Mitochondrial Genetic Drift after Nuclear Transfer in Oocytes


Abstract
1. Introduction
2. Nuclear Transfer for Mitochondrial Replacement
3. Developmental Potential and Mitochondrial Carryover after Nuclear Transfer
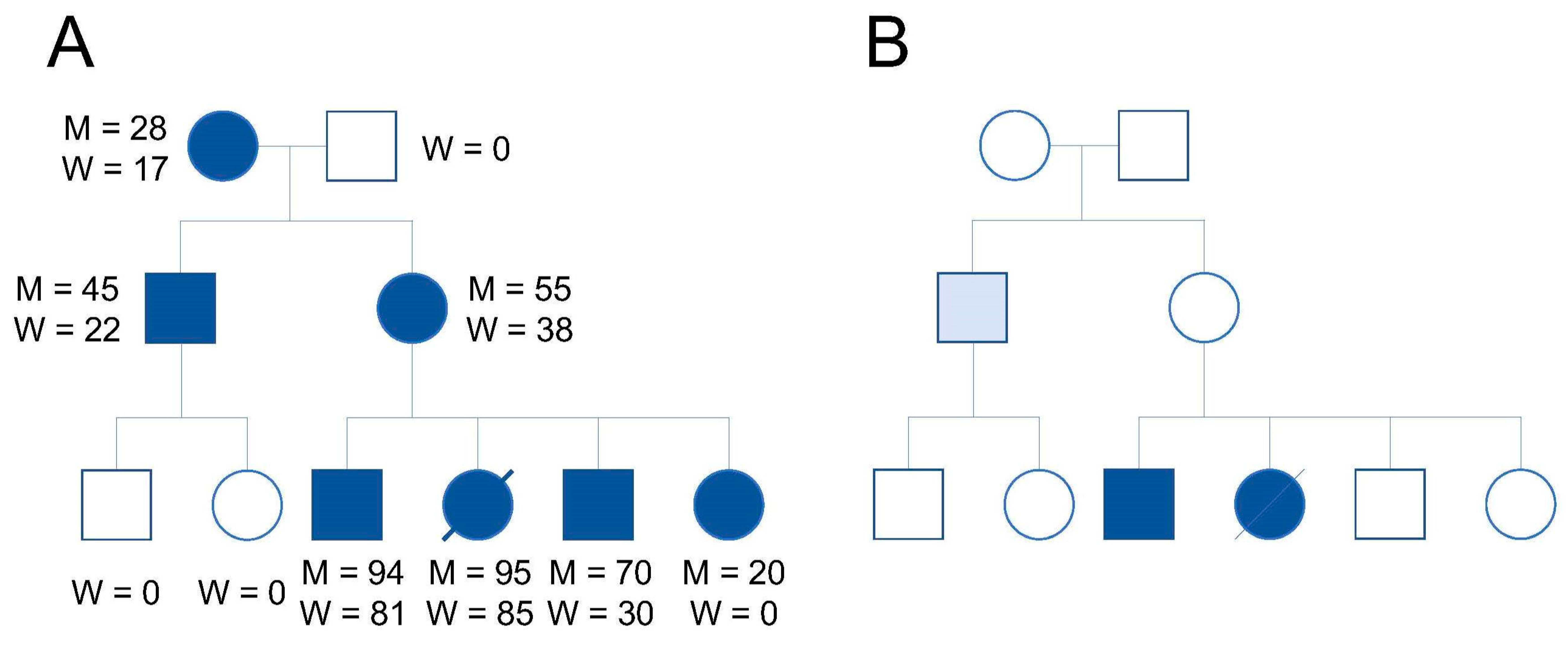
4. mtDNA Genetic Drift after Mitochondria Replacement
5. Mechanistic Insights for mtDNA Replication Bias
6. mtDNA–nDNA Mismatch
7. Mitochondrial Genome Alterations
8. Mitochondrial Replacement for Reproduction
9. Conclusions
Funding
Conflicts of Interest
References
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010, 9, 829–840. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Sutovsky, P.; Moreno, R.D.; Ramalho-Santos, J.; Dominko, T.; Simerly, C.; Schatten, G. Ubiquitin tag for sperm mitochondria. Nature 1999, 402, 371–372. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, A.; Braude, P.; Flinter, F.; Lovell-Badge, R.; Ogilvie, C.; Perry, A.C.F. Assisted reproductive technologies to prevent human mitochondrial disease transmission. Nat. Biotechnol. 2017, 35, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.R.; Samuels, D.C.; Eden, J.A.; Relton, C.L.; Chinnery, P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Shibata, D.; Soong, N.W.; Arnheim, N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 7370–7374. [Google Scholar] [CrossRef]
- Craven, L.; Tang, M.X.; Gorman, G.S.; De Sutter, P.; Heindryckx, B. Novel reproductive technologies to prevent mitochondrial disease. Hum. Reprod. Update 2017, 23, 501–519. [Google Scholar] [CrossRef]
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genom. 2009, 36, 125–131. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- DiMauro, S.; Moraes, C.T. Mitochondrial encephalomyopathies. Arch. Neurol. 1993, 50, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; Group, S.I.-E.B.W.; Kokkali, G.; Coticchio, G.; Bronet, F.; Celebi, C.; Cimadomo, D.; Goossens, V.; Liss, J.; Nunes, S.; et al. ESHRE PGT Consortium and SIG Embryology good practice recommendations for polar body and embryo biopsy for PGT. Hum. Reprod. Update 2020, 2020, hoaa020. [Google Scholar] [CrossRef]
- Surani, M.A.; Barton, S.C.; Norris, M.L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Kono, T.; Nakada, K.; Ishikawa, K.; Inoue, S.; Yonekawa, H.; Hayashi, J. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc. Natl. Acad. Sci. USA 2005, 102, 16765–16770. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Tuppen, H.A.; Greggains, G.D.; Harbottle, S.J.; Murphy, J.L.; Cree, L.M.; Murdoch, A.P.; Chinnery, P.F.; Taylor, R.W.; Lightowlers, R.N.; et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010, 465, 82–85. [Google Scholar] [CrossRef]
- Hyslop, L.A.; Blakeley, P.; Craven, L.; Richardson, J.; Fogarty, N.M.; Fragouli, E.; Lamb, M.; Wamaitha, S.E.; Prathalingam, N.; Zhang, Q.; et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 2016, 534, 383–386. [Google Scholar] [CrossRef]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef]
