KCND3-Related Neurological Disorders: From Old to Emerging Clinical Phenotypes

 , , ,
, , ,
Abstract
1. Introduction
2. Results
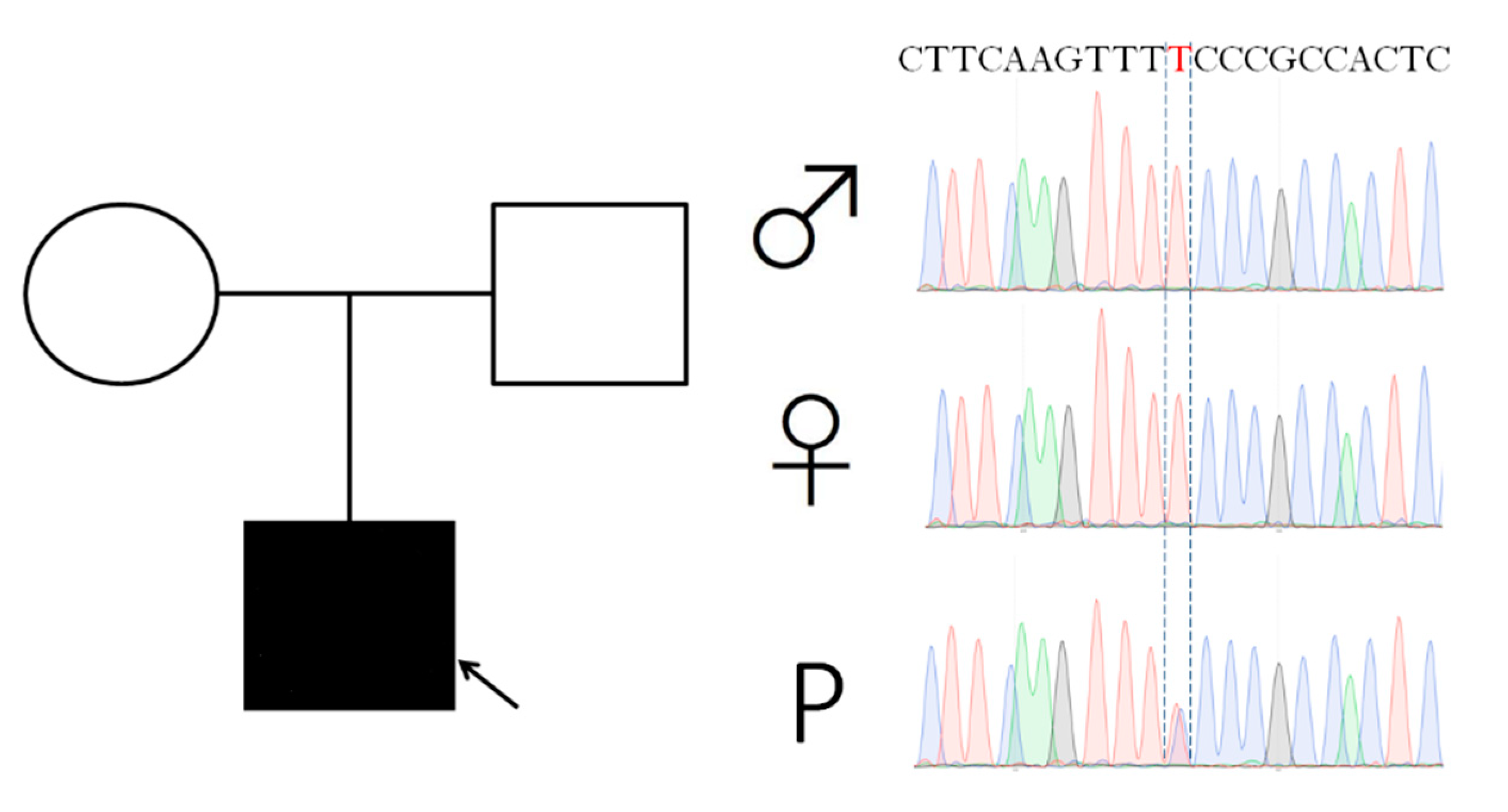
2.1. Case Report
2.2. Age at Onset, Presentation, and Disease Course
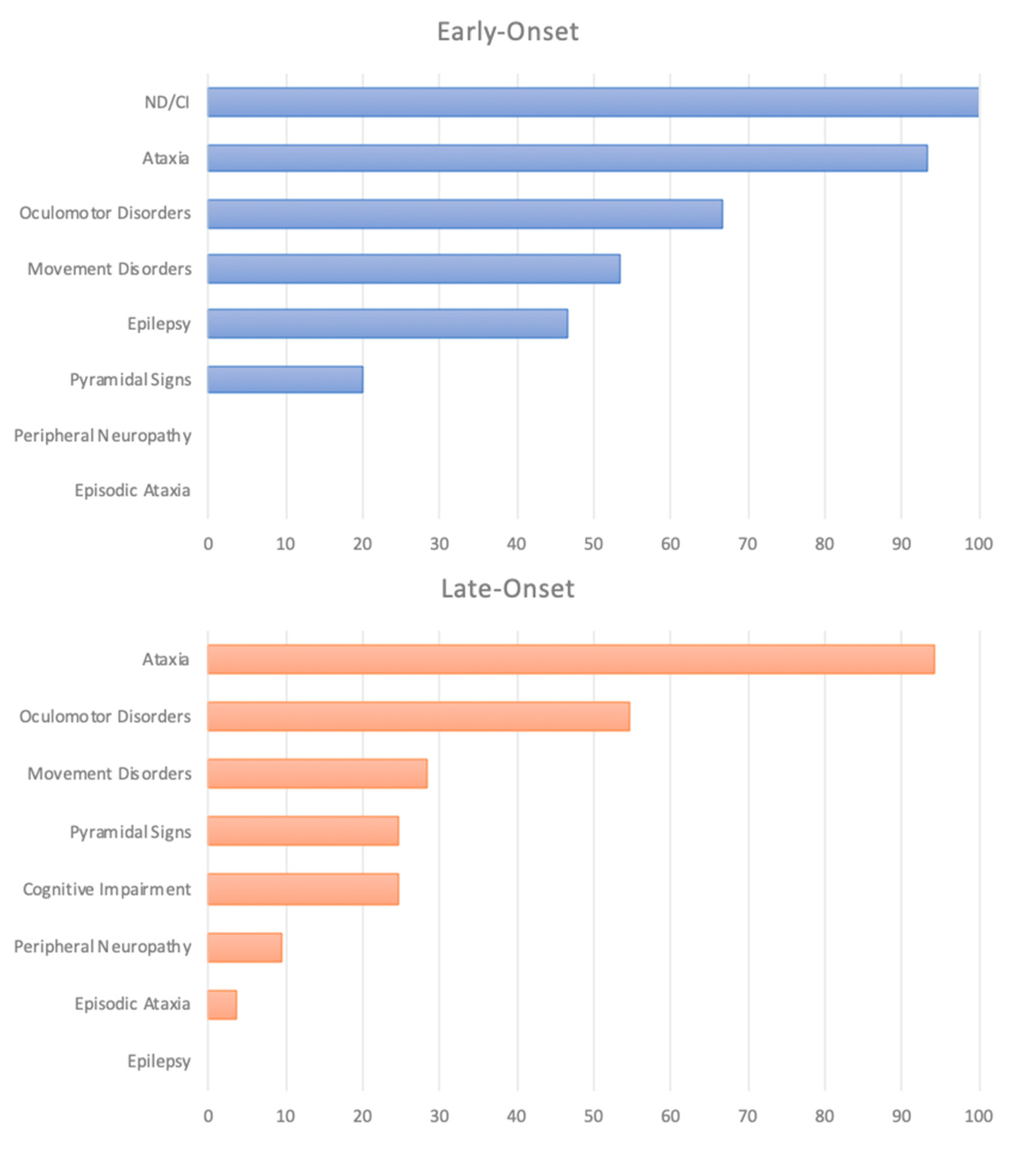
2.3. Clinical Phenotype According to the Age of Onset
2.4. Ataxia
2.5. Oculomotor Disorders
2.6. Episodic Ataxia and Other Paroxysmal Motor and Non-Motor Disorders
2.7. Cognition and Behavioral Disorders
2.8. Movement Disorders
2.9. Epilepsy
2.10. Other Features: Pyramidal Signs and Peripheral Neuropathy
2.11. Neuroimaging
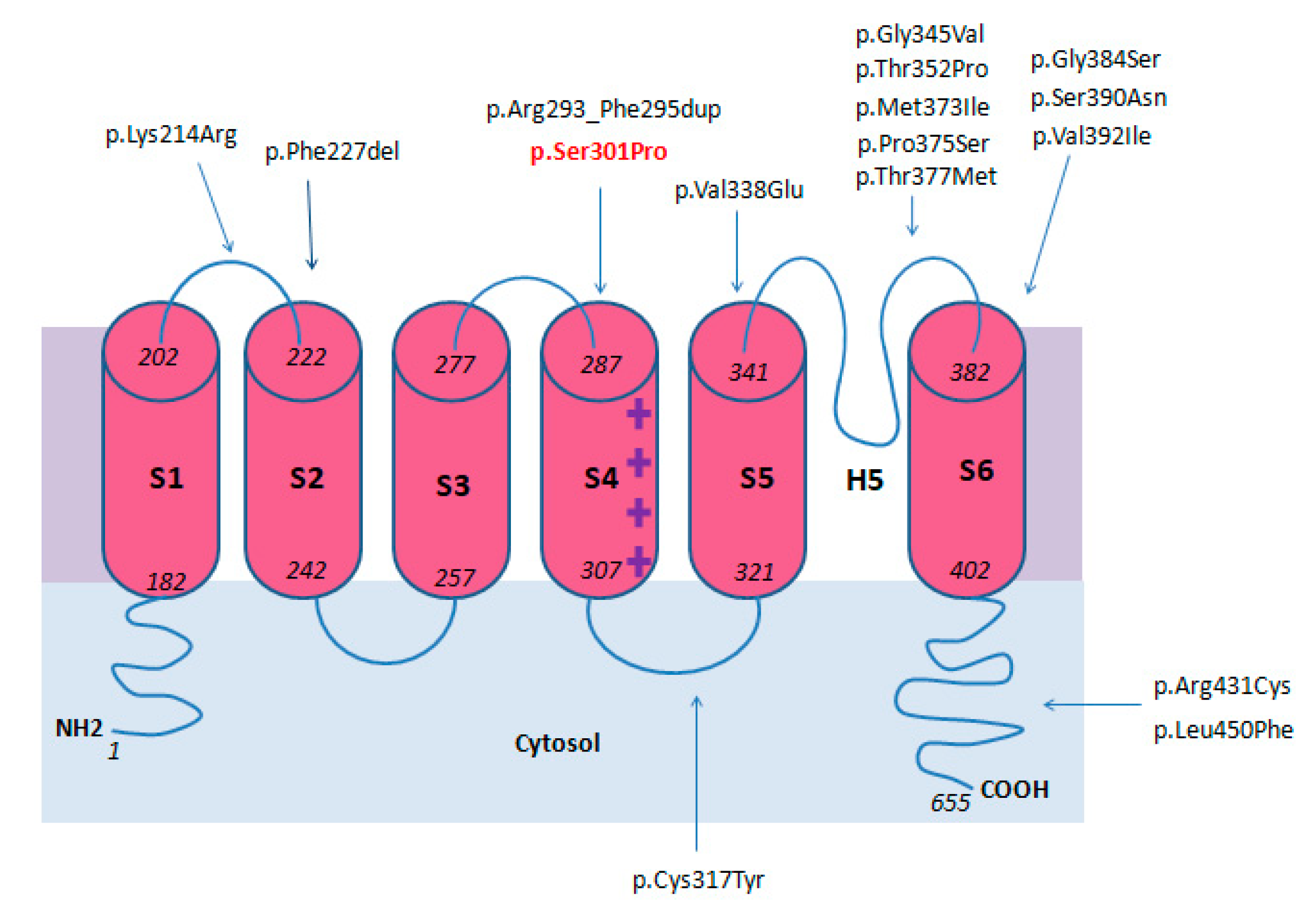
2.12. Genotype
3. Discussion
4. Methods and Materials
4.1. Case Report
4.2. Molecular Studies
4.3. Literature Review
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, D.M.; Nimigean, C.M. Voltage-Gated Potassium Channels: A Structural Examination of Selectivity and Gating. Cold Spring Harb. Perspect. Biol. 2016, 8, a029231. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.W.C.; Leung, Y.-M. The Versatile Kv Channels in the Nervous System: Actions beyond Action Potentials. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Grizel, A.V.; Glukhov, G.S.; Sokolova, O.S. Mechanisms of Activation of Voltage-Gated Potassium Channels. Acta Nat. 2014, 6, 10–26. [Google Scholar] [CrossRef]
- Birnbaum, S.G.; Varga, A.W.; Yuan, L.-L.; Anderson, A.E.; Sweatt, J.D.; Schrader, L.A. Structure and Function of Kv4-Family Transient Potassium Channels. Physiol. Rev. 2004, 84, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-C.; Durr, A.; Majczenko, K.; Huang, Y.-H.; Liu, Y.-C.; Lien, C.-C.; Tsai, P.-C.; Ichikawa, Y.; Goto, J.; Monin, M.-L.; et al. Mutations in KCND3 Cause Spinocerebellar Ataxia Type 22. Ann. Neurol. 2012, 72, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.; Fu, S.; Liu, Y.; Lu, Y.; Zhong, C.; Tang, C.; Soong, B.; Jeng, C. Novel SCA19/22-associated KCND3 Mutations Disrupt Human K V 4.3 Protein Biosynthesis and Channel Gating. Hum. Mutat. 2019, 40, 2088–2107. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Huang, H.-Y.; Tsaur, M.-L. Contrasting Expression of Kv4.3, an A-Type K Channel, in Migrating Purkinje Cells and Other Post-Migratory Cerebellar Neurons. Eur. J. Neurosci. 2003, 18, 601–612. [Google Scholar] [CrossRef]
