, J.M.; Rungta, R.L.; Hefendehl, J.K.; Choi, H.B.; MacVicar, B.A. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 10511–10527. [Google Scholar] [CrossRef]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.E.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Stevens, B. Phagocytic glial cells: Sculpting synaptic circuits in the developing nervous system. Curr. Opin. Neurobiol. 2013, 23, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Bernardinelli, Y.; Nikonenko, I.; Muller, D. Structural plasticity: Mechanisms and contribution to developmental psychiatric disorders. Front. Neuroanat. 2014, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Haberl, M.; Zerbi, V.; Veltien, A.; Ginger, M.; Heerschap, A.; Frick, A. Structural-functional connectivity deficits of neocortical circuits in the Fmr1−/y mouse model of autism. Funct. Neuroimaging 2015, 1, e1500775. [Google Scholar] [CrossRef]
- Schreiner, M.J.; Karlsgodt, K.H.; Uddin, L.Q.; Chow, C.; Congdon, E.; Jalbrzikowski, M.; Bearden, C.E. Default mode network connectivity and reciprocal social behavior in 22q11.2 deletion syndrome. Soc. Cognit. Affect. Neurosci. 2014, 9, 1261–1267. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Anderson, S.R.; Zhang, J.; Steele, M.R.; Romero, C.O.; Kautzman, A.G.; Schafer, D.P.; Vetter, M.L. Complement targets newborn retinal ganglion cells for phagocytic elimination by microglia. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 2025–2040. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; Mcnamara, R.K.; Streiti, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Neurobiology 1998, 95, 10896–10901. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 2018, 48, 979–991.e8. [Google Scholar] [CrossRef]
- Casano, A.M.; Albert, M.; Peri, F. Developmental apoptosis mediates entry and positioning of microglia in the Zebrafish Brain. Cell Rep. 2016, 16, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Blume, Z.I.; Lambert, J.M.; Lovel, A.G.; Mitchell, D.M. Microglia in the developing retina couple phagocytosis with the progression of apoptosis via P2RY12 signaling. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2020, 249, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Fourgeaud, L.; Traves, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagorska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Aparicio, I.; Sierra, A. C1q is related to microglial phagocytosis in the hippocampus in physiological conditions. Matters 2019, 5, e201904000013. [Google Scholar] [CrossRef]
- Perez-Pouchoulen, M.; Yu, S.J.; Roby, C.R.; Bonsavage, N.; McCarthy, M.M. Regulatory control of microglial phagocytosis by estradiol and prostaglandin E2 in the developing rat cerebellum. Cerebellum 2019, 18, 882–895. [Google Scholar] [CrossRef]
- Chen, M.S.; Huber, A.B.; van der Haar, M.E.; Frank, M.; Schnell, L.; Spillmann, A.A.; Christ, F.; Schwab, M.E. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 2000, 403, 434–439. [Google Scholar] [CrossRef]
- Plemel, J.R.; Manesh, S.B.; Sparling, J.S.; Tetzlaff, W. Myelin inhibits oligodendroglial maturation and regulates oligodendrocytic transcription factor expression. Glia 2013, 61, 1471–1487. [Google Scholar] [CrossRef]
- McKerracher, L.; David, S.; Jackson, D.L.; Kottis, V.; Dunn, R.J.; Braun, P.E. Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 1994, 13, 805–811. [Google Scholar] [CrossRef]
- Brück, W.; Friede, R.L. Anti-macrophage CR3 antibody blocks myelin phagocytosis by macrophages in vitro. Acta Neuropathol. 1990, 80, 415–418. [Google Scholar] [CrossRef]
- Reichert, F.; Rotshenker, S. Complement-receptor-3 and scavenger-receptor-AI/II mediated myelin phagocytosis in microglia and macrophages. Neurobiol. Dis. 2003, 12, 65–72. [Google Scholar] [CrossRef]
