Complete Chloroplast Genomes and Comparative Analyses of L. chinensis, L. anhuiensis, and L. aurea (Amaryllidaceae)

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
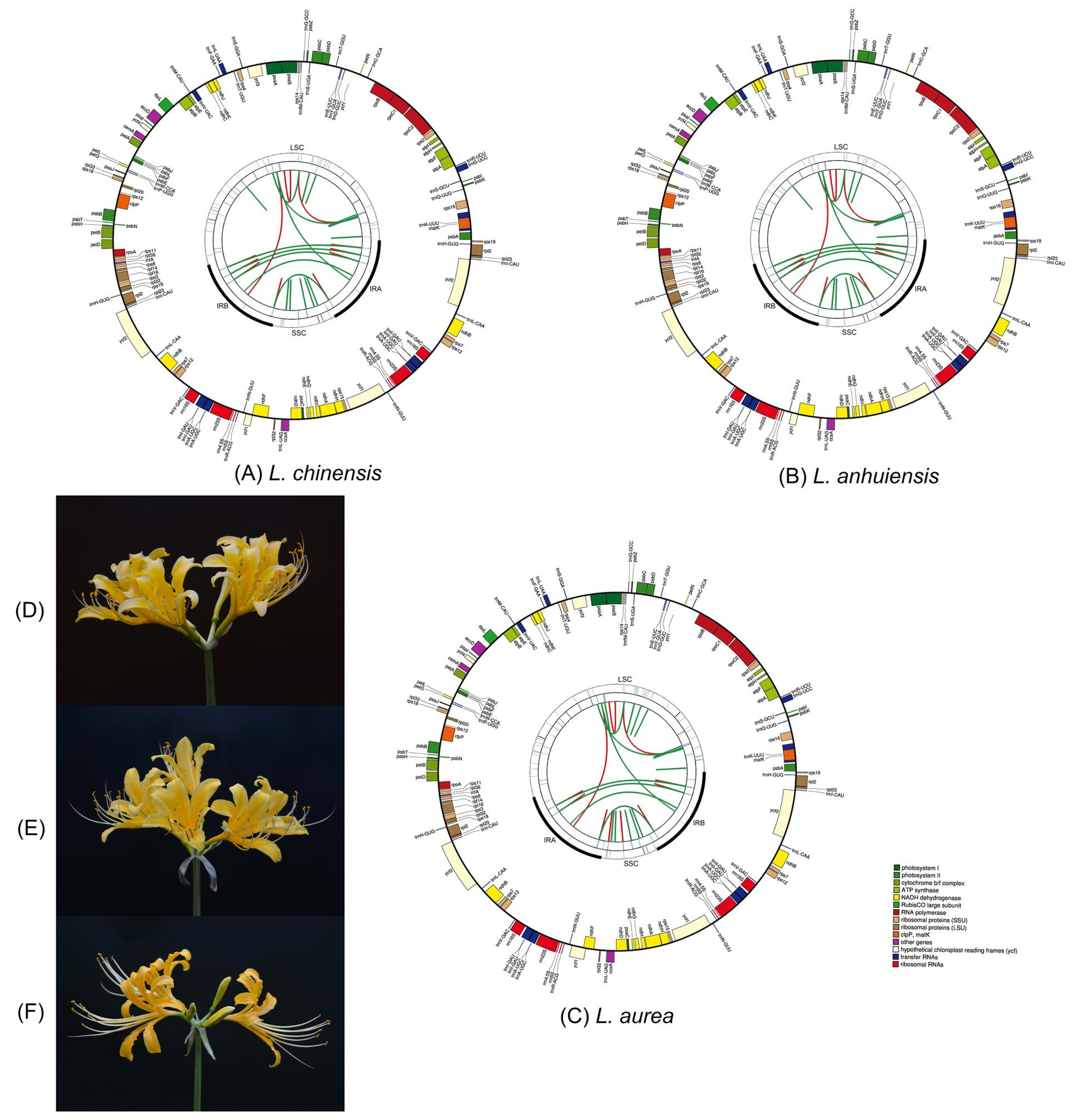
2.1. General Features of the Lycoris Chloroplast Genomes
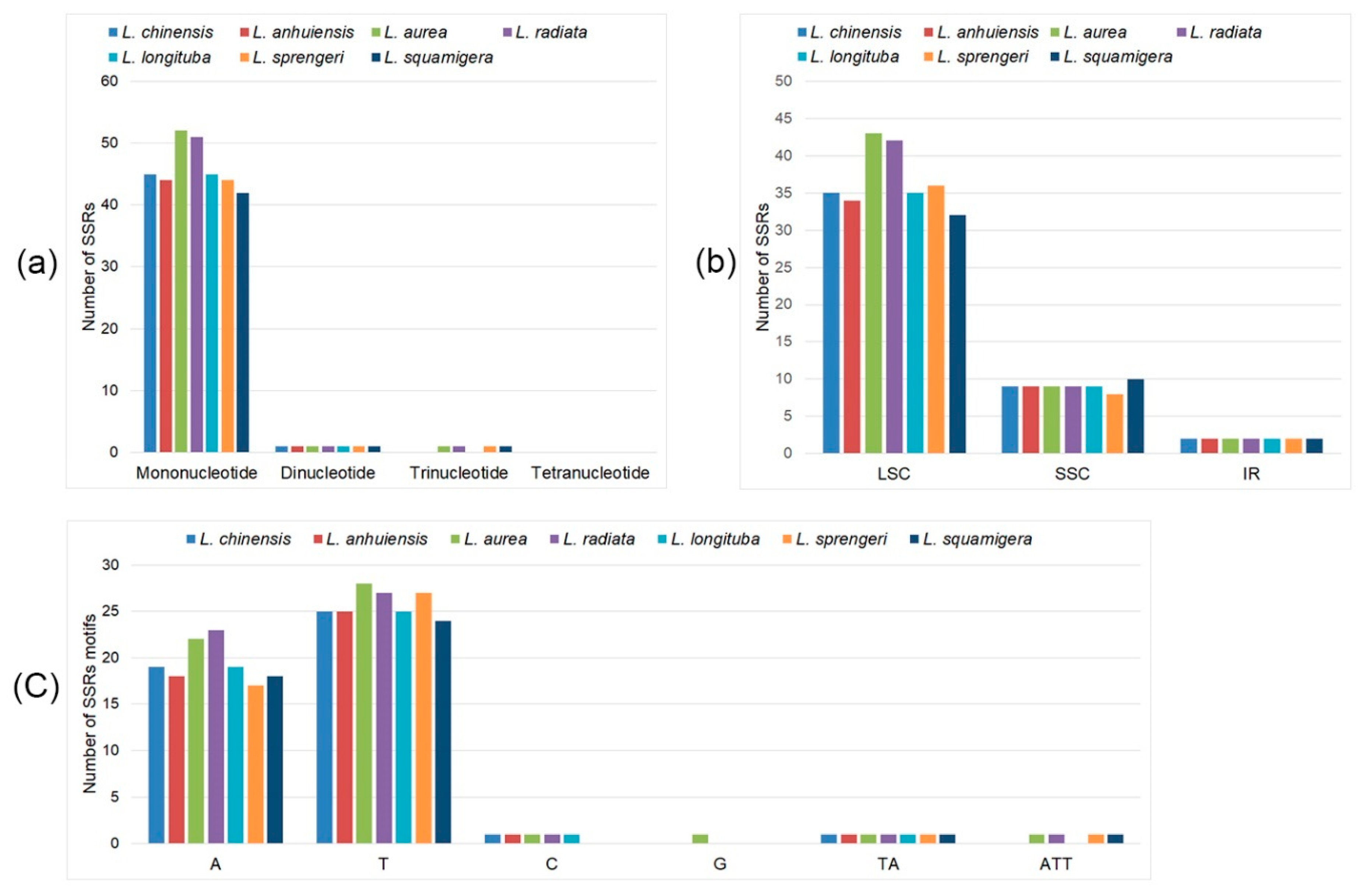
2.2. cpSSRs Analyses and Repeat Structures
2.3. Codon Usage
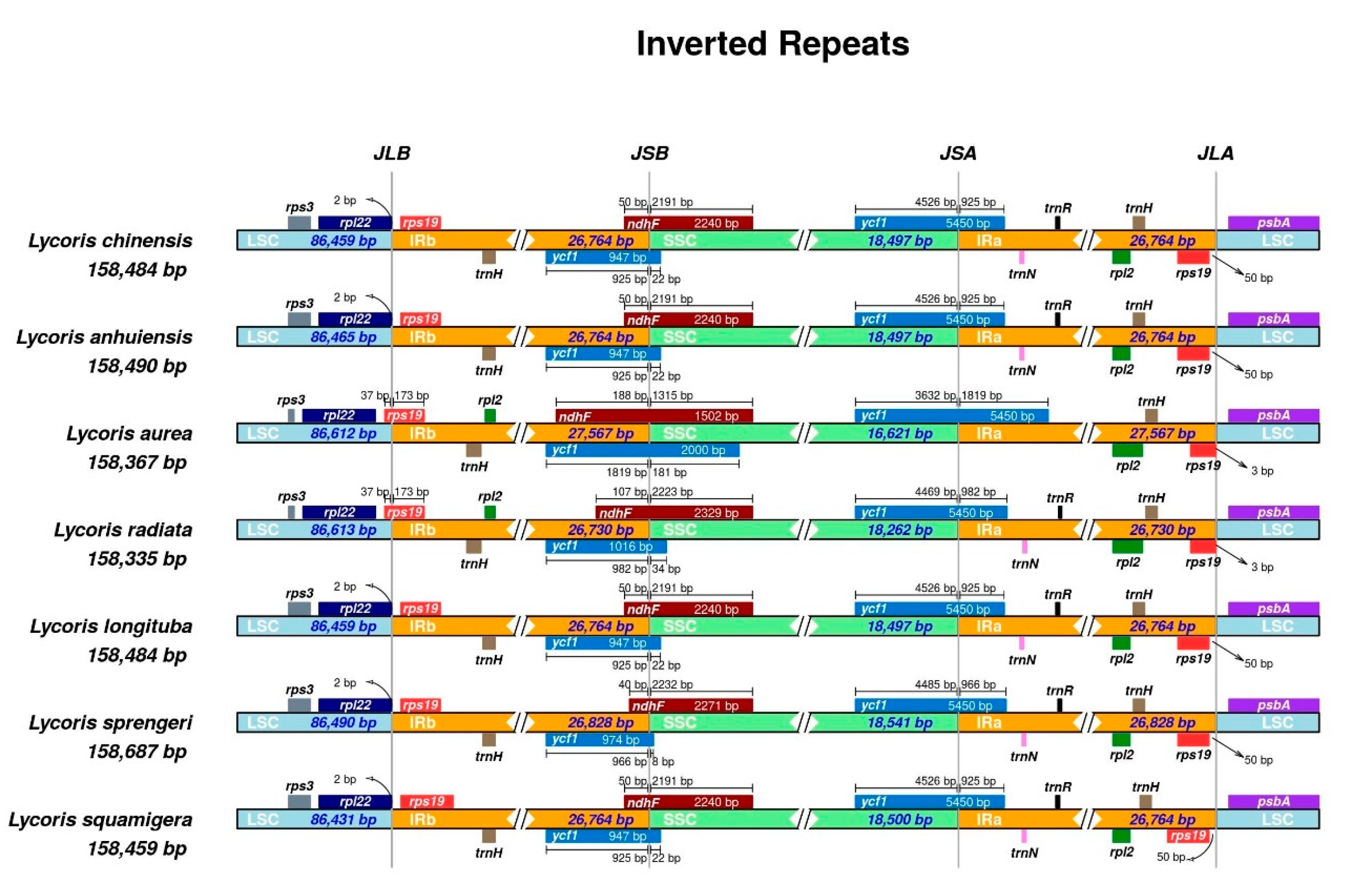
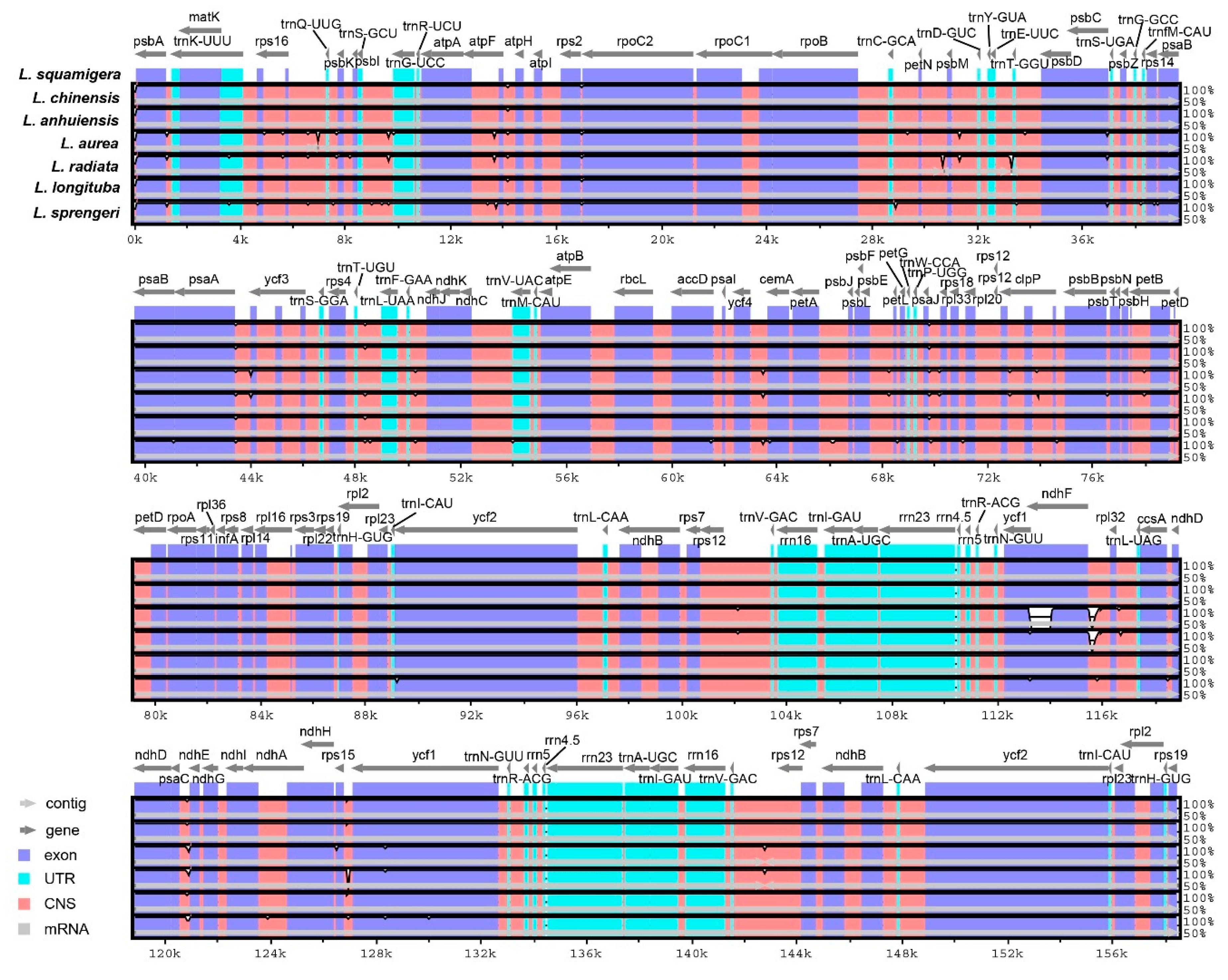
2.4. Inverted Repeats Contraction, Expansion, and Interspecific Comparison
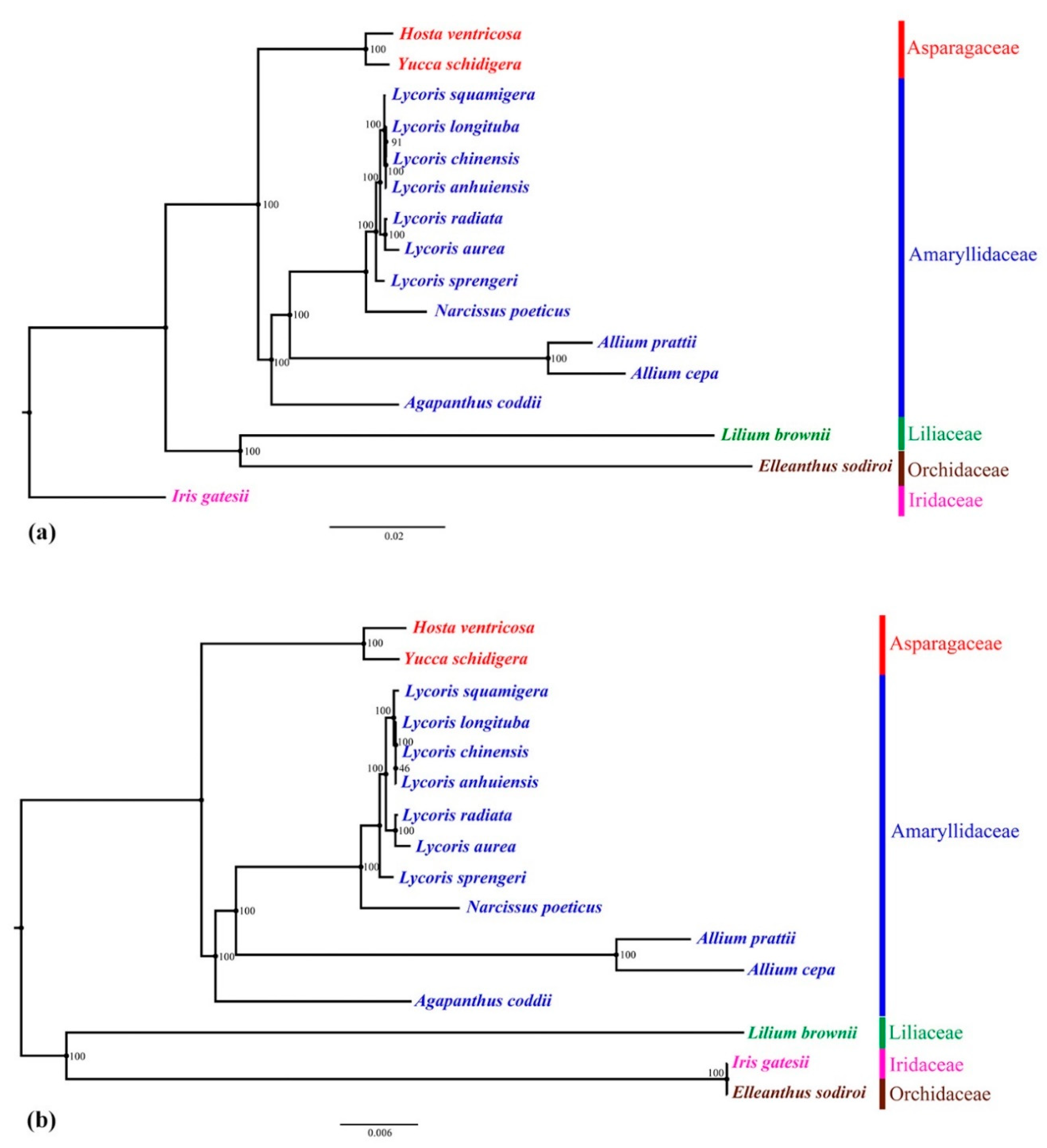
2.5. Phylogenetic Analysis
3. Materials and Methods
3.1. Sample Collection, DNA Extraction, and Sequencing
3.2. Cp Genome Assembly, Annotation and Structure Analysis
3.3. Interspecific Genome Comparison
3.4. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CP | Chloroplast |
| NGS | Next-generation sequencing |
| LSC | Large single copy |
| SSC | Small single copy |
| IR | Inverted repeat |
| GC | Guanine-cytosine |
| PCGs | Protein-coding genes |
| cpSSRs | Chloroplast simple sequence repeats |
| ISSR | inter-simple sequence repeat |
| RSCU | Relative synonymous codon usage |
| CNS | Conserved non-coding sequences |
| RAPD | Random amplification of polymorphic DNA |
| SCoT | Start codon targeted polymorphism |
| ML | Maximum likelihood |
| BIC | Bayesian information criterion |
References
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2003, 20, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, S.; Wang, N.; Xia, B.; Jiang, Y.; Wang, R. Transcriptome analysis of secondary metabolism pathway, transcription factors, and transporters in response to methyl jasmonate in Lycoris aurea. Front. Plant Sci. 2017, 7, 1971. [Google Scholar] [CrossRef] [PubMed]