- Duarri, A.; Jezierska, J.; Fokkens, M.; Meijer, M.; Schelhaas, H.J.; den Dunnen, W.F.A.; van Dijk, F.; Verschuuren-Bemelmans, C.; Hageman, G.; van de Vlies, P.; et al. Mutations in Potassium Channel Kcnd3 Cause Spinocerebellar Ataxia Type 19. Ann. Neurol. 2012, 72, 870–880. [Google Scholar] [CrossRef]
- Schelhaas, H.J.; Ippel, P.F.; Hageman, G.; Sinke, R.J.; van der Laan, E.N.; Beemer, F.A. Clinical and Genetic Analysis of a Four-Generation Family with a Distinct Autosomal Dominant Cerebellar Ataxia. J. Neurol. 2001, 248, 113–120. [Google Scholar] [CrossRef]
- Chung, M.; Lu, Y.; Cheng, N.; Soong, B. A Novel Autosomal Dominant Spinocerebellar Ataxia (SCA22) Linked to Chromosome 1p21-q23. Brain 2003, 126, 1293–1299. [Google Scholar] [CrossRef]
- Harding, A.E. The clinical features and classification of the late onset autosomal dominant cerebellar ataxias: A study of 11 families, including descendants of ‘the drew family of walworth. Brain 1982, 105, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Duarri, A.; Nibbeling, E.; Fokkens, M.R.; Meijer, M.; Boddeke, E.; Lagrange, E.; Stevanin, G.; Brice, A.; Durr, A.; Verbeek, D.S. The L450P Mutation in KCND3 Brings Spinocerebellar Ataxia and Brugada Syndrome Closer Together. Neurogenetics 2013, 14, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Smets, K.; Duarri, A.; Deconinck, T.; Ceulemans, B.; van de Warrenburg, B.P.; Züchner, S.; Gonzalez, M.A.; Schüle, R.; Synofzik, M.; Van der Aa, N.; et al. First de Novo KCND3 Mutation Causes Severe Kv4.3 Channel Dysfunction Leading to Early Onset Cerebellar Ataxia, Intellectual Disability, Oral Apraxia and Epilepsy. BMC Med. Genet. 2015, 16, 51. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Koh, K.; Namekawa, M.; Takiyama, Y. Whole-Exome Sequencing Reveals a Missense Mutation in the KCND3 Gene in a Patient with SCA19/22. Neurol. Clin. Neurosci. 2015, 3, 197–199. [Google Scholar] [CrossRef]
- Choi, K.-D.; Kim, J.-S.; Kim, H.-J.; Jung, I.; Jeong, S.-H.; Lee, S.-H.; Kim, D.U.; Kim, S.-H.; Choi, S.Y.; Shin, J.-H.; et al. Genetic Variants Associated with Episodic Ataxia in Korea. Sci. Rep. 2017, 7, 13855. [Google Scholar] [CrossRef]
- Huin, V.; Strubi-Vuillaume, I.; Dujardin, K.; Brion, M.; Delliaux, M.; Dellacherie, D.; Cuvellier, J.-C.; Cuisset, J.-M.; Riquet, A.; Moreau, C.; et al. Expanding the Phenotype of SCA19/22: Parkinsonism, Cognitive Impairment and Epilepsy. Parkinsonism Relat. Disord. 2017, 45, 85–89. [Google Scholar] [CrossRef]