- Poliani, P.L.; Wang, Y.; Fontana, E.; Robinette, M.L.; Yamanishi, Y.; Gilfillan, S.; Colonna, M. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Investig. 2015, 125, 2161–2170. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Buonsanti, C.; Mariani, M.; Cella, M.; Gilfillan, S.; Cross, A.H.; Colonna, M.; Panina-Bordignon, P. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2007, 37, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.M.; Perron, G.; Won, S.Y.; Michell-Robinson, M.A.; Rezk, A.; Ludwin, S.K.; Moore, C.S.; Hall, J.A.; Bar-Or, A.; Antel, J.P. MerTK is a functional regulator of myelin phagocytosis by human myeloid cells. J. Immunol. 2016, 196, 3375–3384. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.M.; Jang, J.H.; Won, S.Y.; Lin, Y.H.; Touil, H.; Aljarallah, S.; Bar-Or, A.; Antel, J.P. MerTK-mediated regulation of myelin phagocytosis by macrophages generated from patients with MS. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e402. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, H.; Pu, H.; Wang, G.; Li, W.; Leak, R.K.; Chen, J.; Liou, A.K.; Hu, X. n-3 PUFA supplementation benefits microglial responses to myelin pathology. Sci. Rep. 2014, 4, 7458. [Google Scholar] [CrossRef]
- Zorina, Y.; Stricker, J.; Caggiano, A.O.; Button, D.C. Human IgM antibody rHIgM22 promotes phagocytic clearance of myelin debris by microglia. Sci. Rep. 2018, 8, 9392. [Google Scholar] [CrossRef]
- Warrington, A.E.; Bieber, A.J.; Ciric, B.; Pease, L.; Van Keulen, V.P.; Rodriguez, M. A recombinant human IgM promotes myelin repair after a single, very low dose. J. Neurosci. Res. 2007, 85, 967–976. [Google Scholar] [CrossRef]
- Mullin, A.P.; Cui, C.; Wang, Y.; Wang, J.; Troy, E.; Caggiano, A.O.; Parry, T.J.; Colburn, R.W.; Pavlopoulos, E. rHIgM22 enhances remyelination in the brain of the cuprizone mouse model of demyelination. Neurobiol. Dis. 2017, 105, 142–155. [Google Scholar] [CrossRef]
- Cui, C.; Wang, J.; Mullin, A.P.; Caggiano, A.O.; Parry, T.J.; Colburn, R.W.; Pavlopoulos, E. The antibody rHIgM22 facilitates hippocampal remyelination and ameliorates memory deficits in the cuprizone mouse model of demyelination. Brain Res. 2018, 1694, 73–86. [Google Scholar] [CrossRef]
- Mecha, M.; Yanguas-Casas, N.; Feliu, A.; Mestre, L.; Carrillo-Salinas, F.; Azcoitia, I.; Yong, V.W.; Guaza, C. The endocannabinoid 2-AG enhances spontaneous remyelination by targeting microglia. Brain Behav. Immun. 2019, 77, 110–126. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, C.; Hou, Z.; Fu, X.; Yuan, L.; Sun, S.; Zhang, H.; Yang, D.; Yao, X.; Yang, J. Pseudoginsenoside-F11 accelerates microglial phagocytosis of myelin debris and attenuates cerebral ischemic injury through complement receptor 3. Neuroscience 2020, 426, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.F.J.; Jorissen, W.; Mailleux, J.; Nijland, P.G.; Zelcer, N.; Vanmierlo, T.; Van Horssen, J.; Stinissen, P.; Hellings, N.; Hendriks, J. Myelin alters the inflammatory phenotype of macrophages by activating PPARs. Acta Neuropathol. Commun. 2013, 1, 43. [Google Scholar] [CrossRef]
- Kopper, T.J.; Gensel, J.C. Myelin as an inflammatory mediator: Myelin interactions with complement, macrophages, and microglia in spinal cord injury. J. Neurosci. Res. 2018, 96, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Ishikawa, T.; Griep, A.; Axt, D.; Kummer, M.P.; Heneka, M.T. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 17321–17331. [Google Scholar] [CrossRef]
- Yuan Zhang, Y.; Li, X.; Ciric, B.; Curtis, M.; Chen, W.-J.; Rostami, A.; Zhang, G. A dual effect of ursolic acid to the treatment of multiple sclerosis through both immunomodulation and direct remyelination. Proc. Natl. Acad. Sci. USA 2020, 117, 9082–9093. [Google Scholar] [CrossRef]
- Olin, J.; Schneider, L. Galantamine for Dementia due to Alzheimer’s Disease; Wiley: Hoboken, NJ, USA, 2002. [Google Scholar]
- Takata, K.; Kitamura, Y.; Saeki, M.; Terada, M.; Kagitani, S.; Kitamura, R.; Fujikawa, Y.; Maelicke, A.; Tomimoto, H.; Taniguchi, T.; et al. Galantamine-induced amyloid-{beta} clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J. Biol. Chem. 2010, 285, 40180–40191. [Google Scholar] [CrossRef]
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F.J. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 124. [Google Scholar] [CrossRef]
