Abstract
Busulfan is an alkylating agent routinely used in conditioning regimens prior to allogeneic hematopoietic cell transplantation (HCT) for various nonmalignant disorders, including inborn errors of metabolism. The combination of model-based dosing and therapeutic drug monitoring (TDM) of busulfan pharmacokinetics (PK) to a lower exposure target has the potential to reduce the regimen-related toxicity while opening marrow niches sufficient for engraftment in diseases such as mucopolysaccharidosis type I (MPS I). We present four cases of the severe form of MPS I or Hurler syndrome, demonstrating successful and stable CD14/15 donor chimerism following the prospective application of model-based dosing and TDM aimed to achieve lower busulfan exposure. All patients received a busulfan-based conditioning regimen with a median cumulative area-under-the-curve (cAUC) target of 63.7 mg h/L (range, 62.4 to 65.0) in protocol-specific combination of chemotherapeutic regimen. The donor source was unrelated umbilical cord blood for three patients and matched sibling donor bone marrow for one patient. The observed median busulfan cAUC was 66.1 mg h/L (range, 65.2 to 70.6) and was within 10% of the intended target. Stable, full donor myeloid chimerism was achieved for three patients, while one patient achieved a stable mixed chimerism (76% donor CD14/15 at 53 months) without a recurring need for enzyme replacement. The normalization of α-L-iduronidase enzyme levels followed the attainment of successful donor myeloid chimerism in all patients. Regimen-related toxicity remained low with no evidence of acute graft-versus-host disease (GVHD) grades II to IV and chronic GVHD.
1. Introduction
Mucopolysaccharidosis type I (MPS I) is a progressive multi-organ disorder categorized in three distinct MPS I subtypes based on the severity of the disease (Hurler, Hurler-Scheie, and Scheie). The Hurler syndrome is considered the more severe type of MPS I [1,2,3,4,5]. Allogeneic hematopoietic cell transplantation (HCT) at early postnatal age (<2 years) is the treatment of choice for patients with severe MPS I (Hurler syndrome) [1,2,3,4,5,6]. MPS I is an inherited lysosomal storage disorder characterized by the deficiency or a complete absence of α-l-iduronidase enzyme caused by mutations in the lysosomal α-l-iduronidase (IDUA) gene [5]. The lysosomal α-l-iduronidase enzyme degrades glycosaminoglycans (GAGs), including dermatan sulfate and heparan sulfate and a deficiency of this enzyme, resulting in excessive accumulation of GAGs, causing multi-organ dysfunction, including progressive neurological disease, hepatosplenomegaly, corneal clouding, upper airway obstruction, skeletal deformity, and cardiomyopathy, with a high rate of mortality in the first decade of life [1,2,3,4,5].
Combination pretransplant conditioning regimen prior to allogeneic HCT with variable degrees of myeloablation have been used in the treatment of MPS I, with the intensity of conditioning associated with both HCT success and regimen-related toxicity [3,5]. One of the most common conditioning regimens for MPS I includes myeloablative busulfan aimed to achieve a cumulative area-under-the-curve (cAUC) of 90 mg h/L [3,5,7,8] used in combination with fludarabine. The largest study to date in pediatric HCT demonstrates that the busulfan exposure leading to optimal event-free survival for both malignant and nonmalignant disease is within a range cAUC of approximately 75–100 mg h/L [9], suggesting busulfan exposure less than 90 mg h/L has potential to further reduce the regimen-related toxicity, while still providing sufficient myeloablation for stable myeloid engraftment in nonmalignant diseases like MPS I. Therefore, the standard procedure at our center has been to administer busulfan, pharmacokinetically (PK) targeted to achieve a low cumulative exposure (cAUC of 65–70 mg h/L), plus fludarabine in patients with MPS I. We present four patients with severe MPS I (Hurler syndrome) who received intravenous busulfan aimed to achieve a lower cAUC of 65–70 mg h/L, resulting in stable myeloid (CD14/15) engraftment and normalization of α-l-iduronidase enzyme levels.