- Kurihara, M.; Ishiura, H.; Sasaki, T.; Otsuka, J.; Hayashi, T.; Terao, Y.; Matsukawa, T.; Mitsui, J.; Kaneko, J.; Nishiyama, K.; et al. Novel De Novo KCND3 Mutation in a Japanese Patient with Intellectual Disability, Cerebellar Ataxia, Myoclonus, and Dystonia. Cerebellum 2018, 17, 237–242. [Google Scholar] [CrossRef]
- Paucar, M.; Bergendal, Å.; Gustavsson, P.; Nordenskjöld, M.; Laffita-Mesa, J.; Savitcheva, I.; Svenningsson, P. Novel Features and Abnormal Pattern of Cerebral Glucose Metabolism in Spinocerebellar Ataxia 19. Cerebellum 2018, 17, 465–476. [Google Scholar] [CrossRef]
- Wang, J.; Wen, Y.; Zhang, Q.; Yu, S.; Chen, Y.; Wu, X.; Zhang, Y.; Bao, X. Gene Mutational Analysis in a Cohort of Chinese Children with Unexplained Epilepsy: Identification of a New KCND3 Phenotype and Novel Genes Causing Dravet Syndrome. Seizure 2019, 66, 26–30. [Google Scholar] [CrossRef]
- Coutelier, M.; Hammer, M.B.; Stevanin, G.; Monin, M.-L.; Davoine, C.-S.; Mochel, F.; Labauge, P.; Ewenczyk, C.; Ding, J.; Gibbs, J.R.; et al. Efficacy of Exome-Targeted Capture Sequencing to Detect Mutations in Known Cerebellar Ataxia Genes. JAMA Neurol. 2018, 75, 591. [Google Scholar] [CrossRef]
- Olesen, M.S.; Refsgaard, L.; Holst, A.G.; Larsen, A.P.; Grubb, S.; Haunsø, S.; Svendsen, J.H.; Olesen, S.-P.; Schmitt, N.; Calloe, K. A Novel KCND3 Gain-of-Function Mutation Associated with Early-Onset of Persistent Lone Atrial Fibrillation. Cardiovasc. Res. 2013, 98, 488–495. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Bendl, J.; Musil, M.; Štourač, J.; Zendulka, J.; Damborský, J.; Brezovský, J. PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PLoS Comput. Biol. 2016, 12, e1004962. [Google Scholar] [CrossRef]
- Venselaar, H.; te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein Structure Analysis of Mutations Causing Inheritable Diseases. An e-Science Approach with Life Scientist Friendly Interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Kheradmand, A.; Zee, D.S. Cerebellum and Ocular Motor Control. Front. Neurol. 2011. [Google Scholar] [CrossRef]
- Manto, M.; Bower, J.M.; Conforto, A.B.; Delgado-García, J.M.; da Guarda, S.N.F.; Gerwig, M.; Habas, C.; Hagura, N.; Ivry, R.B.; Mariën, P.; et al. Consensus Paper: Roles of the Cerebellum in Motor Control—The Diversity of Ideas on Cerebellar Involvement in Movement. Cerebellum 2012, 11, 457–487. [Google Scholar] [CrossRef]