- Lee, H.S.; Ma, H.; Juanes, R.C.; Tachibana, M.; Sparman, M.; Woodward, J.; Ramsey, C.; Xu, J.; Kang, E.J.; Amato, P.; et al. Rapid mitochondrial DNA segregation in primate preimplantation embryos precedes somatic and germline bottleneck. Cell Rep. 2012, 1, 506–515. [Google Scholar] [CrossRef]
- Paull, D.; Emmanuele, V.; Weiss, K.A.; Treff, N.; Stewart, L.; Hua, H.; Zimmer, M.; Kahler, D.J.; Goland, R.S.; Noggle, S.A.; et al. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 2013, 493, 632–637. [Google Scholar] [CrossRef]
- Tachibana, M.; Amato, P.; Sparman, M.; Woodward, J.; Sanchis, D.M.; Ma, H.; Gutierrez, N.M.; Tippner-Hedges, R.; Kang, E.; Lee, H.S.; et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature 2013, 493, 627–631. [Google Scholar] [CrossRef]
- Yamada, M.; Johannesson, B.; Sagi, I.; Burnett, L.C.; Kort, D.H.; Prosser, R.W.; Paull, D.; Nestor, M.W.; Freeby, M.; Greenberg, E.; et al. Human oocytes reprogram adult somatic nuclei of a type 1 diabetic to diploid pluripotent stem cells. Nature 2014, 510, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; Martinez-Redondo, P.; Ma, H.; Lee, Y.; et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 2016, 540, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Costa-Borges, N.; Spath, K.; Miguel-Escalada, I.; Mestres, E.; Balmaseda, R.; Serafín, A.; Garcia-Jiménez, M.; Vanrell, I.; González, J.; Rink, K.; et al. Maternal spindle transfer overcomes embryo developmental arrest caused by ooplasmic defects in mice. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Emmanuele, V.; Sanchez-Quintero, M.J.; Sun, B.; Lallos, G.; Paull, D.; Zimmer, M.; Pagett, S.; Prosser, R.W.; Sauer, M.V.; et al. Genetic Drift Can Compromise Mitochondrial Replacement by Nuclear Transfer in Human Oocytes. Cell Stem Cell 2016, 18, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Sharpley, M.S.; Marciniak, C.; Eckel-Mahan, K.; McManus, M.; Crimi, M.; Waymire, K.; Lin, C.S.; Masubuchi, S.; Friend, N.; Koike, M.; et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 2012, 151, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.G.; Wellesley, F.C.; Savery, N.J.; Szczelkun, M.D. Length heterogeneity at conserved sequence block 2 in human mitochondrial DNA acts as a rheostat for RNA polymerase POLRMT activity. Nucleic Acids Res. 2016, 44, 7817–7829. [Google Scholar] [CrossRef]
- Hudson, G.; Takeda, Y.; Herbert, M. Reversion after replacement of mitochondrial DNA. Nature 2019, 574, E8–E11. [Google Scholar] [CrossRef]
- Kang, E.; Koski, A.; Amato, P.; Temiakov, D.; Mitalipov, S. Reply to: Reversion after replacement of mitochondrial DNA. Nature 2019, 574, E12–E13. [Google Scholar] [CrossRef]
- Fetterman, J.L.; Zelickson, B.R.; Johnson, L.W.; Moellering, D.R.; Westbrook, D.G.; Pompilius, M.; Sammy, M.J.; Johnson, M.; Dunham-Snary, K.J.; Cao, X.; et al. Mitochondrial genetic background modulates bioenergetics and susceptibility to acute cardiac volume overload. Biochem. J. 2013, 455, 157–167. [Google Scholar] [CrossRef]