2. Case Presentation
2.1. Patient Characteristics
Four pediatric patients (2 boys and 2 girls) with newly diagnosed severe MPS I who met the inclusion criteria were enrolled (Table 1). Diagnosis was established based on key sign and symptoms (including but not limited to macrocephaly, lumbar kyphosis, sensorineural deafness, corneal clouding, and coarse facies), undetectable level of α-l-iduronidase enzyme, and confirmation of pathogenic mutations (Table 2). Three patients received cord-blood transplants from unrelated donors and one patient received matched sibling donor transplant. The median weight of the patients was 11 kg (range, 9 to 13). The median age was 0.9 years (range, 0.4 to 1.3) at diagnosis and 1.3 years (range, 0.7 to 2.2) at transplantation (Table 1). All patients received standard prophylaxis for graft-versus-host disease (GVHD), supportive care for SOS, as well as Pneumocystis jiroveci pneumonia and antifungal and antiviral prophylaxis.

Table 1.
Patient characteristics and transplant outcome.
Table 2.
Specific pathogenic variants in the α-l-iduronidase (IDUA) gene.
2.2. Busulfan Pharmacokinetics
The patient-specific conditioning protocols are depicted in Figure 1. All patients received individualized doses of busulfan according to a PK model for busulfan clearance with an observed median busulfan cAUC of 66.1 mg h/L (range, 65.2 to 70.6), and overall exposure was within 10% of the intended target (Table 3).
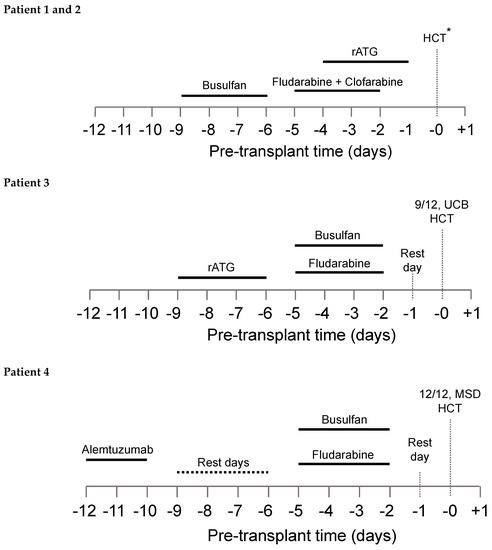
Figure 1.
Conditioning regimen for all four patients. *8/8 UCB HCT and 7/8 UCB HCT for patients 1 and 2, respectively.
Table 3.
Busulfan (Bu) pharmacokinetic parameters.
2.3. Donor Chimerism and Enzyme Levels
The donor myeloid chimerism reflected in CD14/15 in peripheral blood and peripheral blood α-l iduronidase levels was followed to assess engraftment and correction of enzyme deficiency following HCT, respectively. Patients 1, 2, and 3 achieved early and stable engraftment as indicated by 99% donor myeloid chimerism both at 1-month and last recorded (Figure 2). Patient 2 initially achieved 99% myeloid chimerism at 1-month and transitioned to a state of stable mixed chimerism (76% at 53 months) without a drop in enzyme levels below the normal range. In all four patients, successful engraftment led to normalization of α-l iduronidase enzyme level (Table 4). The α-l iduronidase enzyme levels increased slowly from undetectable levels prior to HCT to normal levels within first six months of HCT.
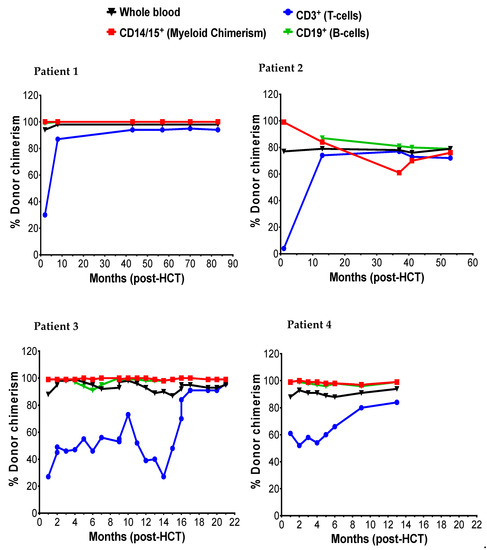
Figure 2.