- Strupp, M.; Hüfner, K.; Sandmann, R.; Zwergal, A.; Dieterich, M.; Jahn, K.; Brandt, T. Central Oculomotor Disturbances and Nystagmus. Dtsch. Aerzteblatt Online 2011. [Google Scholar] [CrossRef]
- Piarroux, J.; Riant, F.; Humbertclaude, V.; Remerand, G.; Hadjadj, J.; Rejou, F.; Coubes, C.; Pinson, L.; Meyer, P.; Roubertie, A. FGF14-related Episodic Ataxia: Delineating the Phenotype of Episodic Ataxia Type 9. Ann. Clin. Transl. Neurol. 2020, 7, 565–572. [Google Scholar] [CrossRef]
- Gennarino, V.A.; Palmer, E.E.; McDonell, L.M.; Wang, L.; Adamski, C.J.; Koire, A.; See, L.; Chen, C.-A.; Schaaf, C.P.; Rosenfeld, J.A.; et al. A Mild PUM1 Mutation Is Associated with Adult-Onset Ataxia, Whereas Haploinsufficiency Causes Developmental Delay and Seizures. Cell 2018, 172, 924–936.e11. [Google Scholar] [CrossRef]
- Waters, M.F.; Minassian, N.A.; Stevanin, G.; Figueroa, K.P.; Bannister, J.P.A.; Nolte, D.; Mock, A.F.; Evidente, V.G.H.; Fee, D.B.; Müller, U.; et al. Mutations in Voltage-Gated Potassium Channel KCNC3 Cause Degenerative and Developmental Central Nervous System Phenotypes. Nat. Genet. 2006, 38, 447–451. [Google Scholar] [CrossRef]
- Kullmann, D.M.; Waxman, S.G. Neurological Channelopathies: New Insights into Disease Mechanisms and Ion Channel Function: Neurological Channelopathies. J. Physiol. 2010, 588, 1823–1827. [Google Scholar] [CrossRef]
- Spillane, J.; Kullmann, D.M.; Hanna, M.G. Genetic Neurological Channelopathies: Molecular Genetics and Clinical Phenotypes. J. Neurol. Neurosurg. Psychiatry 2015. [Google Scholar] [CrossRef]
- Seidel, K.; Küsters, B.; den Dunnen, W.F.A.; Bouzrou, M.; Hageman, G.; Korf, H.-W.; Schelhaas, H.J.; Verbeek, D.; Rüb, U. First Patho-Anatomical Investigation of the Brain of a SCA19 Patient. Neuropathol. Appl. Neurobiol. 2014, 40, 640–644. [Google Scholar] [CrossRef]
- Bostan, A.C.; Strick, P.L. The Basal Ganglia and the Cerebellum: Nodes in an Integrated Network. Nat. Rev. Neurosci. 2018, 19, 338–350. [Google Scholar] [CrossRef]
- Bologna, M.; Berardelli, A. Cerebellum: An Explanation for Dystonia? Cerebellum Ataxias 2017, 4, 6. [Google Scholar] [CrossRef]
- Wu, T.; Hallett, M. The Cerebellum in Parkinson’s Disease. Brain 2013, 136, 696–709. [Google Scholar] [CrossRef]