- Betancourt, A.M.; King, A.L.; Fetterman, J.L.; Millender-Swain, T.; Finley, R.D.; Oliva, C.R.; Crowe, D.R.; Ballinger, S.W.; Bailey, S.M. Mitochondrial-nuclear genome interactions in non-alcoholic fatty liver disease in mice. Biochem. J. 2014, 461, 223–232. [Google Scholar] [CrossRef]
- Ma, H.; Marti Gutierrez, N.; Morey, R.; Van Dyken, C.; Kang, E.; Hayama, T.; Lee, Y.; Li, Y.; Tippner-Hedges, R.; Wolf, D.P.; et al. Incompatibility between Nuclear and Mitochondrial Genomes Contributes to an Interspecies Reproductive Barrier. Cell Metab. 2016, 24, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Ocampo, A.; Suzuki, K.; Luo, J.; Bacman, S.R.; Williams, S.L.; Sugawara, A.; Okamura, D.; Tsunekawa, Y.; Wu, J.; et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 2015, 161, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Viscomi, C.; Simard, M.L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, M.V.; Xu, J.; Mitchell, C.; Marin, D.; Zimmerman, R.; Rana, B.; Weinstein, E.; King, R.T.; Smith, M.; Tsang, S.H.; et al. Reading frame restoration at the EYS locus, and allele-specific chromosome removal after Cas9 cleavage in human embryos. BioRxiv 2020. [Google Scholar] [CrossRef]
- Adashi, E.Y.; Cohen, I.G. Preventing Mitochondrial Diseases: Embryo-Sparing Donor-Independent Options. Trends Mol. Med. 2018, 24, 449–457. [Google Scholar] [CrossRef]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020. [Google Scholar] [CrossRef]
- Baltz, J.M.; Tartia, A.P. Cell volume regulation in oocytes and early embryos: Connecting physiology to successful culture media. Hum. Reprod. Update 2010, 16, 166–176. [Google Scholar] [CrossRef]
- Biggers, J.D.; Summers, M.C. Choosing a culture medium: Making informed choices. Fertil. Steril. 2008, 90, 473–483. [Google Scholar] [CrossRef]
- Lane, M.; Gardner, D.K. Embryo culture medium: Which is the best? Best Pract. Res. Clin. Obstet. Gynaecol. 2007, 21, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Floros, V.I.; Pyle, A.; Dietmann, S.; Wei, W.; Tang, W.C.W.; Irie, N.; Payne, B.; Capalbo, A.; Noli, L.; Coxhead, J.; et al. Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat. Cell Biol. 2018, 20, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Cree, L.M.; Samuels, D.C.; de Sousa Lopes, S.C.; Rajasimha, H.K.; Wonnapinij, P.; Mann, J.R.; Dahl, H.H.; Chinnery, P.F. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 2008, 40, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Spath, K.; Alfarawati, S.; Kaper, F.; Craig, A.; Michel, C.E.; Kokocinski, F.; Cohen, J.; Munne, S.; Wells, D. Altered levels of mitochondrial DNA are associated with female age, aneuploidy, and provide an independent measure of embryonic implantation potential. PLoS Genet. 2015, 11, e1005241. [Google Scholar] [CrossRef]
- Leese, H.J. Quiet please, do not disturb: A hypothesis of embryo metabolism and viability. Bioessays 2002, 24, 845–849. [Google Scholar] [CrossRef]