The post-HCT dynamic of myeloid (CD14/15), T-cells (CD3), and B-cells.
Table 4.
The post-HCT α-l-iduronidase levels.
2.4. Transplant Outcomes
The key transplant-related outcomes are presented in Table 1. Overall, all patients tolerated conditioning well. Neutrophil and platelet engraftment occurred at a median of 19 days (range, 18 to 20) and 32.5 days (range, 21 to 122), respectively, after transplantation. No patients developed seizures attributable to busulfan administration or hepatic SOS. Mucositis occurred in three patients (Patients 1, 2 and 4), although the cases were mild and resolved with standard supportive care. No patients experienced grade III–IV acute or chronic GVHD.
Only patient 3 developed autoimmune cytopenias as a significant complication, which necessitated the use of high dose steroids and multiple immunosuppressive agents. At the time of this report, he remained on two immunosuppressive agents with ongoing steroid taper, and significantly improved lymphoid chimerism (99% donor myeloid chimerism at 21-months).
As of the last follow-up evaluation, all four patients continued to have stable donor chimerism and normal peripheral-blood α-l-iduronidase activity. At present, all patients are receiving long-term multi-disciplinary care with close follow-up of donor chimerism and routine monitoring of α-l iduronidase enzyme levels.
3. Discussion
We describe the successful application of model-based dosing and PK-guided low-exposure busulfan-based conditioning in four pediatric patients who underwent allogeneic HCT for newly diagnosed severe MPS I. This approach led to stable CD14/15 donor chimerism, which was associated with normalization of the α-l-iduronidase enzyme levels, the key enzyme deficient in MPS I. None of the patients experienced severe regimen-related toxicity as well as no acute or chronic GVHD. To our knowledge, this is the first report demonstrating that prospective targeting of a lower busulfan cAUC of 65–70 mg h/L is sufficient to achieve a stable donor chimerism and the normalization of α-l-iduronidase enzyme levels in severe MPS I patients undergoing HCT. While these patients did not experience unexpected acute regimen-related toxicity at the time of preparing this report, the need for follow-up to assess long-term regimen-related toxicity and late graft failure [10] requires a multidisciplinary approach.
The baseline developmental status before transplantation, age at transplantation, and α-l-iduronidase enzyme levels are three of the most critical factors contributing to the overall long-term success following allogeneic HCT for MPS I [3,5,6,7,8,11,12]. The intensity of combination of myeloablative conditioning and effective immunosuppression are other important factors [1,3,5,6,11,12]. Diagnosis of MPS I early in life with minimal or no impairment in baseline cognitive development at the time of HCT, along with a suitable donor source, has been shown to offer the best chance of long-term, favorable cognitive and developmental prognosis [3,4]. Consistent with these observations, three patients received a relatively early diagnosis (Table 1), thus undergoing HCT at ages 0.7, 1.1, and 1.5 years, thereby minimizing the progression of MPS I. Only patient 1 underwent HCT at age 2.2 years.
Retrospective studies of large patient cohorts with MPS-1 showed preference for HLA-matched unrelated UCB units based on a relatively faster availability of UCB, better donor chimerism than those engrafted with bone marrow [6,8,11] and with no difference in the event-free survival compared to fully-matched sibling donor and 6/6 human leukocyte antigen (HLA)-UCB (81% at 5 years) [1,11,13]. Despite these advantages, primary or secondary aplastic type graft failure was more commonly observed following UCB transplantation [8]. In the present report, all patients showed early and rapid engraftment following well-matched UCB donor and MSD. Furthermore, donor CD14/15 chimerism is stable and has resulted in functional normalization of enzyme levels.