- Zani, I.A.; Stephen, S.L.; Mughal, N.A.; Russell, D.; Homer-Vanniasinkam, S.; Wheatcroft, S.B.; Ponnambalam, S. Scavenger receptor structure and function in health and disease. Cells 2015, 4, 178–201. [Google Scholar] [CrossRef]
- Sun, X.; Wang, X.; Chen, T.; Li, T.; Cao, K.; Lu, A.; Chen, Y.; Sun, D.; Luo, J.; Fan, J.; et al. Myelin activates FAK/Akt/NF-kappaB pathways and provokes CR3-dependent inflammatory response in murine system. PLoS ONE 2010, 5, e9380. [Google Scholar] [CrossRef]
- Cantoni, C.; Bollman, B.; Licastro, D.; Xie, M.; Mikesell, R.; Schmidt, R.; Yuede, C.M.; Galimberti, D.; Olivecrona, G.; Klein, R.S.; et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 2015, 129, 429–447. [Google Scholar] [CrossRef]
- Diemel, L.T.; Jackson, S.J.; Cuzner, M.L. Role for TGF-beta1, FGF-2 and PDGF-AA in a myelination of CNS aggregate cultures enriched with macrophages. J. Neurosci. Res. 2003, 74, 858–867. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, R.D.; Piras, G.; Ida, J.A., Jr.; Dubois-Dalcq, M. A role for TGF-beta in oligodendrocyte differentiation. J. Cell. Biol. 1993, 121, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.F.; Timmermans, S.; Huynh-Thu, V.A.; Irrthum, A.; Smeets, H.J.; Gustafsson, J.A.; Steffensen, K.R.; Mulder, M.; Stinissen, P.; Hellings, N.; et al. Myelin-derived lipids modulate macrophage activity by liver X receptor activation. PLoS ONE 2012, 7, e44998. [Google Scholar] [CrossRef] [PubMed]
- Raine, C.S. Multiple sclerosis: The resolving lesion revealed. J. Neuroimmunol. 2017, 304, 2–6. [Google Scholar] [CrossRef] [PubMed]
- van Zwam, M.; Samsom, J.N.; Nieuwenhuis, E.E.; Melief, M.J.; Wierenga-Wolf, A.F.; Dijke, I.E.; Talens, S.; van Meurs, M.; Voerman, J.S.; Boven, L.A.; et al. Myelin ingestion alters macrophage antigen-presenting function in vitro and in vivo. J. Leukoc. Biol. 2011, 90, 123–132. [Google Scholar] [CrossRef]
- Loving, B.A.; Bruce, K.D. Lipid and lipoprotein metabolism in microglia. Front. Physiol. 2020, 11, 393. [Google Scholar] [CrossRef]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Junker, A.; Krumbholz, M.; Eisele, S.; Mohan, H.; Augstein, F.; Bittner, R.; Lassmann, H.; Wekerle, H.; Hohlfeld, R.; Meinl, E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain J. Neurol. 2009, 132, 3342–3352. [Google Scholar] [CrossRef]
- Koning, N.; Bo, L.; Hoek, R.M.; Huitinga, I. Downregulation of macrophage inhibitory molecules in multiple sclerosis lesions. Ann. Neurol. 2007, 62, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, D.A.; Koning, N.; Schuurman, K.G.; van Strien, M.E.; van Eden, C.G.; Hamann, J.; Huitinga, I. Selective upregulation of scavenger receptors in and around demyelinating areas in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2013, 72, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Gitik, M.; Liraz-Zaltsman, S.; Oldenborg, P.A.; Reichert, F.; Rotshenker, S. Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPalpha (signal regulatory protein-alpha) on phagocytes. J. Neuroinflamm. 2011, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Dahlman-Wright, K. Liver X receptor in cholesterol metabolism. J. Endocrinol. 2010, 204, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.-T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lütjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684–688. [Google Scholar] [CrossRef]
- Kratzer, A.; Buchebner, M.; Pfeifer, T.; Becker, T.M.; Uray, G.; Miyazaki, M.; Miyazaki-Anzai, S.; Ebner, B.; Chandak, P.G.; Kadam, R.S.; et al. Synthetic LXR agonist attenuates plaque formation in apoE-/- mice without inducing liver steatosis and hypertriglyceridemia. J. Lipid Res. 2009, 50, 312–326. [Google Scholar] [CrossRef]
- Tangirala, R.K.; Bischoff, E.D.; Joseph, S.B.; Wagner, B.L.; Walczak, R.; Laffitte, B.A.; Daige, C.L.; Thomas, D.; Heyman, R.A.; Mangelsdorf, D.J.; et al. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc. Natl. Acad. Sci. USA 2002, 99, 11896–11901. [Google Scholar] [CrossRef]