- Bareš, M.; Apps, R.; Avanzino, L.; Breska, A.; D’Angelo, E.; Filip, P.; Gerwig, M.; Ivry, R.B.; Lawrenson, C.L.; Louis, E.D.; et al. Consensus Paper: Decoding the Contributions of the Cerebellum as a Time Machine. From Neurons to Clinical Applications. Cerebellum 2019, 18, 266–286. [Google Scholar] [CrossRef]
- Choi, S.-M. Movement Disorders Following Cerebrovascular Lesions in Cerebellar Circuits. JMD 2016, 9, 80–88. [Google Scholar] [CrossRef]
- Levy, A.; Lang, A.E. Ataxia-Telangiectasia: A Review of Movement Disorders, Clinical Features, and Genotype Correlations: Ataxia-Telangiectasia. Mov. Disord. 2018, 33, 1238–1247. [Google Scholar] [CrossRef]
- Rossi, M.; Balint, B.; Millar Vernetti, P.; Bhatia, K.P.; Merello, M. Genetic Dystonia-Ataxia Syndromes: Clinical Spectrum, Diagnostic Approach, and Treatment Options: Genetic Dystonia-Ataxia Syndromes. Mov. Disord. Clin. Pract. 2018, 5, 373–382. [Google Scholar] [CrossRef]
- Kuo, P.-H.; Gan, S.-R.; Wang, J.; Lo, R.Y.; Figueroa, K.P.; Tomishon, D.; Pulst, S.M.; Perlman, S.; Wilmot, G.; Gomez, C.M.; et al. Dystonia and Ataxia Progression in Spinocerebellar Ataxias. Parkinsonism Relat. Disord. 2017, 45, 75–80. [Google Scholar] [CrossRef]
- Park, H.; Kim, H.-J.; Jeon, B.S. Parkinsonism in Spinocerebellar Ataxia. Biomed Res. Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Boesch, S.M.; Muller, J.; Wenning, G.K.; Poewe, W. Cervical Dystonia in Spinocerebellar Ataxia Type 2: Clinical and Polymyographic Findings. J. Neurol. Neurosurg. Psychiatry 2006, 78, 520–522. [Google Scholar] [CrossRef]
- Muglan, J.A.; Menon, S.; Jog, M.S. Pearls & Oy-Sters: Spinocerebellar Ataxia Type 3 Presenting with Cervical Dystonia without Ataxia. Neurology 2016, 86, e1–e3. [Google Scholar] [CrossRef]
- Charlesworth, G.; Mohire, M.D.; Schneider, S.A.; Stamelou, M.; Wood, N.W.; Bhatia, K.P. Ataxia Telangiectasia Presenting as Dopa-Responsive Cervical Dystonia. Neurology 2013, 81, 1148–1151. [Google Scholar] [CrossRef]
- Becker, A.E.; Vargas, W.; Pearson, T.S. Ataxia with Vitamin E Deficiency May Present with Cervical Dystonia. Tremor Other Hyperkinetic Mov. 2016. [Google Scholar] [CrossRef]
- Klier, E.M. Midbrain Control of Three-Dimensional Head Orientation. Science 2002, 295, 1314–1316. [Google Scholar] [CrossRef]
- Shaikh, A.G.; Zee, D.S.; Crawford, J.D.; Jinnah, H.A. Cervical Dystonia: A Neural Integrator Disorder. Brain 2016, 139, 2590–2599. [Google Scholar] [CrossRef]