Autoimmune cytopenias are a well described complication of UCB HCT [14,15]. A recent publication reported that development of autoimmune cytopenia following UCB HCT in children with MPS I is a significant risk factor for graft rejection [16]. The authors reported busulfan plus fludarabine as conditioning (in comparison to busulfan plus cyclophosphamide) and higher patient absolute lymphocyte count at the time of transplantation were the two factors associated with increased risk of cytopenias, concluding that this may be secondary to inadequate immunoablation. In our report, Patient 3, the recipient of UCB HCT developed autoimmune cytopenia at day 66 requiring the use of multiple immunosuppressive medications for a prolonged period. Additionally, he demonstrated persistent mixed lymphoid chimerism, suggesting that the autoimmunity was potentially the result of inadequate immunoablation of the host immune system rather than myeloablation. However, not all patients with mixed lymphoid chimerism develop autoimmune cytopenia, and causal mechanism of immune dysregulation is not well characterized [17].
The long-term toxicities of myeloablative busulfan exposure in pediatric patients are well established [18,19]. While the current literature suggests targeting a high busulfan cAUC of 90 mg h/L in children with MPS I [3,5,7,8], we aimed for a lower cAUC to support myeloid engraftment but limit drug-related toxicity [9,20,21] based on our center’s experience with other nonmalignant disorders [22,23]. We hypothesized that since busulfan crosses the blood–brain barrier, a reduction in overall busulfan exposure by ~27% may help preserve long-term neurocognitive outcomes in pediatric patients. This may be even more important, as widespread implementation of newborn screening may lead to identification of pediatric patient eligible for HCT at a very early age.
As demonstrated by high levels of donor CD14/15 chimerism and normalization of α-l iduronidase enzyme levels early following HCT, a lower busulfan exposure appears sufficient to effectively facilitate engraftment of donor hematopoietic stem cells. Limiting busulfan exposure to decrease toxicity while maintaining the efficacy of the regimen is the ideal goal in transplanting children with inherited metabolic disorders. Our results in these four cases seem to support the notion that a lower busulfan exposure used in combination with fludarabine may be a suitable alternative to higher busulfan exposure HCT for children with MPS I.
The normalization of α-l-iduronidase enzyme level is a good predictor of outcomes following allogeneic HCT [1,7,12,24], including neurodevelopmental outcomes [5,25]. Factors contributing to correction of enzyme levels post HCT include receipt of cells from a non-carrier donor and full donor chimerism [1,3,7,11,12,24,26]. The patients included in this report received cells from either non-carrier donor or UCB unit with normal enzyme level. These findings are particularly important, because high donor chimerism and enzyme levels improve many clinical manifestations of MPS I, including obstructive airway disease, hepatosplenomegaly, cardiovascular function, hearing, vision, linear growth, and others [1,5,12,27].
4. Methods
4.1. Patient Characteristics
This was a single-center retrospective study comprised of 4 children affected by MPS I who underwent allogeneic HCT at the University of California San Francisco Benioff Children’s Hospital between January 2012 and August 2018. The diagnosis of Hurler’s syndrome was confirmed on the basis of the activity of α-l-iduronidase in peripheral-blood leukocytes, genetic testing for pathogenic mutation, and the clinical phenotype. All patients and/or guardians provided written informed consent to participate in the routine TDM of busulfan as part of their specific transplant protocol. Consent for participation in the PK analysis was waived as part of the University of California San Francisco Committee on Human Subjects’ Research approval process. Eligibility criteria for the inclusion of individual busulfan PK data in this study included (1) subjects 1–26 years of age at the time of HCT; (2) met institutional and protocol specific eligibility criteria for allogeneic HCT that included intravenous busulfan therapy and; (3) busulfan plasma time-concentration data were available for analysis.
