Y44A Mutation in the Acidic Domain of HIV-2 Tat Impairs Viral Reverse Transcription and LTR-Transactivation


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Mapping Destabilizing Mutations
2.2. In Silico Analysis
2.3. In Vitro Characterization of HIV-2 Tat Mutations
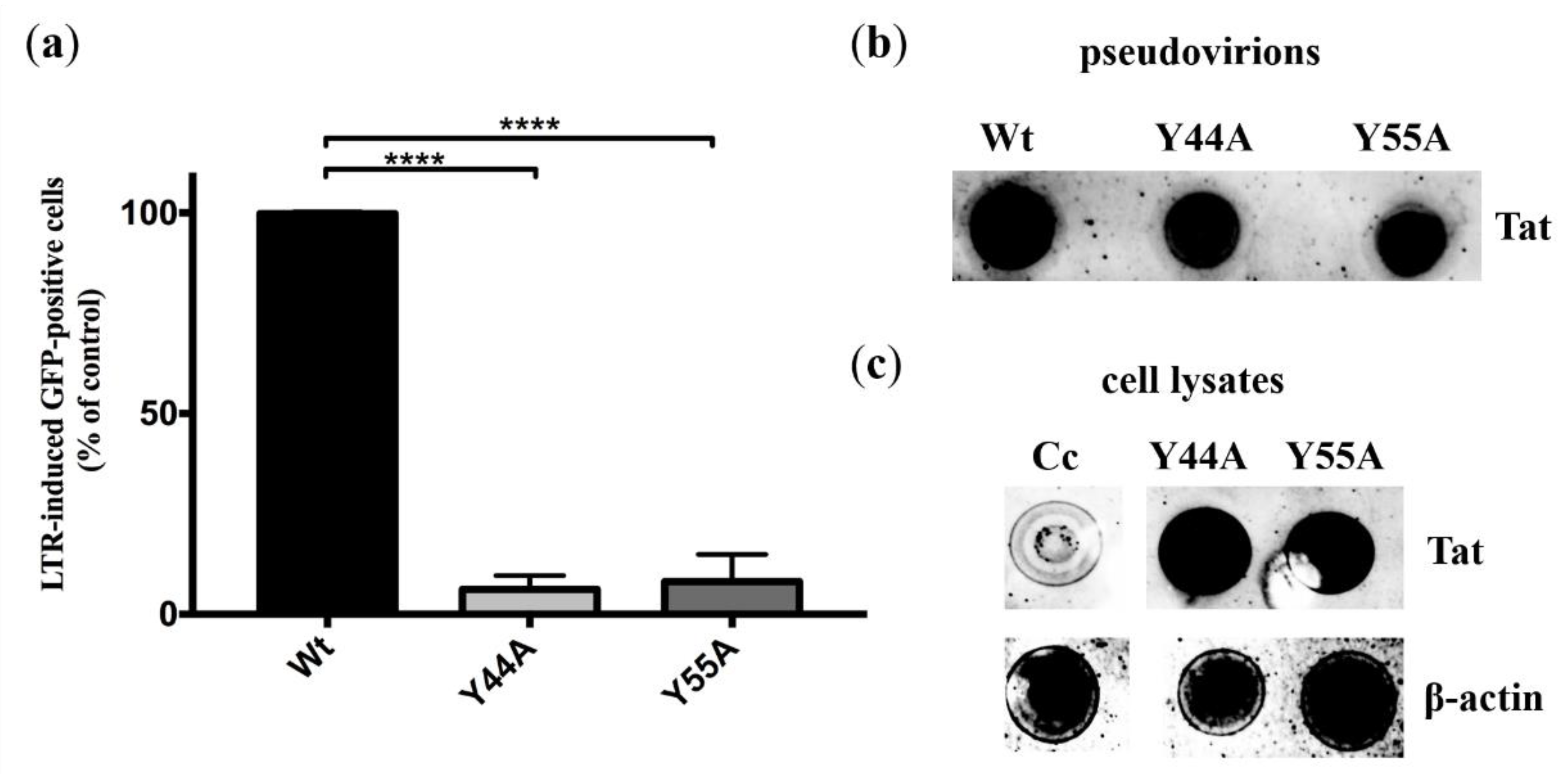
2.3.1. Experiments in HIV Indicator Cells
2.3.2. Effects of Y44A and Y55A Mutations on RT Activity
2.3.3. Detection of Tat in Pseudovirions
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and In Silico Predictions
4.2. HIV-2 Vector System
4.3. Mutagenesis
4.4. Experiments on HEK293T Cells
4.4.1. Production of Pseudovirions
4.4.2. Transduction of HEK293T Cells
4.4.3. Detection of HIV-2 Tat and RT by Western Blot
4.4.4. Proximity Ligation Assay
4.5. Experiments on GHOST(3) Cells
4.5.1. Production of Viral Particles for Transduction of GHOST(3) Cells
4.5.2. Transduction of GHOST(3) Cells
4.5.3. Dot-Blotting
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDS | acquired immunodeficiency syndrome |
| CMV | cytomegalovirus |
| HIV | human immunodeficiency virus |
| HIV-2 | human immunodeficiency virus type 2 |
| GFP | green fluorescent protein |
| LTR | RNA element on the long terminal repeat |
| MHC | major histocompatibility complex |
| PLA | proximity ligation assay |
| P-TEFb | positive transcription elongation factor b |
| RNAPII | RNA polymerase II |
| RT | reverse transcriptase |
| SIV | simian immunodeficiency virus |
| TAR | transactivation response |
| Tat | transactivator protein |
| Vpx | Viral protein X |
References
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. Retroviruses, the Interactions of Retroviruses and Their Hosts Retroviruses; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1997. [Google Scholar]
- Li, G.; De Clercq, E. HIV Genome-Wide Protein Associations, a Review of 30 Years of Research. Microbiol. Mol. Biol. Rev. 2016, 80, 679–731. [Google Scholar] [CrossRef] [PubMed]
- Damond, F.; Gueudin, M.; Pueyo, S.; Farfara, I.; Robertson, D.L.; Descamps, D.; Chène, G.; Matheron, S.; Campa, P.; Brun-Vézinet, F.; et al. Plasma RNA viral load in human immunodeficiency virus type 2 subtype A and subtype B infections. J. Clin. Microbiol. 2002, 40, 3654–3659. [Google Scholar] [CrossRef]
- Soares, R.S.; Tendeiro, R.; Foxall, R.B.; Baptista, A.P.; Cavaleiro, R.; Gomes, P.; Camacho, R.; Valadas, E.; Doroana, M.; Lucas, M.; et al. Cell-associated viral burden provides evidence of ongoing viral replication in aviremic HIV-2-infected patients. J. Virol. 2011, 85, 2429–2438. [Google Scholar] [CrossRef] [PubMed]
