BrassicaEDB: A Gene Expression Database for Brassica Crops


 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
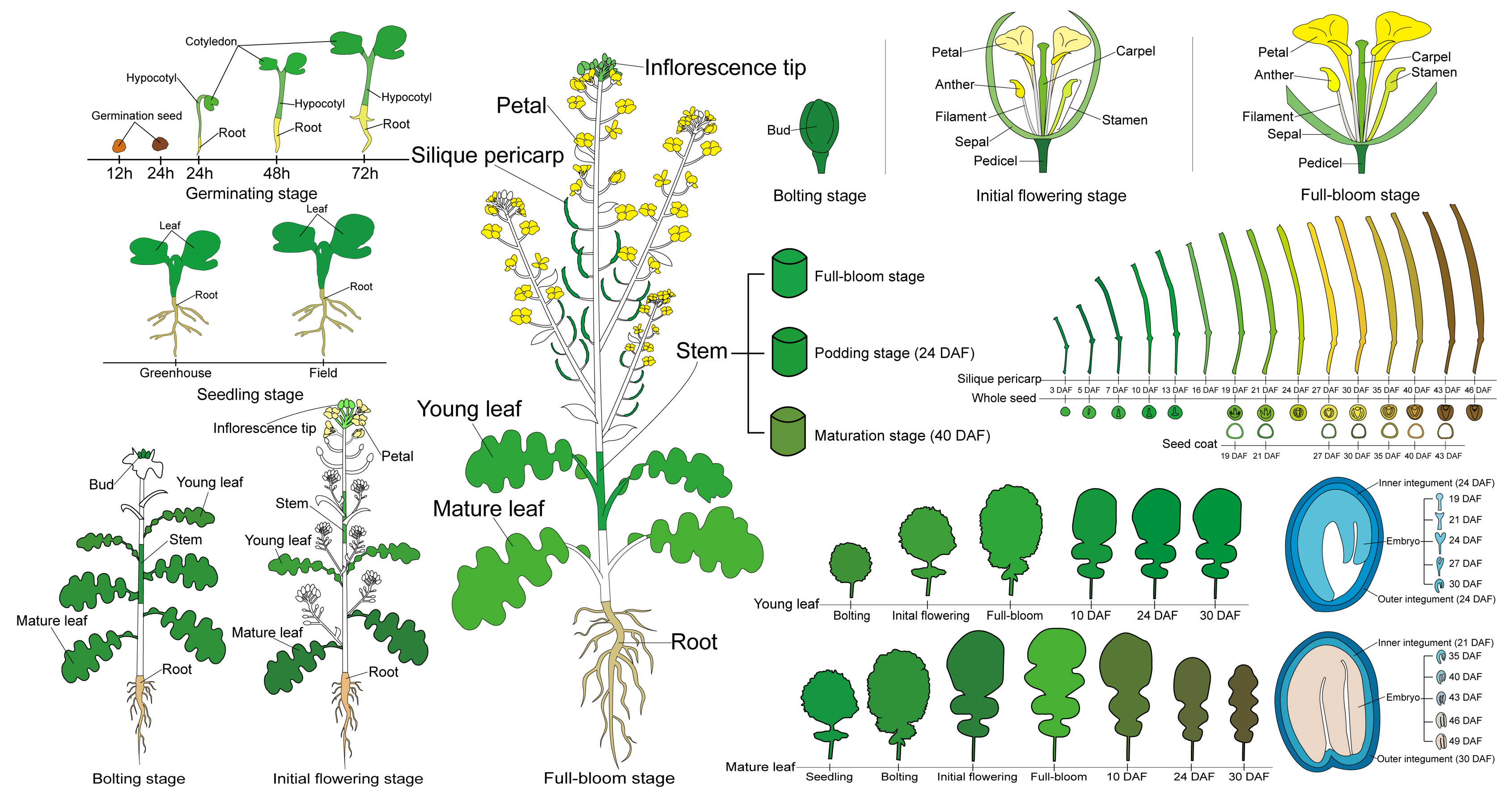
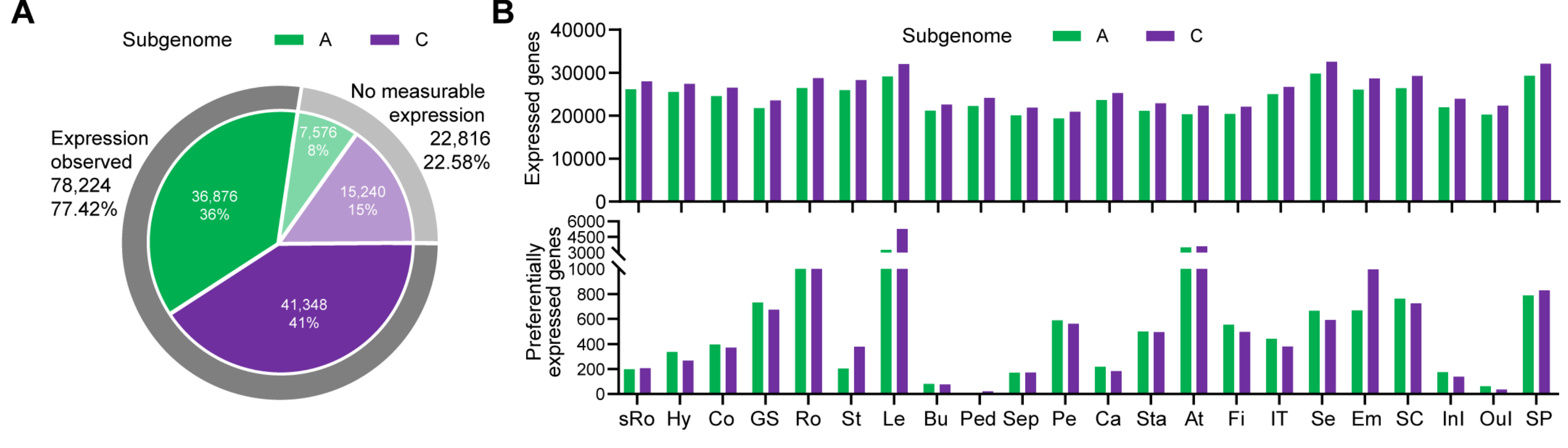
2.1. RNA-Seq-Based Global Expression Data from 206 Rapeseed Samples
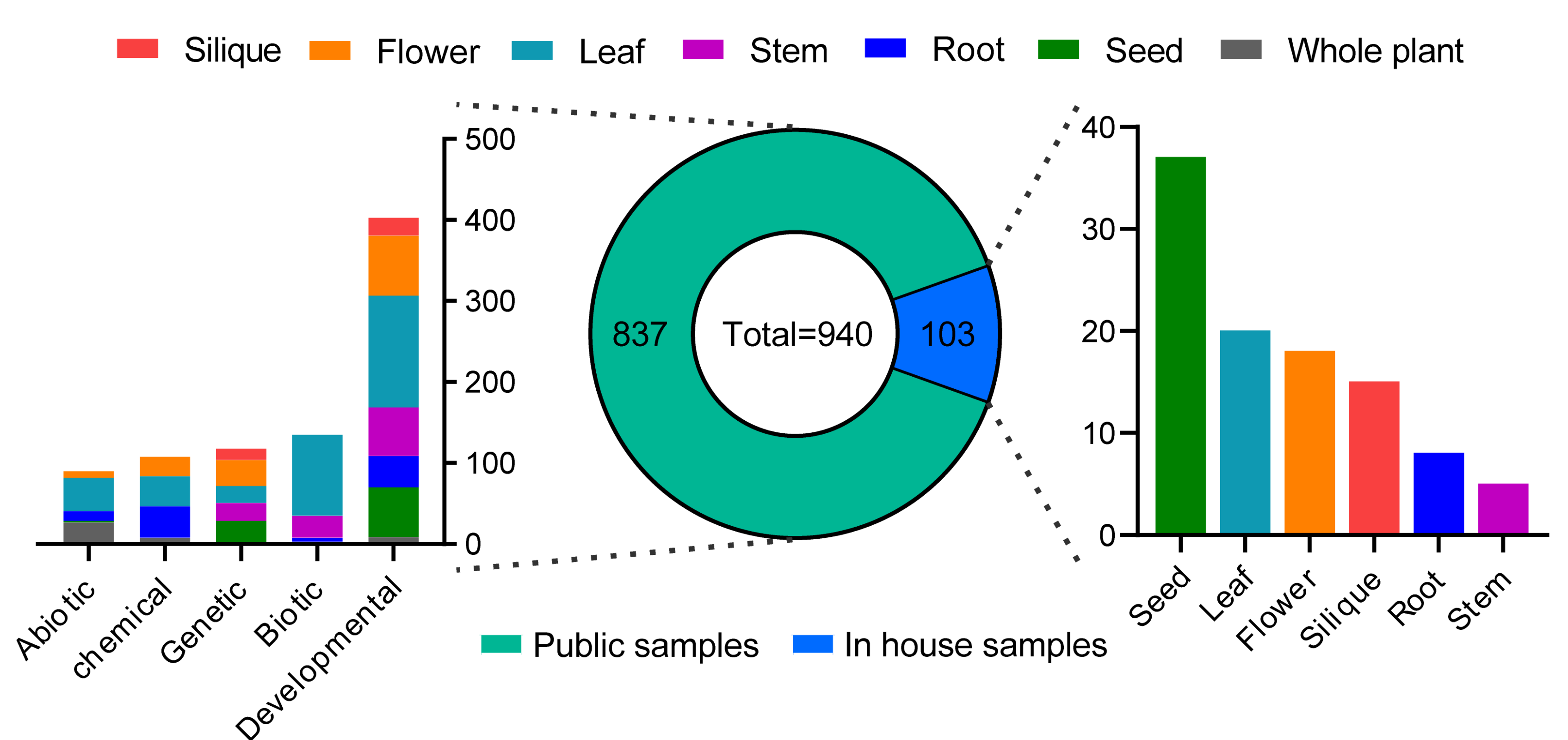
2.2. RNA-Seq-Based Global Expression Data of 837 Samples from SRA Database
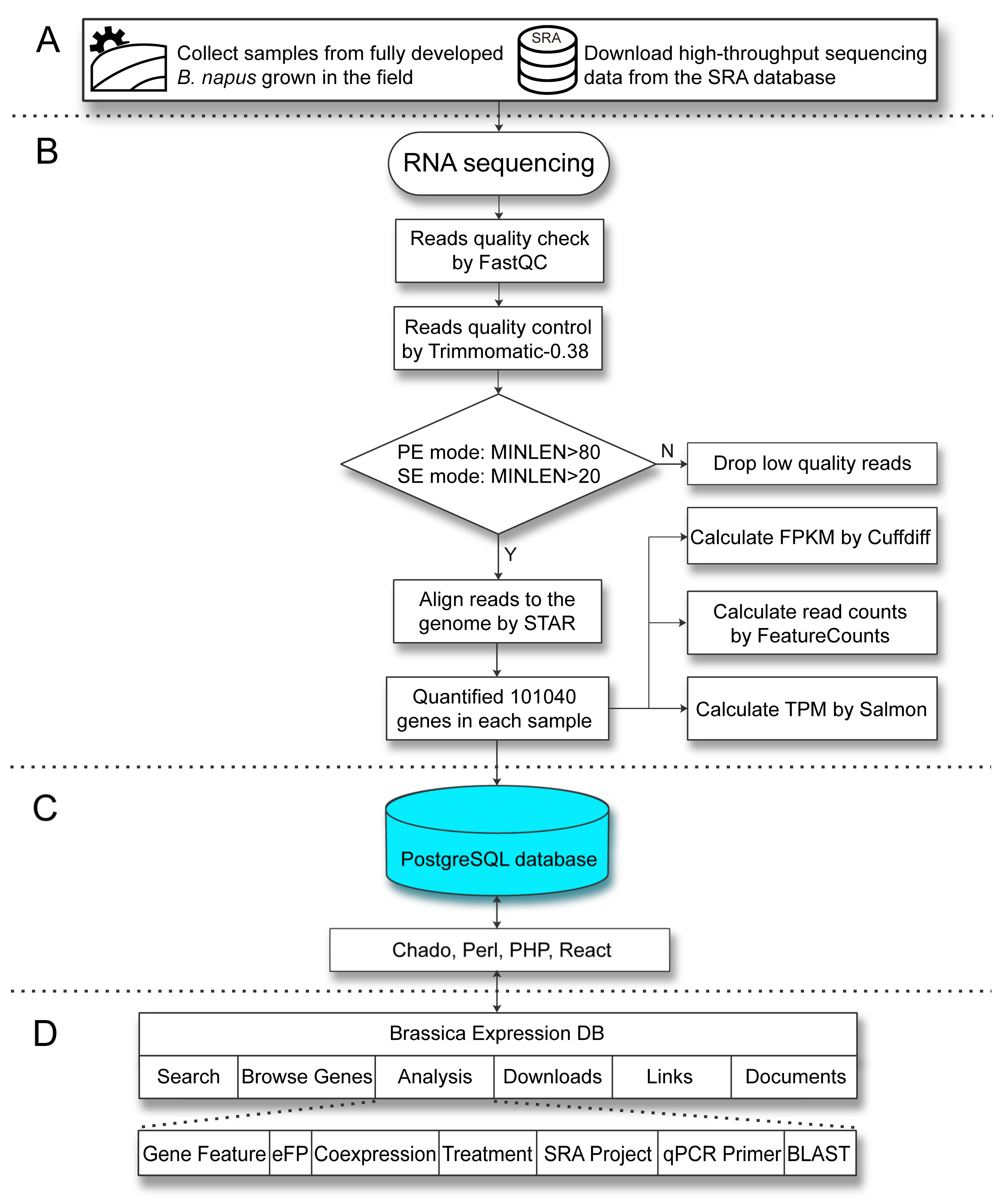
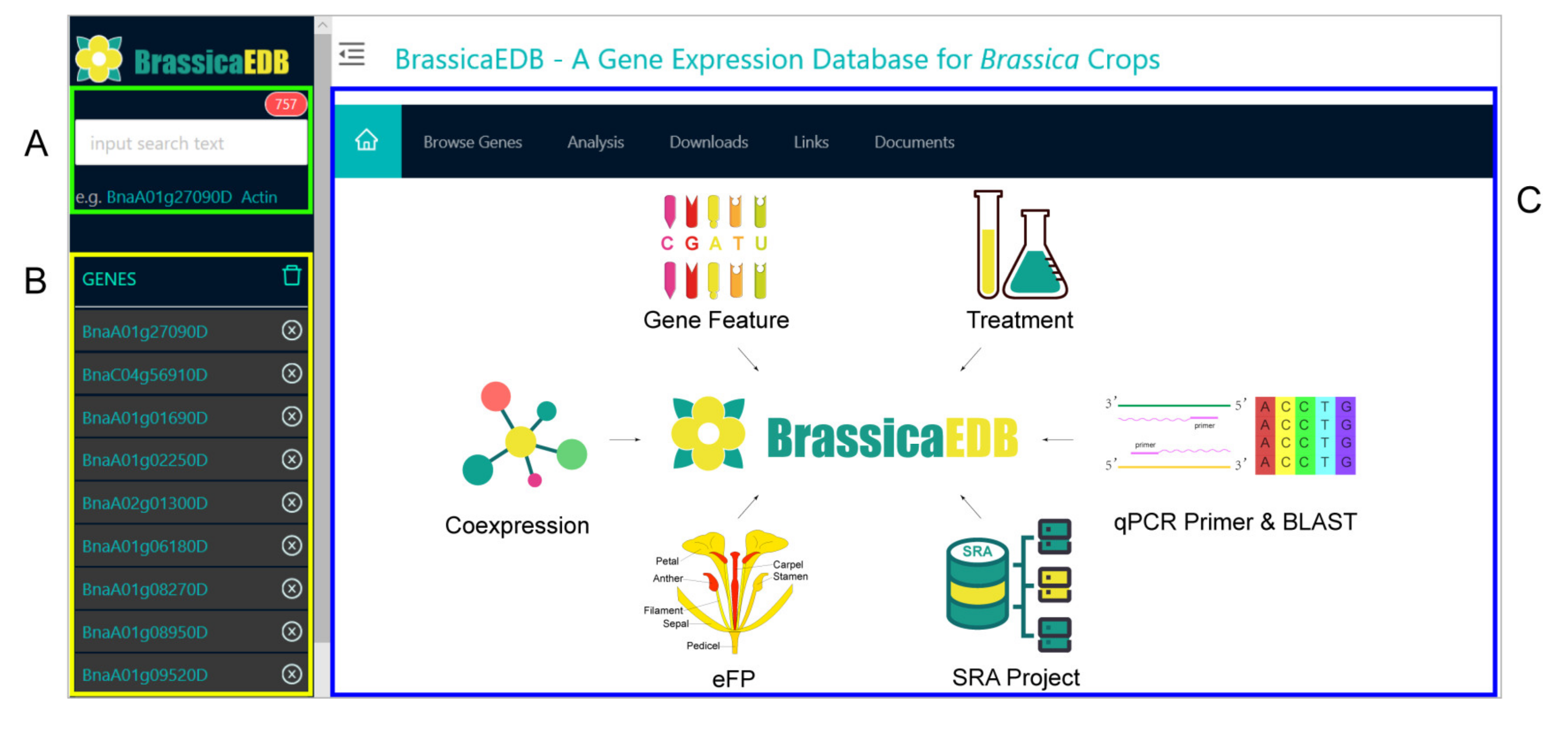
2.3. System Architecture and User Interface
2.3.1. Gene Feature Module
2.3.2. eFP Module
2.3.3. Coexpression Module
2.3.4. Treatment Module
2.3.5. SRA Project Module
2.3.6. BLAST and qPCR Primer Modules
2.3.7. Other Modules
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. RNA Isolation and Transcriptome Sequencing
4.3. Public Data Sources
4.4. RNA-Seq Data Analysis
4.5. System Architecture and Software for Database Construction
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ZS11 | ZhongShuang11 |
| RNA-Seq | RNA Sequencing |
| FPKM | Fragments Per Kilobase Million |
| TPM | Transcripts Per Million |
| eFP | Electronic Fluorescent Pictograph |
| SRA | Sequence Read Archive |