- Benqi, Y.; Shoubiao, Z.; Qi, L.; Weihua, Q. Medicinal and ornamental values of genus Lycoris. Chin. Wild Plant Resour. 2006, 2, 29–32. [Google Scholar]
- Tsi, Z.; Meerow, A. Amaryllidaceae. Flora of China; Beijing Science Press & Missouri Botanical Garden Press: Beijing, China, 2000; Volume 24, p. 264. [Google Scholar]
- Quan, M.; Ou, L.; She, C. A new species of Lycoris (Amaryllidaceae) from Hunan, China. Novon 2013, 22, 307–310. [Google Scholar] [CrossRef]
- Meng, W.; Zheng, L.; Shao, J.; Zhou, S.; Liu, K. A new natural allotriploid, Lycoris × hubeiensis hybr. nov. (Amaryllidaceae), identified by morphological, karyological and molecular data. Nord. J. Bot. 2018, 36. [Google Scholar] [CrossRef]
- Shi, S.; Qiu, Y.; Li, E.; Wu, L.; Fu, C. Phylogenetic relationships and possible hybrid origin of Lycoris species (Amaryllidaceae) revealed by ITS sequences. Biochem. Genet. 2006, 44, 198–208. [Google Scholar] [CrossRef]
- Hori, T.; Hayashi, A.; Sasanuma, T.; Kurita, S. Genetic variations in the chloroplast genome and phylogenetic clustering of Lycoris species. Genes. Genet. Syst. 2006, 81, 243–253. [Google Scholar] [CrossRef]
- Tae, K.H.; Ko, S.C. A taxonomic study of the genus Lycoris (Amaryllidaceae) based on morphological characters. Trop. Med. Int. Health 1995, 9, 41–46. [Google Scholar] [CrossRef]
- Yuan, J.; Sun, S.; Peng, F.; Feng, X.; Zheng, Y.; Xia, B. Genetic variations in trnL-F sequence and phylogenetic clustering of Lycoris species. China J. Chin. Mater. Med. 2008, 33, 1523–1527. [Google Scholar]
- Shi, S.; Sun, Y.; Wei, L.; Lei, X.; Cameron, K.M.; Fu, C. Plastid DNA sequence data help to clarify phylogenetic relationships and reticulate evolution in Lycoris (Amaryllidaceae). Bot. J. Linn. Soc. 2014, 176, 115–126. [Google Scholar]