- Popko, B.; Chen, Y. Cholesterol crystals impede nerve repair. Science 2018, 359, 635–636. [Google Scholar]
- Chawla, A.; Boisvert, W.A.; Lee, C.-H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPARg-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Chinetti, G.; Lestavel, S.; Bocher, V.; Remaley, A.T.; Neve, B.; Torra, I.P.; Teissier, E.; Minnich, A.; Jaye, M.; Duverger, N.; et al. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001, 7, 53–58. [Google Scholar] [CrossRef]
- Pipalia, N.; Cosner, C.C.; Huang, A.; Chatterjee, A.; Bourbon, P.; Farley, N.; Helquist, P.; Wiest, O.; Maxfield, F.R. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. USA 2011, 108, 5620–5625. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.; Rauniyar, N.; Lavallee-Adam, M.; Yates, J.R., 3rd; Balch, W.E. Quantitative analysis of the proteome response to the Histone Deacetylase Inhibitor (HDACi) Vorinostat in Niemann-Pick Type C1 disease. Mol. Cell. Proteom. MCP 2017, 16, 1938–1957. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Hait, N.C.; Maceyka, M.; Colaco, A.; Maczis, M.; Wassif, C.A.; Cougnoux, A.; Porter, F.D.; Milstien, S.; Platt, N.; et al. FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibroblasts. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Blom, T.; Back, N.; Mutka, A.L.; Bittman, R.; Li, Z.; de Lera, A.; Kovanen, P.T.; Diczfalusy, U.; Ikonen, E. FTY720 stimulates 27-hydroxycholesterol production and confers atheroprotective effects in human primary macrophages. Circ. Res. 2010, 106, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Ludwin, S.K.; Darlington, P.J.; Jarjour, A.A.; Soliven, B.; Kennedy, T.E.; Antel, J.P. Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am. J. Pathol. 2010, 176, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Sucksdorff, M.; Rissanen, E.; Tuisku, J.; Nuutinen, S.; Paavilainen, T.; Rokka, J.; Rinne, J.; Airas, L. Evaluation of the effect of fingolimod treatment on microglial activation using serial PET imaging in multiple sclerosis. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 1646–1651. [Google Scholar] [CrossRef]
- Cardozo, P.L.; de Lima, I.B.Q.; Maciel, E.M.A.; Silva, N.C.; Dobransky, T.; Ribeiro, F.M. Synaptic elimination in neurological disorders. Curr. Neuropharmacol. 2019, 17, 1071–1095. [Google Scholar] [CrossRef]
- Di Filippo, M.; Portaccio, E.; Mancini, A.; Calabresi, P. Multiple sclerosis and cognition: Synaptic failure and network dysfunction. Nat. Rev. Neurosci. 2018, 19, 599–609. [Google Scholar] [CrossRef]
- Di Filippo, M.; Sarchielli, P.; Picconi, B.; Calabresi, P. Neuroinflammation and synaptic plasticity: Theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol. Sci. 2008, 29, 402–412. [Google Scholar] [CrossRef]
- Rossi, S.; Motta, C.; Studer, V.; Barbieri, F.; Buttari, F.; Bergami, A.; Sancesario, G.; Bernardini, S.; De Angelis, G.; Martino, G.; et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult. Scler. 2014, 20, 304–312. [Google Scholar] [CrossRef]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Klaus, F.R.; Kollias, G.; Fontana, A.; Pryce, C.R.; et al. Neuroinflammatory TNFalpha impairs memory via astrocyte signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Geurts, J.; Bö, L.; Roosendaal, S.; Hazes, T.; Danie, R.; Barkhof, F.; Witter, M.P.; Huitinga, I.; van der Valk, P. Extensive hippocampal demyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.; Dukes, S.; Patel, R.; Nicholas, R.; Vora, A.; Reynolds, R. Substantial archaeocortical atrophy and neuronal loss in multiple sclerosis. Brain Pathol. 2009, 19, 238–253. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Chang, A.; Doud, M.K.; Kidd, G.J.; Ribaudo, M.V.; Young, E.A.; Fox, R.J.; Staugaitis, S.M.; Trapp, B.D. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann. Neurol. 2011, 69, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Sicotte, N.L.; Kern, K.C.; Giesser, B.S.; Arshanapalli, A.; Schultz, A.; Montag, M.; Wang, H.; Bookheimer, S.Y. Regional hippocampal atrophy in multiple sclerosis. Brain 2008, 131, 1134–1141. [Google Scholar] [CrossRef]