| NGS | Next-generation sequencing |
| GEO | Gene Expression Omnibus |
| NCBI | National Center for Biotechnology Information |
| EBI | European Bioinformatics Institute |
| HAG | Hours after germination |
| DAF | Days after flowering |
| GO | Gene Ontology |
| SVG | Scalable Vector Graphics |
| WGCNA | Weighted Gene Coexpression Netwoek Analysis |
| PCC | Pearson correlation coefficient |
| BLAST | Basic Local Alignment Search Tool |
| API | Application programming interfaces |
References
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The gene expression omnibus database. Stat. Sci. 2016, 93, 110. [Google Scholar]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update–from bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.K.K.; Tirnaz, S.; Dolatabadian, A.; Pires, J.C. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2018, 16, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Song, J.M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Xie, W.Z. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Ohyanagi, H.; Takano, T.; Terashima, S.; Kobayashi, M.; Kanno, M.; Morimoto, K.; Kanegae, H.; Sasaki, Y.; Saito, M.; Asano, S.; et al. Plant Omics Data Center: An integrated web repository for interspecies gene expression networks with NLP-based curation. Plant Cell Physiol. 2015, 56, e9. [Google Scholar] [CrossRef]
- Kudo, T.; Terashima, S.; Takaki, Y.; Tomita, K.; Saito, M.; Kanno, M.; Yokoyama, K.; Yano, K. PlantExpress: A database integrating OryzaExpress and ArthaExpress for single-species and cross-species gene expression network analyses with microarray-based transcriptome data. Plant Cell Physiol. 2017, 58, e1. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef]
- Lu, F.; Wei, Z.; Luo, Y.; Guo, H.; Zhang, G.; Xia, Q.; Wang, Y. SilkDB 3.0: Visualizing and exploring multiple levels of data for silkworm. Nucleic Acids Res. 2020, 48, D749–D755. [Google Scholar] [CrossRef] [PubMed]
- Yano, R.; Nonaka, S.; Ezura, H. Melonet-DB, a grand RNA-Seq gene expression atlas in melon (Cucumis melo L.). Plant Cell Physiol. 2018, 59, e4. [Google Scholar] [CrossRef] [PubMed]
- Waese, J.; Fan, J.; Pasha, A.; Yu, H.; Fucile, G.; Shi, R.; Cumming, M.; Kelley, L.A.; Sternberg, M.J.; Krishnakumar, V.; et al. ePlant: Visualizing and exploring multiple levels of data for hypothesis generation in plant biology. Plant Cell 2017, 29, 1806–1821. [Google Scholar] [CrossRef] [PubMed]