- Vidyavijayan, K.K.; Hassan, S.; Precilla, L.K.; Ashokkumar, M.; Chandrasekeran, P.; Swaminathan, S.; Hanna, L.E. Biased Nucleotide Composition and Differential Codon Usage Pattern in HIV-1 and HIV-2. AIDS Res. Hum. Retrovir. 2017, 33, 298–307. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, A.; Sarr, A.D.; Sankale, J.L.; Meloni, S.T.; Mboup, S.; Kanki, P. Direct evidence of lower viral replication rates in vivo in human immunodeficiency virus type 2 (HIV-2) infection than in HIV-1 infection. J. Virol. 2007, 81, 5325–5330. [Google Scholar] [CrossRef]
- Mahlknecht, U.; Dichamp, I.; Varin, A.; Van Lint, C.; Herbein, G. NF-kappaB-dependent control of HIV-1 transcription by the second coding exon of Tat in T cells. J. Leukoc. Biol. 2008, 83, 718–727. [Google Scholar] [CrossRef]
- Seelamgari, A.; Maddukuri, A.; Berro, R.; de la Fuente, C.; Kehn, K.; Deng, L.; Dadgar, S.; Bottazzi, M.E.; Ghedin, E.; Pumfery, A.; et al. Role of viral regulatory and accessory proteins in HIV-1 replication. Front. Biosci. 2004, 9, 2388–2413. [Google Scholar] [CrossRef]
- Bose, D.; Gagnon, J.; Chebloune, Y. Comparative Analysis of Tat-Dependent and Tat-Deficient Natural Lentiviruses. Vet. Sci. 2015, 2, 293–348. [Google Scholar] [CrossRef]
- Jones, K.A.; Peterlin, B.M. Control of RNA initiation and elongation at the HIV-1 promoter. Annu. Rev. Biochem. 1994, 63, 717–743. [Google Scholar] [CrossRef]
- Lata, S.; Mishra, R.; Banerjea, A.C. Proteasomal Degradation Machinery: Favorite Target of HIV-1 Proteins. Front. Microbiol. 2018, 9, 2738. [Google Scholar] [CrossRef]
- Karn, J.; Stoltzfus, C.M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2012, 2, a006916. [Google Scholar] [CrossRef] [PubMed]
- Karn, J. Tackling Tat. J. Mol. Biol. 1999, 293, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 2011, 6, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Harrich, D.; Ulich, C.; García-Martínez, L.F.; Gaynor, R.B. Tat is required for efficient HIV-1 reverse transcription. EMBO J. 1997, 16, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, A.; Meredith, L.W.; Suhrbier, A.; Kiernan, R.; Harrich, D. The HIV-1 Tat protein stimulates reverse transcription in vitro. Curr. HIV Res. 2007, 5, 473–483. [Google Scholar] [CrossRef]
- Apolloni, A.; Hooker, C.W.; Mak, J.; Harrich, D. Human immunodeficiency virus type 1 protease regulation of tat activity is essential for efficient reverse transcription and replication. J. Virol. 2003, 77, 9912–9921. [Google Scholar] [CrossRef][Green Version]
- Meredith, L.W.; Sivakumaran, H.; Major, L.; Suhrbier, A.; Harrich, D. Potent inhibition of HIV-1 replication by a Tat mutant. PLoS ONE 2009, 4, e7769. [Google Scholar] [CrossRef]
- Apolloni, A.; Lin, M.H.; Sivakumaran, H.; Li, D.; Kershaw, M.H.; Harrich, D. A mutant Tat protein provides strong protection from HIV-1 infection in human CD4+ T cells. Hum. Gene Ther. 2013, 24, 270–282. [Google Scholar] [CrossRef]
- Lin, M.H.; Sivakumaran, H.; Jones, A.; Li, D.; Harper, C.; Wei, T.; Jin, H.; Rustanti, L.; Meunier, F.A.; Spann, K.; et al. A HIV-1 Tat mutant protein disrupts HIV-1 Rev function by targeting the DEAD-box RNA helicase DDX1. Retrovirology 2014, 11, 121. [Google Scholar] [CrossRef]