4.2. Donor Selection and Transplantation Procedure
If a HLA-matched sibling donor (MSD) who was not a carrier of the disease was available, this was the donor of choice. Otherwise, a unit of cord blood (UCB) with the highest number of nucleated cells (minimum, 3 × 107 per kilogram of body weight) and with high-normal leukocyte α-l-iduronidase activity that matched at least six of eight HLA loci was selected. Standard clinical laboratory procedure was used to prepare UCB cells for transplantation, including thawing, quantifying, and assessing the viability of the nucleated cells, clonal hematopoietic progenitor cells, and CD34+ cells; bone marrow was processed as per the cell therapeutic laboratory standard operating procedure.
4.3. Busulfan Therapeutic Drug Monitoring (TDM)
Busulfan was administered intravenously over 2 or 3 h at dose intervals of 6 (patient 1 and 2) or 24 h (patient 3 and 4) as outlined in the predefined protocol-specific combination pre-transplant conditioning regimen. The timing for collection of busulfan PK samples was based on the dose interval and methodology for AUC estimation as previously described [28]. Briefly, for every 6-h dosing, the first PK collection (PK1) occurred with administration of dose 1 or dose 3, followed by repeat assessment with any dose modification. For every 24-h dosing, PK1 was collected following dose 1, with repeat collections occurring with dose 2 and 3, if clinically indicated. For patients 1 and 2, initial doses for busulfan were based on a first-generation PK model as previously described [28,29] and non-compartmental (NCA) estimation of AUC using the trapezoidal rule following TDM. Updated individualized doses for days 2–4 were calculated by scaling the previous dose with the ratio of obtained AUCobs and predefined AUCtarget using the equation and solving for new dose as follows:
Dose administered in mg/AUCobs = new dose in mg/desired AUCtarget.
For patients 3 and patient 4, the first dose and cAUC was estimated using a validated population pharmacokinetic model and nonlinear mixed effects modeling using model-informed precision dosing platform [9,28,29]. Calculation of doses for days 2–4 were determined by simulation of concentration time course and cumulative exposure from the individualized model. From these simulations, the regimen for the remaining days that would result in cumulative exposure closest to the desired target concentration was identified and recommended. Medication with a known, suspected, or theoretical interaction with busulfan based on drug class or shared metabolic pathways was strictly avoided as part of standard of care policies.
4.4. Donor Chimerism and Transplant Outcome
The primary outcome of interest was a donor CD14/15 chimerism and normalization of the α-l-iduronidase enzyme level post-HCT. Chimerism of peripheral blood (PB) samples was performed for all patients at regular intervals following HCT using Short Tandem Repeat (STR) markers, as previously described [30]. The cell lineages analyzed included CD3, CD19, and CD14/15. The first chimerism test was done at the absolute neutrophil count reached 500 for 3 consecutive days. Thereafter, the frequency of chimerism testing was based on the stability of donor chimerism across three lineages or as clinically indicated following HCT.
Neutrophil engraftment was defined as the first of three consecutive days with an absolute neutrophil count (ANC) ≥ 500/μL. Hepatic sinusoidal obstruction syndrome (SOS) was scored according to EBMT Criteria [31]. Mucositis was graded according to CTCAEv.5.0 criteria. Acute and chronic GVHD were graded based on standard MAGIC and NIH criteria [32]. The estimation of α-l-iduronidase enzyme levels in patient’s blood was performed by the lysosomal diseases testing laboratory at the Thomas Jefferson University, Philadelphia, and activity is expressed in nmol/h/mg protein.
4.5. Data Analysis
GraphPad Prism (GraphPad Prism Software Inc., San Diego, CA, USA) software was used to perform descriptive statistics and generate graphs.
5. Conclusions
In conclusion, lower exposure busulfan in combination with fludarabine provided stable multi-lineage engraftment, with prompt and durable normalization of the α-l-iduronidase enzyme and with no significant regimen-related acute toxicity in four pediatric patients with newly diagnosed severe MPS I. Long-term follow-up will be required to assess the durability of the donor chimerism and the persistence of the normal levels of α-l-iduronidase enzyme and to monitor potential late effects of busulfan conditioning.