- Nagaharu, U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 1935, 7, 389–452. [Google Scholar]
- Becker, M.G.; Zhang, X.; Walker, P.L.; Wan, J.C.; Millar, J.L.; Khan, D.; Belmonte, M.F. Transcriptome analysis of the Brassica napus–Leptosphaeria maculans pathosystem identifies receptor, signaling and structural genes underlying plant resistance. Plant J. 2017, 90, 573–586. [Google Scholar] [CrossRef]
- Wei, L.; Jian, H.; Lu, K.; Filardo, F.; Yin, N.; Liu, L.; Li, J. Genome-wide association analysis and differential expression analysis of resistance to Sclerotinia stem rot in Brassica napus. Plant Biotechnol. J. 2016, 14, 1368–1380. [Google Scholar] [CrossRef]
- Hong, M.; Hu, K.; Tian, T.; Li, X.; Chen, L.; Zhang, Y.; Fu, T. Transcriptomic analysis of seed coats in yellow-seeded Brassica napus reveals novel genes that influence proanthocyanidin biosynthesis. Front. Plant Sci. 2017, 8, 1674. [Google Scholar] [CrossRef]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS 11′ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. [Google Scholar] [CrossRef]
- Winter, D.; Vinegar, B.; Nahal, H.; Ammar, R.; Wilson, G.V.; Provart, N.J. An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2007, 2, e718. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Lu, K.; Li, T.; He, J.; Chang, W.; Zhang, R.; Liu, M.; Yu, M.; Fan, Y.; Ma, J.; Sun, W.; et al. qPrimerDB: A thermodynamics-based gene-specific qPCR primer database for 147 organisms. Nucleic Acids Res. 2018, 46, D1229–D1236. [Google Scholar] [CrossRef] [PubMed]
- Mungall, C.J.; Emmert, D.B.; FlyBase, C. A Chado case study: An ontology-based modular schema for representing genome-associated biological information. Bioinformatics 2007, 23, i337–i346. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Lowe, A.J.; Jones, A.E.; Raybould, A.F.; Trick, M.; Moule, C.L.; Edwards, K.J. Transferability and genome specificity of a new set of microsatellite primers among Brassica species of the U triangle. Mol. Ecol. Notes 2002, 2, 7–11. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, H.H.; Wang, X. Machine learning for big data analytics in plants. Trends Plant Sci. 2014, 19, 798–808. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, H.; Li, T.; Luo, C.; Huang, H.; Ruan, Y.; Li, X.; Niu, Y.; Fan, Y.; Sun, W.; Zhang, K.; et al. BrassicaEDB: A Gene Expression Database for Brassica Crops. Int. J. Mol. Sci. 2020, 21, 5831. https://doi.org/10.3390/ijms21165831
Chao H, Li T, Luo C, Huang H, Ruan Y, Li X, Niu Y, Fan Y, Sun W, Zhang K, et al. BrassicaEDB: A Gene Expression Database for Brassica Crops. International Journal of Molecular Sciences. 2020; 21(16):5831. https://doi.org/10.3390/ijms21165831
Chicago/Turabian StyleChao, Haoyu, Tian Li, Chaoyu Luo, Hualei Huang, Yingfei Ruan, Xiaodong Li, Yue Niu, Yonghai Fan, Wei Sun, Kai Zhang, and et al. 2020. "BrassicaEDB: A Gene Expression Database for Brassica Crops" International Journal of Molecular Sciences 21, no. 16: 5831. https://doi.org/10.3390/ijms21165831
APA StyleChao, H., Li, T., Luo, C., Huang, H., Ruan, Y., Li, X., Niu, Y., Fan, Y., Sun, W., Zhang, K., Li, J., Qu, C., & Lu, K. (2020). BrassicaEDB: A Gene Expression Database for Brassica Crops. International Journal of Molecular Sciences, 21(16), 5831. https://doi.org/10.3390/ijms21165831