- Guyader, M.; Emerman, M.; Sonigo, P.; Clavel, F.; Montagnier, L.; Alizon, M. Genome organization and transactivation of the human immunodeficiency virus type 2. Nature 1987, 326, 662–669. [Google Scholar] [CrossRef]
- Jeang, K.T.; Xiao, H.; Rich, E.A. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J. Biol. Chem. 1999, 274, 28837–28840. [Google Scholar] [CrossRef] [PubMed]
- Kurnaeva, M.A.; Sheval, E.V.; Musinova, Y.R.; Vassetzky, Y.S. Tat basic domain: A “Swiss army knife” of HIV-1 Tat? Rev. Med. Virol. 2019, 29, e2031. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dahiya, S.; Kortagere, S.; Aiamkitsumrit, B.; Cunningham, D.; Pirrone, V.; Nonnemacher, M.R.; Wigdahl, B. Impact of Tat Genetic Variation on HIV-1 Disease. Adv. Virol. 2012, 2012, 123605. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.K. Human immunodeficiency virus type 2 (HIV-2) trans-activator (Tat): Functional domains and the search for trans-dominant negative mutants. AIDS Res. Hum. Retrovir. 1993, 9, 839–848. [Google Scholar] [CrossRef]
- Ruben, S.; Perkins, A.; Purcell, R.; Joung, K.; Sia, R.; Burghoff, R.; Haseltine, W.A.; Rosen, C.A. Structural and functional characterization of human immunodeficiency virus tat protein. J. Virol. 1989, 63, 1–8. [Google Scholar] [CrossRef]
- Weissman, J.D.; Brown, J.A.; Howcroft, T.K.; Hwang, J.; Chawla, A.; Roche, P.A.; Schiltz, L.; Nakatani, Y.; Singer, D.S. HIV-1 tat binds TAFII250 and represses TAFII250-dependent transcription of major histocompatibility class I genes. Proc. Natl. Acad. Sci. USA 1998, 95, 11601–11606. [Google Scholar] [CrossRef]
- Clark, E.; Nava, B.; Caputi, M. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 2017, 8, 27569–27581. [Google Scholar] [CrossRef]
- Smith, S.M.; Pentlicky, S.; Klase, Z.; Singh, M.; Neuveut, C.; Lu, C.Y.; Reitz, M.S., Jr.; Yarchoan, R.; Marx, P.A.; Jeang, K.T. An in vivo replication-important function in the second coding exon of Tat is constrained against mutation despite cytotoxic T lymphocyte selection. J. Biol. Chem. 2003, 278, 44816–44825. [Google Scholar] [CrossRef]
- Pagtakhan, A.S.; Tong-Starksen, S.E. Interactions between Tat of HIV-2 and transcription factor Sp1. Virology 1997, 238, 221–230. [Google Scholar] [CrossRef][Green Version]
- Kalantari, P.; Narayan, V.; Natarajan, S.K.; Muralidhar, K.; Gandhi, U.H.; Vunta, H.; Henderson, A.J.; Prabhu, K.S. Thioredoxin reductase-1 negatively regulates HIV-1 transactivating protein Tat-dependent transcription in human macrophages. J. Biol. Chem. 2008, 283, 33183–33190. [Google Scholar] [CrossRef]