- Yoo, Y.K.; Yuan, T.; Lee, J.S.; Lee, A.K.; Roh, M.S.; Kurita, S.; Suh, J.K. Species relationships of Lycoris endemic to Korea evaluated by RAPD and SNPs of nrDNA-ITS regions. Hortic. Environ. Biote. 2011, 52, 145–151. [Google Scholar] [CrossRef]
- Shi, S.; Qiu, Y.; Wu, L.; Fu, C. Interspecific relationships of Lycoris (Amaryllidaceae) inferred from inter-simple sequence repeat data. Sci. Hortic-amsterdam 2006, 110, 285–291. [Google Scholar] [CrossRef]
- Gao, Y.; Zhu, Y.; Tong, Z.; Xu, Z.; Jiang, X.; Huang, C. Analysis of genetic diversity and relationships among genus Lycoris based on start codon targeted (SCoT) marker. Biochem. Syst. Ecol. 2014, 57, 221–226. [Google Scholar] [CrossRef]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Li, D.; Der Bank, M.V.; Twyford, A.D. Telling plant species apart with DNA: From barcodes to genomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150338. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D. Chloroplast DNA evolution and biosystematic uses of chloroplast DNA variation. Am. Nat. 1987, 130. [Google Scholar] [CrossRef]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.S.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef]
- Lu, Q.; Ye, W.; Lu, R.; Xu, W.; Qiu, Y. Phylogenomic and comparative analyses of complete plastomes of Croomia and Stemona (Stemonaceae). Int. J. Mol. Sci. 2018, 19, 2383. [Google Scholar] [CrossRef]
- Xue, S.; Shi, T.; Luo, W.; Ni, X.; Iqbal, S.; Ni, Z.; Huang, X.; Yao, D.; Shen, Z.; Gao, Z. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic. Res. Engl. 2019, 6, 89. [Google Scholar] [CrossRef]
- Li, X.; Zuo, Y.; Zhu, X.; Liao, S.; Ma, J. Complete chloroplast genomes and comparative analysis of sequences evolution among seven Aristolochia (Aristolochiaceae) medicinal species. Int. J. Mol. Sci. 2019, 20, 1045. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yang, J.; Park, J.; Yamada, T.; Maki, M.; Kim, S. Comparison of whole plastome sequences between thermogenic skunk cabbage Symplocarpus renifolius and Nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. Int. J. Mol. Sci. 2019, 20, 4678. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.; Yu, M.; Chang, W. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Jin, S.W.; Park, J.Y.; Kang, S.-J.; Park, H.-S.; Shim, H.; Lee, T.J.; Kang, J.H.; Sung, S.H.; Yang, T.-J. The complete chloroplast genome sequence of Magic Lily (Lycoris squamigera). Mitochondrial DNA B 2018, 3, 1210–1211. [Google Scholar] [CrossRef]
- Zhang, F.; Shu, X.; Wang, T.; Zhuang, W.; Wang, Z.L. The complete chloroplast genome sequence of Lycoris radiata. Mitochondrial DNA B 2019, 4, 2886–2887. [Google Scholar] [CrossRef]
- Zhang, F.; Tong, H.; Yang, H.; Wang, T.; Zhuang, W.; Shu, X.; Wang, Z.L. Characterisation of the complete chloroplast genome of Lycoris longituba (Amaryllidaceae). Mitochondrial DNA B 2019, 4, 3782–3783. [Google Scholar] [CrossRef]
- Zhang, F.; Zhuang, W.; Shu, X.; Wang, T.; Wang, Z.L. Complete chloroplast genome of Lycoris sprengeri (Amaryllidaceae) and genetic comparison. Mitochondrial DNA B 2019, 4, 3577–3578. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar]
- Xie, D.-F.; Tan, J.-B.; Yu, Y.; Gui, L.-J.; Su, D.-M.; Zhou, S.-D.; He, X.-J. Insights into phylogeny, age and evolution of Allium (Amaryllidaceae) based on the whole plastome sequences. Ann. Bot. Lond. 2020, 125, 1039–1055. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, X.; Nie, L.; Sun, W.; Hu, H.; Lin, Y.; Li, H.; Zheng, X.; Song, J.; Yao, H. Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible Amomum species. Int. J. Mol. Sci. 2019, 20, 4040. [Google Scholar] [CrossRef]