Author Contributions
P.S., C.C.D., J.L.-B., and S.K.: study conception, and design; P.S., C.C.D., J.L.-B., and S.K.: patient management, data collection, analysis, and interpretation of data. All authors were involved in drafting and critical evaluation and/or revising the article. All authors have read and agree to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Authors acknowledge clinical staff involved in the care of patients presented in this case report.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ANC | Absolute neutrophil count |
| cAUC | Cumulative area-under-the-curve |
| ERT | Enzyme replacement therapy |
| GAGs | Glycosaminoglycans |
| GVHD | Graft-versus-host disease |
| HCT | Hematopoietic cell transplantation |
| HLA | Human leukocyte antigen |
| IDUA | α-L-iduronidase |
| MPS I | Mucopolysaccharidosis type I |
| MSD | Matched sibling donor |
| PK | Pharmacokinetics |
| SOS | Sinusoidal obstruction syndrome |
| TDM | Therapeutic drug monitoring |
| UCB | Umbilical cord blood |
References
- Van den Broek, B.; van Doorn, J.; Hegeman, C.V.; Nierkens, S.; Lindemans, C.A.; Verhoeven, N.M.; Boelens, J.J.; van Hasselt, P. Hurdles in treating Hurler disease: Potential routes to achieve a “real” cure. Blood Adv. 2020, 4, 2837–2849. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.Y.; Boelens, J.J.; Jones, S.A.; Wynn, R.F. Early treatment is associated with improved cognition in Hurler syndrome. Front. Pediatr. 2019, 7, 433. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef]
- Poe, M.D.; Chagnon, S.L.; Escolar, M.L. Early treatment is associated with improved cognition in H urler syndrome. Ann. Neurol. 2014, 76, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses; past, present, and future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef] [PubMed]
- Aldenhoven, M.; Jones, S.A.; Bonney, D.; Borrill, R.E.; Coussons, M.; Mercer, J.; Bierings, M.B.; Versluys, B.; van Hasselt, P.M.; Wijburg, F.A.; et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: Results after implementation of international guidelines. Biol. Blood Marrow Transplant. 2015, 21, 1106–1109. [Google Scholar] [CrossRef] [PubMed]
- Boelens, J.J.; Wynn, R.F.; O’meara, A.; Veys, P.; Bertrand, Y.; Souillet, G.; Wraith, J.E.; Fischer, A.; Cavazzana-Calvo, M.; Sykora, K.W. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: A risk factor analysis for graft failure. Bone Marrow Transplant. 2007, 40, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Lum, S.H.; Miller, W.P.; Jones, S.; Poulton, K.; Ogden, W.; Lee, H.; Logan, A.; Bonney, D.; Lund, T.C.; Orchard, P.J.; et al. Changes in the incidence, patterns and outcomes of graft failure following hematopoietic stem cell transplantation for Hurler syndrome. Bone Marrow Transplant. 2017, 52, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Bartelink, I.H.; Lalmohamed, A.; van Reij, E.M.; Dvorak, C.C.; Savic, R.M.; Zwaveling, J.; Bredius, R.G.; Egberts, A.C.G.; Bierings, M.; Kletzel, M.; et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: A multicentre, retrospective cohort analysis. Lancet Haematol. 2016, 3, e526–e536. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. Mucopolysaccharidosis I: Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef]
- Boelens, J.J.; Aldenhoven, M.; Purtill, D.; Ruggeri, A.; DeFor, T.; Wynn, R.; Wraith, E.; Cavazzana-Calvo, M.; Rovelli, A.; Fischer, A.; et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood 2013, 121, 3981–3987. [Google Scholar] [CrossRef] [PubMed]