- Verhoef, K.; Koper, M.; Berkhout, B. Determination of the minimal amount of Tat activity required for human immunodeficiency virus type 1 replication. Virology 1997, 237, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Klaver, B.; Harwig, A.; Vink, M.; Ooms, M.; Centlivre, M.; Berkhout, B. Construction of a doxycycline-dependent simian immunodeficiency virus reveals a nontranscriptional function of tat in viral replication. J. Virol. 2007, 81, 11159–11169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mahdi, M.; Szojka, Z.; Mótyán, J.A.; Tőzsér, J. Inhibitory Effects of HIV-2 Vpx on Replication of HIV-1. J. Virol. 2018, 92, e00554-18. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.; Kraft, M.; Ejchart, A.; Westendorp, M.; Frank, R.; Rösch, P. Structural studies of HIV-1 Tat protein. J. Mol. Biol. 1995, 247, 529–535. [Google Scholar] [CrossRef]
- Kameoka, M.; Rong, L.; Götte, M.; Liang, C.; Russell, S.R.; Wainberg, A.M. Role for Human Immunodeficiency Virus Type 1 Tat Protein in Suppression of Viral Reverse Transcriptase Activity during Late Stages of Viral Replication. J. Virol. 2001, 75, 2675–2683. [Google Scholar] [CrossRef]
- Baldauf, H.M.; Stegmann, L.; Schwarz, S.M.; Ambiel, I.; Trotard, M.; Martin, M.; Burggraf, M.; Lenzi, G.M.; Lejk, H.; Pan, X.; et al. Vpx overcomes a SAMHD1-independent block to HIV reverse transcription that is specific to resting CD4 T cells. Proc. Natl. Acad. Sci. USA 2017, 114, 2729–2734. [Google Scholar] [CrossRef]
- Foley, B.; Leitner, T.; Apetrei, C.; Hahn, B.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S.; Korber, B. HIV Sequence Compendium; Theoretical Biology and Biophysics Group, Los Alamos National Laboratory: Los Alamos, NM, USA, 2018; LA-UR 18-25673. [Google Scholar]
- Ulich, C.; Dunne, A.; Parry, E.; Hooker, C.W.; Gaynor, R.B.; Harrich, D. Functional domains of Tat required for efficient human immunodeficiency virus type 1 reverse transcription. J. Virol. 1999, 73, 2499–2508. [Google Scholar] [CrossRef]
- Lin, M.H.; Apolloni, A.; Cutillas, V.; Sivakumaran, H.; Martin, S.; Li, D.; Wei, T.; Wang, R.; Jin, H.; Spann, K.; et al. A mutant tat protein inhibits HIV-1 reverse transcription by targeting the reverse transcription complex. J. Virol. 2015, 89, 4827–4836. [Google Scholar] [CrossRef][Green Version]
- Ensoli, B.; Buonaguro, L.; Barillari, G.; Fiorelli, V.; Gendelman, R.; Morgan, R.A.; Wingfield, P.; Gallo, R.C. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J. Virol. 1993, 67, 277–28741. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdos, G.; Dosztányi, Z. IUPred2A context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0 predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [PubMed]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef] [PubMed]
- Pandurangan, A.P.; Ochoa-Montano, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [PubMed]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes, a study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Mahdi, M.; Matúz, K.; Tóth, F.; Tőzsér, J. A modular system to evaluate the efficacy of protease inhibitors against HIV-2. PLoS ONE 2014, 9, e113221. [Google Scholar] [CrossRef]