- Jurica, M.S.; Stoddard, B.L. Homing endonucleases: Structure, function and evolution. Cell. Mol. Life Sci. 1999, 55, 1304–1326. [Google Scholar] [CrossRef] [PubMed]
- Kelchner, S.A. Group II introns as phylogenetic tools: Structure, function, and evolutionary constraints. Am. J. Bot 2002, 89, 1651–1669. [Google Scholar] [CrossRef] [PubMed]
- Hoot, S.B.; Palmer, J.D. Structural rearrangements, including parallel inversions, within the chloroplast genome of Anemone and related genera. J. Mol. Evol. 1994, 38, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Palmer, J.D. A chloroplast DNA phylogeny of the Caryophyllales based on structural and inverted repeat restriction site variation. Syst. Bot. 1994, 19, 236–252. [Google Scholar] [CrossRef]
- Campagna, M.; Downie, S. The intron in chloroplast gene rpl16 is missing from the flowering plant families Geraniaceae, Goodeniaceae, and Plumbaginaceae. T. Illinois State Acad. Sci. 1998, 91, 1–11. [Google Scholar]
- Katayama, H.; Ogihara, Y. Phylogenetic affinities of the grasses to other monocots as revealed by molecular analysis of chloroplast DNA. Curr. Genet. 1996, 29, 572–581. [Google Scholar] [CrossRef]
- Downie, S.R.; Llanas, E.; Katzdownie, D.S. Multiple independent losses of the rpoC1 intron in angiosperm chloroplast DNA’s. Syst. Bot. 1996, 21, 135–151. [Google Scholar] [CrossRef]
- Thiede, J.; Schmidt, S.A.; Rudolph, B. Phylogenetic implication of the chloroplast rpoC1 intron loss in the Aizoaceae (Caryophyllales). Biochem. Syst. Ecol. 2007, 35, 372–380. [Google Scholar] [CrossRef]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef]
- Wheeler, G.L.; Dorman, H.E.; Buchanan, A.T.; Challagundla, L.; Wallace, L.E. A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Appl. Plant Sci. 2014, 2, 1400059. [Google Scholar] [CrossRef]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Zhang, J.; Yao, X.; Huang, H. Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot 2011, 98. [Google Scholar] [CrossRef] [PubMed]