- Fragouli, E.; McCaffrey, C.; Ravichandran, K.; Spath, K.; Grifo, J.A.; Munné, S.; Wells, D. Clinical implications of mitochondrial DNA quantification on pregnancy outcomes: A blinded prospective non-selection study. Hum. Reprod. 2017, 32, 2340–2347. [Google Scholar] [CrossRef]
- Yamada, M.; Egli, D. Genome Transfer Prevents Fragmentation and Restores Developmental Potential of Developmentally Compromised Postovulatory Aged Mouse Oocytes. Stem Cell Rep. 2017, 8, 576–588. [Google Scholar] [CrossRef]
- Lister, L.M.; Kouznetsova, A.; Hyslop, L.A.; Kalleas, D.; Pace, S.L.; Barel, J.C.; Nathan, A.; Floros, V.; Adelfalk, C.; Watanabe, Y.; et al. Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr. Biol. 2010, 20, 1511–1521. [Google Scholar] [CrossRef]
- Yamada-Fukunaga, T.; Yamada, M.; Hamatani, T.; Chikazawa, N.; Ogawa, S.; Akutsu, H.; Miura, T.; Miyado, K.; Tarin, J.J.; Kuji, N.; et al. Age-associated telomere shortening in mouse oocytes. Reprod. Biol. Endocrinol. 2013, 11, 108. [Google Scholar] [CrossRef]
- Udagawa, O.; Ishihara, T.; Maeda, M.; Matsunaga, Y.; Tsukamoto, S.; Kawano, N.; Miyado, K.; Shitara, H.; Yokota, S.; Nomura, M.; et al. Mitochondrial fission factor Drp1 maintains oocyte quality via dynamic rearrangement of multiple organelles. Curr. Biol. 2014, 24, 2451–2458. [Google Scholar] [CrossRef]
- Van Blerkom, J.; Davis, P.W.; Lee, J. ATP content of human oocytes and developmental potential and outcome after in-vitro fertilization and embryo transfer. Hum. Reprod. 1995, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.C.; Liu, V.W.; Lau, E.Y.; Yeung, W.S.; Ng, E.H.; Ho, P.C. Mitochondrial DNA content and 4977 bp deletion in unfertilized oocytes. Mol. Hum. Reprod. 2005, 11, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.T.; Ren, Z.; Yeung, W.S.; Shu, Y.M.; Xu, Y.W.; Zhuang, G.L.; Liang, X.Y. Low mitochondrial DNA and ATP contents contribute to the absence of birefringent spindle imaged with PolScope in in vitro matured human oocytes. Hum. Reprod. 2007, 22, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Scott, R.; Schimmel, T.; Levron, J.; Willadsen, S. Birth of infant after transfer of anucleate donor oocyte cytoplasm into recipient eggs. Lancet 1997, 350, 186–187. [Google Scholar] [CrossRef]
- Barritt, J.; Willadsen, S.; Brenner, C.; Cohen, J. Cytoplasmic transfer in assisted reproduction. Hum. Reprod. Update 2001, 7, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, A.; Yoshizawa, M.; Matsumoto, H.; Fukui, E. Improvement of embryonic development and production of offspring by transferring meiosis-II chromosomes of senescent mouse oocytes into cytoplasts of young mouse oocytes. J. Assist. Reprod. Genet. 2009, 26, 35–39. [Google Scholar] [CrossRef][Green Version]
- Coghlan, A. First Baby Born Using 3-parent Technique to Treat Infertility. Available online: https://www.newscientist.com/article/2118334-first-baby-born-using-3-parent-technique-to-treat-infertility/ (accessed on 30 June 2020).