- Batla, A.; Sánchez, M.C.; Erro, R.; Ganos, C.; Stamelou, M.; Balint, B.; Brugger, F.; Antelmi, E.; Bhatia, K.P. The Role of Cerebellum in Patients with Late Onset Cervical/Segmental Dystonia?–Evidence from the Clinic. Parkinsonism Relat. Disord. 2015, 21, 1317–1322. [Google Scholar] [CrossRef]
- Schmahmann, J. The Cerebellar Cognitive Affective Syndrome. Brain 1998, 121, 561–579. [Google Scholar] [CrossRef] [PubMed]
- Hoche, F.; Guell, X.; Vangel, M.G.; Sherman, J.C.; Schmahmann, J.D. The Cerebellar Cognitive Affective/Schmahmann Syndrome Scale. Brain 2018, 141, 248–270. [Google Scholar] [CrossRef] [PubMed]
- Brossard-Racine, M.; du Plessis, A.J.; Limperopoulos, C. Developmental Cerebellar Cognitive Affective Syndrome in Ex-Preterm Survivors Following Cerebellar Injury. Cerebellum 2015, 14, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Bulgheroni, S.; D’Arrigo, S.; Signorini, S.; Briguglio, M.; Di Sabato, M.L.; Casarano, M.; Mancini, F.; Romani, M.; Alfieri, P.; Battini, R.; et al. Cognitive, Adaptive, and Behavioral Features in Joubert Syndrome. Am. J. Med. Genet. 2016, 170, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Teive, H.A.G.; Arruda, W.O. Cognitive Dysfunction in Spinocerebellar Ataxias. Dement. Neuropsychol. 2009, 3, 180–187. [Google Scholar] [CrossRef]
- Moro, A.; Munhoz, R.P.; Moscovich, M.; Arruda, W.O.; Raskin, S.; Silveira-Moriyama, L.; Ashizawa, T.; Teive, H.A.G. Nonmotor Symptoms in Patients with Spinocerebellar Ataxia Type 10. Cerebellum 2017, 16, 938–944. [Google Scholar] [CrossRef]
- Lindsay, E.; Storey, E. Cognitive Changes in the Spinocerebellar Ataxias Due to Expanded Polyglutamine Tracts: A Survey of the Literature. Brain Sci. 2017, 7, 83. [Google Scholar] [CrossRef]
- Bürk, K.; Globas, C.; Bösch, S.; Klockgether, T.; Zühlke, C.; Daum, I.; Dichgans, J. Cognitive Deficits in Spinocerebellar Ataxia Type 1, 2, and 3. J. Neurol. 2003, 250, 207–211. [Google Scholar] [CrossRef]
- Moriarty, A.; Cook, A.; Hunt, H.; Adams, M.E.; Cipolotti, L.; Giunti, P. A Longitudinal Investigation into Cognition and Disease Progression in Spinocerebellar Ataxia Types 1, 2, 3, 6, and 7. Orphanet J. Rare Dis. 2016, 11, 82. [Google Scholar] [CrossRef]
- Swaminathan, A. Epilepsy in Spinocerebellar Ataxia Type 8: A Case Report. J. Med. Case Rep. 2019, 13, 333. [Google Scholar] [CrossRef]