- Hirao, T.; Watanabe, A.; Kurita, M.; Kondo, T.; Takata, K. A frameshift mutation of the chloroplast matK coding region is associated with chlorophyll deficiency in the Cryptomeria japonica virescent mutant Wogon-Sugi. Curr. Genet. 2009, 55, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.; Wu, H.; Wang, Y.; Gao, L.; Zhang, S.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Wu, C.; Huang, Y.; Chaw, S. The complete chloroplast genome of Ginkgo biloba reveals the mechanism of inverted repeat contraction. Genome Biol. Evol. 2012, 4, 374–381. [Google Scholar] [CrossRef]
- Tangphatsornruang, S.; Sangsrakru, D.; Chanprasert, J.; Uthaipaisanwong, P.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. The chloroplast genome sequence of Mungbean (Vigna radiata) determined by high-throughput pyrosequencing: Structural organization and phylogenetic relationships. DNA Res. 2010, 17, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Zhang, Y.; Zeng, C.; Guo, Z.; Li, D. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 2007, 8, 174. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef]
- Silva, S.R.; Pinheiro, D.G.; Penha, H.A.; Plachno, B.J.; Michael, T.P.; Meer, E.J.; Miranda, V.F.O.; Varani, A.M. Intraspecific variation within the Utricularia amethystina Species morphotypes based on chloroplast genomes. Int. J. Mol. Sci. 2019, 20, 6130. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, Y.; Liao, W.; Tong, Z. Analysis on rDNA-ITS sequence and research of intra-specific phylogeny of Lycoris albiflora. J. Plant Resour. Environ. 2009, 18, 25–31. [Google Scholar]