- Costa-Borges, N.; Nikitos, E.; Spath, K.; Wells, D.; Rink, K.; Vasilopoulos, Y.; Zevomanolakis, I.; Konstantinos, D.; Panagiotis, P.; Grigorakis, S.; et al. Preliminary results from the first registered pilot trial with maternal spindle transfer to overcome infertility. Fertil. Steril. 2019, 112, e5–e6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Stem Cell Lines | mtDNA Haplotype | mtDNA Heteroplasmy |
|---|---|---|
| Nucleus:Cytoplasm | ||
| ST-ES1 | H56:H2a | no mtDNA shift |
| ST-ES2 | H2a:H56 | no mtDNA shift |
| ST-ES3 | H2a:H56 | no mtDNA shift |
| ST-ES4 | H44a:H13a | no mtDNA shift |
| ST-ES5 | H1b:U5a | no mtDNA shift |
| ST-ES6 | H1b:U5a | no mtDNA shift |
| ST-ES7 | U5a:H1b | mtDNA shift |
| ST-ES8 | U5a:H1b | mtDNA shift |
| ST-ES9 | U5a:V3 | no mtDNA shift |
| ST-ES10 | V3:U5a | no mtDNA shift |
| ST-ES11 | Hae:D1f | no mtDNA shift |
| ST-ES12 | Hae:D1f | no mtDNA shift |
| ST-ES13 | Hae:D1f | no mtDNA shift |
| ST-ES14 | D4a:A2g | no mtDNA shift |
| ST-ES15 | A2g:D4a | no mtDNA shift |
| 13513ST-ES | T2b:T2 | no mtDNA shift |
| 3243ST-ES1 | H49:B2k | mtDNA shift |
| 3243ST-ES2 | H49:B2k | mtDNA shift |
| 31PNT | K:U | no mtDNA shift |
| 36PNT | H:H | mtDNA shift |
| 45PNT | L0:H | no mtDNA shift |
| 47PNT | J:U | no mtDNA shift |
| 55PNT | H:K | no mtDNA shift |
| MR-PS1 | HV:C | no mtDNA shift |
| MR-PS2 | HV:C | no mtDNA shift |
| MR-PS3 | C:HV | no mtDNA shift |
| MR-PS4 | C:I | no mtDNA shift |
| MR-PS5 | HV:J | no mtDNA shift |
| MR-PS6 | A:L3 | no mtDNA shift |
| MR-PS7 | A:L3 | no mtDNA shift |
| MR-PS8 | L0:L3 | no mtDNA shift |
| MR-PS9 | L3:U | no mtDNA shift |
| MR-PS10 | L3:U | no mtDNA shift |
| MR-PS11 | L3:HV | no mtDNA shift |
| MR-PS12 | HV:L3 | mtDNA shift |
| NT5 | K:L0 | no mtDNA shift |
| NT6 | K:L0 | mtDNA shift |
| NT8 | K:L0 | mtDNA shift |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, M.; Akashi, K.; Ooka, R.; Miyado, K.; Akutsu, H. Mitochondrial Genetic Drift after Nuclear Transfer in Oocytes. Int. J. Mol. Sci. 2020, 21, 5880. https://doi.org/10.3390/ijms21165880
Yamada M, Akashi K, Ooka R, Miyado K, Akutsu H. Mitochondrial Genetic Drift after Nuclear Transfer in Oocytes. International Journal of Molecular Sciences. 2020; 21(16):5880. https://doi.org/10.3390/ijms21165880
Chicago/Turabian StyleYamada, Mitsutoshi, Kazuhiro Akashi, Reina Ooka, Kenji Miyado, and Hidenori Akutsu. 2020. "Mitochondrial Genetic Drift after Nuclear Transfer in Oocytes" International Journal of Molecular Sciences 21, no. 16: 5880. https://doi.org/10.3390/ijms21165880
APA StyleYamada, M., Akashi, K., Ooka, R., Miyado, K., & Akutsu, H. (2020). Mitochondrial Genetic Drift after Nuclear Transfer in Oocytes. International Journal of Molecular Sciences, 21(16), 5880. https://doi.org/10.3390/ijms21165880