- Marcián, V.; Filip, P.; Bareš, M.; Brázdil, M. Cerebellar Dysfunction and Ataxia in Patients with Epilepsy: Coincidence, Consequence, or Cause? Tremor Other Hyperkinetic Mov. 2016. [Google Scholar] [CrossRef]
- Waters, M.F. Spinocerebellar Ataxia Type 13. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Villa, C.; Combi, R. Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Front. Cell. Neurosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Köhling, R.; Wolfart, J. Potassium Channels in Epilepsy. Cold Spring Harb. Perspect Med. 2016, 6, a022871. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.M.; Azzopardi, P.S.; Wickremarathne, D.; Patton, G.C. The Age of Adolescence. Lancet Child Adolesc. Health 2018, 2, 223–228. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Age of Onset | 28 (1–90) |
|---|---|
| First symptom | Ataxia (42) |
| Neurodevelopmental disorders/cognitive impairment (5) | |
| Epilepsy (5) | |
| Episodic ataxia (2) | |
| Head tremor (2) | |
| Intermittent diplopia (1) | |
| Psychiatric symptoms (1) | |
| Ataxia | 64/68 |
| Episodic ataxia | 2/68 |
| Neurodevelopmental disorders/Cognitive impairment | 28/68 |
| Movement disorder | 23/68 |
| Parkinsonism | 12 |
| Tremor | 7 |
| Myoclonus | 5 |
| Dystonia | 3 |
| Epilepsy | 7/68 |
| Focal | 3 |
| Generalized | 2 |
| Mixed | 1 |
| Unspecified | 1 |
| Pyramidal signs | 16/68 |
| Oculomotor disorders | 39/68 |
| Saccadic Pursuit | 27 |
| Nystagmus | 22 |
| GEN | 16 |
| Unspecified | 11 |
| Downbeat Nystagmus | 1 |
| Dysmetric saccades | 3 |
| Vertical ophtalmoplegia | 1 |
| Supranuclear palsy | 1 |
| Slow saccades | 1 |
| Diplopia/Intermittent Diplopia | 2 |
| Peripheral Neuropathy | 5/68 |
| Patients (n./35) | References | |
|---|---|---|
| Normal | 5 | [9,13,16,19] |
| Global cerebellar atrophy | 17 | [5,6,9,14,17] |
| Hemispheric cerebellar atrophy | 2 | [9] |
| Vermian cerebellar atrophy | 11 | [5,8,16,18,20] |
| Cerebral atrophy | 5 | [6,9,17] |
| White matter lesions | 3 | [9,18] |
| Variant | N of Different Families | Location | Pathogenetic Mechanism | References |
|---|---|---|---|---|
| Phe227del rs397515475 | 4 | S2 domain | Impaired protein trafficking | [5,16] |
| Gly345Val rs797045634 | 2 | Pore loop | ND | [5] |
| Thr377Met rs1571636501 | 2 | Pore loop | Increased protein degradation Impaired protein trafficking Reduced outward K+ current | [5,6,18] |