- Mukherjee, S.; Lee, H.L.; Pacchia, A.L.; Ron, Y.; Dougherty, J.P. A HIV-2-based self-inactivating vector for enhanced gene transduction. J. Biotechnol. 2007, 127, 745–757. [Google Scholar] [CrossRef]
- Chesebro, B.; Wehrly, K.; Nishio, J.; Perryman, S. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates, definition of critical amino acids involved in cell tropism. J. Virol. 1992, 66, 6547–6554. [Google Scholar] [CrossRef]
- Echetebu, C.O.; Rice, A.P. Mutational analysis of the amino and carboxy termini of the HIV-2 Tat protein. J. Acquir. Immune Defic. Syndr. 1993, 6, 550–557. [Google Scholar]
- Klutch, M.; Woerner, A.M.; Marcus-Sekura, C.J.; Levin, J.G. Generation of HIV-1/HIV-2 cross-reactive peptide antisera by small sequence changes in HIV-1 reverse transcriptase and integrase immunizing peptides. J. Biomed. Sci. 1998, 5, 192–202. [Google Scholar] [CrossRef]
- Kappes, J.C.; Parkin, J.S.; Conway, J.A.; Kim, J.; Brouillette, C.G.; Shaw, G.M.; Hahn, B.H. Intracellular transport and virion incorporation of vpx requires interaction with other virus type-specific components. Virology 1993, 193, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Mörner, A.; Björndal, A.; Albert, J.; Kewalramani, V.N.; Littman, D.R.; Inoue, R.; Thorstensson, R.; Fenyö, E.M.; Björling, E. Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. J. Virol. 1999, 73, 2343–2349. [Google Scholar] [CrossRef] [PubMed]
- Vödrös, D.; Fenyö, E.M. Quantitative evaluation of HIV and SIV co-receptor use with GHOST(3) cell assay. Methods Mol. Biol. 2005, 304, 333–342. [Google Scholar] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szojka, Z.; Mótyán, J.A.; Miczi, M.; Mahdi, M.; Tőzsér, J. Y44A Mutation in the Acidic Domain of HIV-2 Tat Impairs Viral Reverse Transcription and LTR-Transactivation. Int. J. Mol. Sci. 2020, 21, 5907. https://doi.org/10.3390/ijms21165907
Szojka Z, Mótyán JA, Miczi M, Mahdi M, Tőzsér J. Y44A Mutation in the Acidic Domain of HIV-2 Tat Impairs Viral Reverse Transcription and LTR-Transactivation. International Journal of Molecular Sciences. 2020; 21(16):5907. https://doi.org/10.3390/ijms21165907
Chicago/Turabian StyleSzojka, Zsófia, János András Mótyán, Márió Miczi, Mohamed Mahdi, and József Tőzsér. 2020. "Y44A Mutation in the Acidic Domain of HIV-2 Tat Impairs Viral Reverse Transcription and LTR-Transactivation" International Journal of Molecular Sciences 21, no. 16: 5907. https://doi.org/10.3390/ijms21165907
APA StyleSzojka, Z., Mótyán, J. A., Miczi, M., Mahdi, M., & Tőzsér, J. (2020). Y44A Mutation in the Acidic Domain of HIV-2 Tat Impairs Viral Reverse Transcription and LTR-Transactivation. International Journal of Molecular Sciences, 21(16), 5907. https://doi.org/10.3390/ijms21165907