- Lum, S.H.; Stepien, K.M.; Ghosh, A.; Broomfield, A.; Church, H.; Mercer, J.; Jones, S.; Wynn, R. Long term survival and cardiopulmonary outcome in children with Hurler syndrome after haematopoietic stem cell transplantation. J. Inherit. Metab. Dis. 2017, 40, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luan, Z.; Jiang, H.; Fang, J.; Qin, M.; Lee, V.; Chen, J. Allogeneic Hematopoietic Stem Cell Transplantation in Thirty-Four Pediatric Cases of Mucopolysaccharidosis-A Ten-Year Report from the China Children Transplant Group. Biol. Blood Marrow Transplant. 2016, 22, 2104–2108. [Google Scholar] [CrossRef]
- Daikeler, T.; Labopin, M.; Ruggeri, A.; Crotta, A.; Abinun, M.; Hussein, A.A.; Carlson, K.; Cornillon, J.; Diez-Martin, J.L.; Gandemer, V.; et al. New autoimmune diseases after cord blood transplantation: A retrospective study of EUROCORD and the Autoimmune Disease Working Party of the European Group for Blood and Marrow Transplantation. Blood 2013, 121, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Page, K.M.; Mendizabal, A.M.; Prasad, V.K.; Martin, P.L.; Parikh, S.; Wood, S.; Sempowski, G.D.; Szabolcs, P.; Kurtzberg, J. Posttransplant autoimmune hemolytic anemia and other autoimmune cytopenias are increased in very young infants undergoing unrelated donor umbilical cord blood transplantation. Biol. Blood Marrow Transplant. 2008, 14, 1108–1117. [Google Scholar] [CrossRef]
- Deambrosis, D.; Lum, S.H.; Hum, R.M.; Poulton, K.; Ogden, W.; Jones, S.; Stanworth, S.; Bonney, D.; Hiwarkar, P.; Wynn, R.F. Immune cytopenia post–cord transplant in Hurler syndrome is a forme fruste of graft rejection. Blood Adv. 2019, 3, 570–574. [Google Scholar] [CrossRef]
- Neely, J.A.; Dvorak, C.C.; Pantell, M.S.; Melton, A.; Huang, J.N.; Shimano, K.A. Autoimmune cytopenias in pediatric hematopoietic cell transplant patients. Front. Pediatr. 2019, 7, 171. [Google Scholar] [CrossRef]
- Gyurkocza, B.; Sandmaier, B.M. Conditioning regimens for hematopoietic cell transplantation: One size does not fit all. Blood 2014, 124, 344–353. [Google Scholar] [CrossRef]
- Allewelt, H.; El-Khorazaty, J.; Mendizabal, A.; Taskindoust, M.; Martin, P.L.; Prasad, V.; Page, K.; Sanders, J.; Kurtzberg, J. Late effects after umbilical cord blood transplantation in very young children after busulfan-based, myeloablative conditioning. Biol. Blood Marrow Transplant. 2016, 22, 1627–1635. [Google Scholar] [CrossRef][Green Version]
- Dvorak, C.C.; Long-Boyle, J.; Dara, J.; Melton, A.; Shimano, K.A.; Huang, J.N.; Puck, J.M.; Dorsey, M.J.; Facchino, J.; Chang, C.K. Low Exposure Busulfan Conditioning to Achieve Sufficient Multi-lineage Chimerism in Patients with Severe Combined Immunodeficiency. Biol. Blood Marrow Transplant. 2019, 7, 1355–1362. [Google Scholar] [CrossRef]
- Mamcarz, E.; Zhou, S.; Lockey, T.; Abdelsamed, H.; Cross, S.J.; Kang, G.; Ma, Z.; Condori, J.; Dowdy, J.; Triplett, B.; et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N. Engl. J. Med. 2019, 380, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Law, J.; Cowan, M.J.; Dvorak, C.C.; Musick, L.; Long-Boyle, J.R.; Baxter-Lowe, L.A.; Horn, B. Busulfan, fludarabine, and alemtuzumab as a reduced toxicity regimen for children with malignant and nonmalignant diseases improves engraftment and graft-versus-host disease without delaying immune reconstitution. Biol. Blood Marrow Transplant. 2012, 18, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.F.; Long-Boyle, J.R.; Shimano, K.A.; Melton, A.; Kharbanda, S.; Dara, J.; Higham, C.; Huang, J.N.; Cowan, M.J.; Dvorak, C.C. Reduced Toxicity Conditioning for Non-Malignant Hematopoietic Cell Transplants. Biol. Blood Marrow Transplant. 2020, 20, 30351–30357. [Google Scholar]