- Quan, M.; Ou, L.; She, C.; Wu, X.; Chen, D. rDNA internal transcribed spacer sequence analysis of Lycoris Hert. Afr. J. Biotechnol. 2012, 11, 7361–7365. [Google Scholar]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, 22–28. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Bui, M.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | L. chinensis | L. anhuiensis | L. aurea | L. radiata [26] | L. longituba [27] | L. sprengeri [28] | L. squamigera [25] |
|---|---|---|---|---|---|---|---|
| Genome size (bp) | 158,484 | 158,490 | 158,367 | 158,335 | 158,484 | 158,687 | 158,482 |
| LSC size (bp) | 86,458 | 86,464 | 86,611 | 86,612 | 86,458 | 86,489 | 86,454 |
| SSC size (bp) | 18,496 | 18,496 | 16,620 | 18,261 | 18,496 | 18,540 | 18,500 |
| IR size (bp) | 26,765 | 26,765 | 27,568 | 26,731 | 26,765 | 26,829 | 26,764 |
| GC content (%) | 37.8 | 37.8 | 37.8 | 37.8 | 37.8 | 37.8 | 37.8 |
| No. of genes | 137 | 137 | 137 | 137 | 137 | 137 | 159 |
| No. of PCGs | 87 | 87 | 87 | 87 | 87 | 87 | 105 |
| No. of tRNAs | 42 | 42 | 42 | 42 | 42 | 42 | 46 |
| No. of rRNAs | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Category of Genes | Group of Genes | Name of Genes |
|---|---|---|
| Genes for photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH,psbI, psbJ, psbK, psbL, psbN, psbT, psbZ, ycf3 | |
| Subunits of NADH-dehydrogenase | ndhA, ndhB (x2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB, petD, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | |
| Subunit of rubisco | rbcL | |
| Self-replication | Large subunit of ribosome | rpl2 (x2), rpl14, rpl16, rpl20, rpl22, rpl23 (x2), rpl32, rpl33, rpl36 |
| DNA dependent RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7 (x2), rps8, rps11, rps12 (x2), rps14, rps15, rps16, rps18, rps19 (x2) | |
| Ribosomal RNAs | rrn23S (x2), rrn16S (x2), rrn5S (x2), rrn4.5S (x2) | |