- Jurgens, T.; Jafari, M.; Kreutzfeldt, M.; Bahn, E.; Bruck, W.; Merkler, D. Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain 2016, 139, 39–46. [Google Scholar] [CrossRef]
- Vercellino, M.; Merola, A.; Piacentino, C.; Votta, B.; Capello, E.; Mancardi, G.L.; Mutani, R.; Giordana, M.T. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: Correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J. Neuropathol. Exp. Neurol. 2007, 66, 732–739. [Google Scholar] [CrossRef]
- Planche, V.; Panatier, A.; Hiba, B.; Ducourneau, E.G.; Raffard, G.; Dubourdieu, N.; Maitre, M.; Leste-Lasserre, T.; Brochet, B.; Dousset, V.; et al. Selective dentate gyrus disruption causes memory impairment at the early stage of experimental multiple sclerosis. Brain Behav. Immun. 2017, 60, 240–254. [Google Scholar] [CrossRef]
- Rajendran, L.; Paolicelli, R.C. Microglia-mediated synapse loss in Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Lehrman, E.K.; Wilton, D.K.; Litvina, E.Y.; Welsh, C.A.; Chang, S.T.; Frouin, A.; Walker, A.J.; Heller, M.D.; Umemori, H.; Chen, C.; et al. CD47 protects synapses from excess microglia-mediated pruning during development. Neuron 2018, 100, 120–134.e6. [Google Scholar] [CrossRef]
- Michailidou, I.; Willems, J.G.; Kooi, E.J.; van Eden, C.; Gold, S.M.; Geurts, J.J.; Baas, F.; Huitinga, I.; Ramaglia, V. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann. Neurol. 2015, 77, 1007–1026. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.C.; et al. Complement C3-deficient mice fail to display age-related hippocampal decline. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Rong, M.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Modulators of Microglial Phagocytic Phenotype | ||
| Drug | Mechanism | Reference |
| Docosahexaenoic acid and Eicosapentaenoic acid | Enhance myelin phagocytosis by microglia | [87] |
| Endocannabinoid 2-AG | Upregulates the expression levels of phagocytosis associated genes and promotes microglial myelin uptake | [92] |
| Pseudoginsenoside-F11 | Accelerates CR3-dependent myelin phagocytosis by microglial cells | [93] |
| rHIgM22 | Binds to myelin debris and facilitates their internalization by microglia | [88] |
| Ursolic acid * | Agonist of PPARγ signaling, which upregulates the expression of CD36 receptor, involved in the internalization of Aβ and lipids | [97] |
| Galantamine * | Favors microglial Aβ internalization | [99] |
| Modulators of Microglial Lipid Metabolism | ||
| Drug | Mechanism | Reference |
| LBH589 # and Vorinostat # | Both HDAC inhibitors that increase NPC expression in fibroblasts, promoting the release of cholesterol from lysosomes | [124,125] |
| FTY720 | HDAC inhibitor that also promotes lipid efflux from human primary macrophage through the overexpression of both NPC proteins and ABC transporters | [126,127] |
| DMHCA, GW3965, Wy14643, Rosiglitazone and Troglitazone | Activators of LXR/RXR heterodimeric transcription factor which upregulates ABC transporters thus also promoting cholesterol/lipid exit from macrophages | [118,119,120,121,122,123] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, M.V.; Fernandes, A. Microglial Phagocytosis—Rational but Challenging Therapeutic Target in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 5960. https://doi.org/10.3390/ijms21175960
Pinto MV, Fernandes A. Microglial Phagocytosis—Rational but Challenging Therapeutic Target in Multiple Sclerosis. International Journal of Molecular Sciences. 2020; 21(17):5960. https://doi.org/10.3390/ijms21175960
Chicago/Turabian StylePinto, Maria V., and Adelaide Fernandes. 2020. "Microglial Phagocytosis—Rational but Challenging Therapeutic Target in Multiple Sclerosis" International Journal of Molecular Sciences 21, no. 17: 5960. https://doi.org/10.3390/ijms21175960
APA StylePinto, M. V., & Fernandes, A. (2020). Microglial Phagocytosis—Rational but Challenging Therapeutic Target in Multiple Sclerosis. International Journal of Molecular Sciences, 21(17), 5960. https://doi.org/10.3390/ijms21175960