- Bartelink, I.; Van Reij, E.; Gerhardt, C.; Van Maarseveen, E.; De Wildt, A.; Versluys, B.; Lindemans, C.A.; Bierings, M.; Boelens, J.J. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: Maintaining efficacy with less toxicity. Biol. Blood Marrow Transplant. 2014, 20, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Boelens, J.J.; van Hasselt, P.M. Neurodevelopmental outcome after hematopoietic cell transplantation in inborn errors of metabolism: Current considerations and future perspectives. Neuropediatrics 2016, 47, 285–292. [Google Scholar]
- Wynn, R.F.; Wraith, J.E.; Mercer, J.; O’meara, A.; Tylee, K.; Thornley, M.; Church, H.J.; Bigger, B.W. Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J. Pediatr. 2009, 154, 609–611. [Google Scholar] [CrossRef]
- Boelens, J.J.; Rocha, V.; Aldenhoven, M.; Wynn, R.; O’Meara, A.; Michel, G.; Ionescu, I.; Parikh, S.; Prasad, V.K.; Szabolcs, P.; et al. Risk factor analysis of outcomes after unrelated cord blood transplantation in patients with hurler syndrome. Biol. Blood Marrow Transplant. 2009, 15, 618–625. [Google Scholar] [CrossRef]
- Long-Boyle, J.R.; Savic, R.; Yan, S.; Bartelink, I.; Musick, L.; French, D.; Law, J.; Horn, B.; Cowan, M.J.; Dvorak, C.C. Population pharmacokinetics of busulfan in pediatric and young adult patients undergoing hematopoietic cell transplant: A model-based dosing algorithm for personalized therapy and implementation into routine clinical use. Ther. Drug Monit. 2015, 37, 236–245. [Google Scholar] [CrossRef]
- Savic, R.M.; Cowan, M.J.; Dvorak, C.C.; Pai, S.-Y.; Pereira, L.; Bartelink, I.H.; Boelens, J.J.; Bredius, R.G.; Wynn, R.F.; Cuvelier, G.D.; et al. Effect of weight and maturation on busulfan clearance in infants and small children undergoing hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2013, 19, 1608–1614. [Google Scholar] [CrossRef]
- Ozyurek, E.; Cowan, M.J.; Koerper, M.A.; Baxter-Lowe, L.-A.; Dvorak, C.; Horn, B.N. Increasing mixed chimerism and the risk of graft loss in children undergoing allogeneic hematopoietic stem cell transplantation for non-malignant disorders. Bone Marrow Transplant. 2008, 42, 83–91. [Google Scholar] [CrossRef]
- Corbacioglu, S.; Carreras, E.; Ansari, M.; Balduzzi, A.; Cesaro, S.; Dalle, J.-H.; Dignan, F.; Gibson, B.; Guengoer, T.; Gruhn, B.; et al. Diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in pediatric patients: A new classification from the European society for blood and marrow transplantation. Bone Marrow Transplant. 2018, 53, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Schoemans, H.M.; Lee, S.J.; Ferrara, J.L.; Wolff, D.; Levine, J.E.; Schultz, K.R.; Shaw, B.E.; Flowers, M.E.; Ruutu, T.; Greinix, H.; et al. EBMT-NIH-CIBMTR Task Force position statement on standardized terminology & guidance for graft-versus-host disease assessment. Bone Marrow Transplant. 2018, 53, 1401–1415. [Google Scholar] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).