| Ser390Asn rs397515478 | 2 | S6 domain | Increased protein degradation Impaired protein trafficking Reduced outward K+ current | [8,14] |
| Met373Ile rs397515477 | 1 | Pore loop | Increased protein degradation Impaired protein trafficking Reduced outward K+ current | [8] |
| Thr352Pro rs397515476 | 1 | Pore loop | Increased protein degradation Impaired protein trafficking Reduced outward K+ current | [8] |
| Val338Glu rs1571939827 | 1 | S5 Domain | Increased protein degradation Reduced outward K+ current | [5,6] |
| Leu450Phe rs150401343 | 1 | C-terminal tail | Gain of function | [12] |
| Arg293_295PheDup No rsID | 1 | S4 domain | Reduced outward K+ current function | [13] |
| Arg431Cys rs777183510 | 1 | C-terminal tail | ND | [15] |
| Gly384Ser No rsID | 1 | S6 domain | ND | [17] |
| Val392Ile rs786205867 | 1 | S6 domain | Gain of function | [19] |
| Lys214Arg rs142744204 | 1 | First extracellular loop | ND | [20] |
| Cys317Tyr rs1571939905 | 1 | Second intracellular loop | Increased protein degradation Reduced outward K+ current | [6] |
| Pro375Ser rs1571636508 | 1 | Pore loop | Increased protein degradation Impaired protein trafficking Reduced outward K+ current | [6] |
| Ser301Pro rs79821338 | 1 | S4 domain | ND | Current report |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pollini, L.; Galosi, S.; Tolve, M.; Caputi, C.; Carducci, C.; Angeloni, A.; Leuzzi, V. KCND3-Related Neurological Disorders: From Old to Emerging Clinical Phenotypes. Int. J. Mol. Sci. 2020, 21, 5802. https://doi.org/10.3390/ijms21165802
Pollini L, Galosi S, Tolve M, Caputi C, Carducci C, Angeloni A, Leuzzi V. KCND3-Related Neurological Disorders: From Old to Emerging Clinical Phenotypes. International Journal of Molecular Sciences. 2020; 21(16):5802. https://doi.org/10.3390/ijms21165802
Chicago/Turabian StylePollini, Luca, Serena Galosi, Manuela Tolve, Caterina Caputi, Carla Carducci, Antonio Angeloni, and Vincenzo Leuzzi. 2020. "KCND3-Related Neurological Disorders: From Old to Emerging Clinical Phenotypes" International Journal of Molecular Sciences 21, no. 16: 5802. https://doi.org/10.3390/ijms21165802
APA StylePollini, L., Galosi, S., Tolve, M., Caputi, C., Carducci, C., Angeloni, A., & Leuzzi, V. (2020). KCND3-Related Neurological Disorders: From Old to Emerging Clinical Phenotypes. International Journal of Molecular Sciences, 21(16), 5802. https://doi.org/10.3390/ijms21165802