| Transfer RNAs | trnA-UGC (x4), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC, trnH-GUG (x2), trnI-CAU (x2), trnI-GAU (x4), trnK-UUU, trnL-CAA (x2), trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU (x2), trnP-UGG, trnQ-UUG, trnR-ACG (x2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (x2), trnV-UAC, trnW-CCA, trnY-GUA | |
| Other genes | Subunit of Acetyl-CoA-carboxylase | accD |
| c-type cytochrome synthesis gene | ccsA | |
| Envelop membrane protein | cemA | |
| Protease | clpP | |
| Translational initiation factor | infA | |
| Maturase | matK | |
| Unknown | Conserved open reading frames | ycf1 (x3), ycf2 (x2), ycf4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Wang, T.; Shu, X.; Wang, N.; Zhuang, W.; Wang, Z. Complete Chloroplast Genomes and Comparative Analyses of L. chinensis, L. anhuiensis, and L. aurea (Amaryllidaceae). Int. J. Mol. Sci. 2020, 21, 5729. https://doi.org/10.3390/ijms21165729
Zhang F, Wang T, Shu X, Wang N, Zhuang W, Wang Z. Complete Chloroplast Genomes and Comparative Analyses of L. chinensis, L. anhuiensis, and L. aurea (Amaryllidaceae). International Journal of Molecular Sciences. 2020; 21(16):5729. https://doi.org/10.3390/ijms21165729
Chicago/Turabian StyleZhang, Fengjiao, Tao Wang, Xiaochun Shu, Ning Wang, Weibing Zhuang, and Zhong Wang. 2020. "Complete Chloroplast Genomes and Comparative Analyses of L. chinensis, L. anhuiensis, and L. aurea (Amaryllidaceae)" International Journal of Molecular Sciences 21, no. 16: 5729. https://doi.org/10.3390/ijms21165729
APA StyleZhang, F., Wang, T., Shu, X., Wang, N., Zhuang, W., & Wang, Z. (2020). Complete Chloroplast Genomes and Comparative Analyses of L. chinensis, L. anhuiensis, and L. aurea (Amaryllidaceae). International Journal of Molecular Sciences, 21(16), 5729. https://doi.org/10.3390/ijms21165729