Structural Isomerism and Enhanced Lipophilicity of Pyrithione Ligands of Organoruthenium(II) Complexes Increase Inhibition on AChE and BuChE

 ,
,  , and
, and
Abstract

1. Introduction
2. Results and Discussion
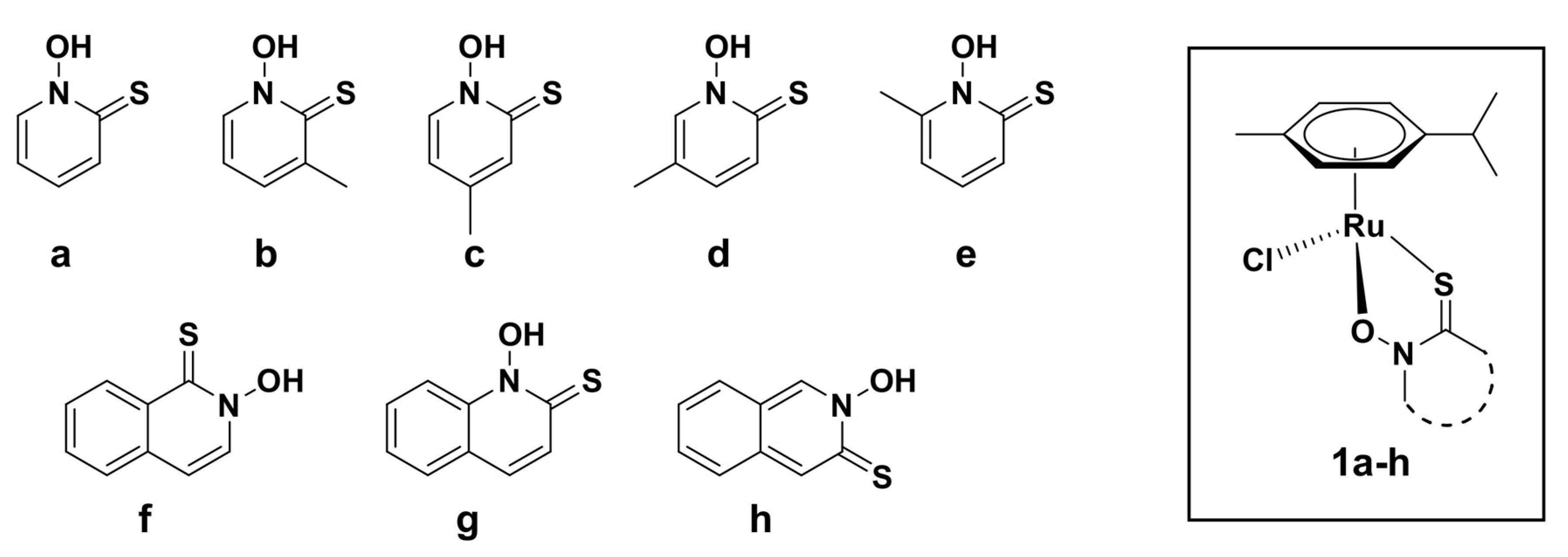
2.1. Synthesis and Crystal Structures
2.2. Stability of Organoruthenium(II) Pyrithione Complexes
2.3. Enzymatic Assays
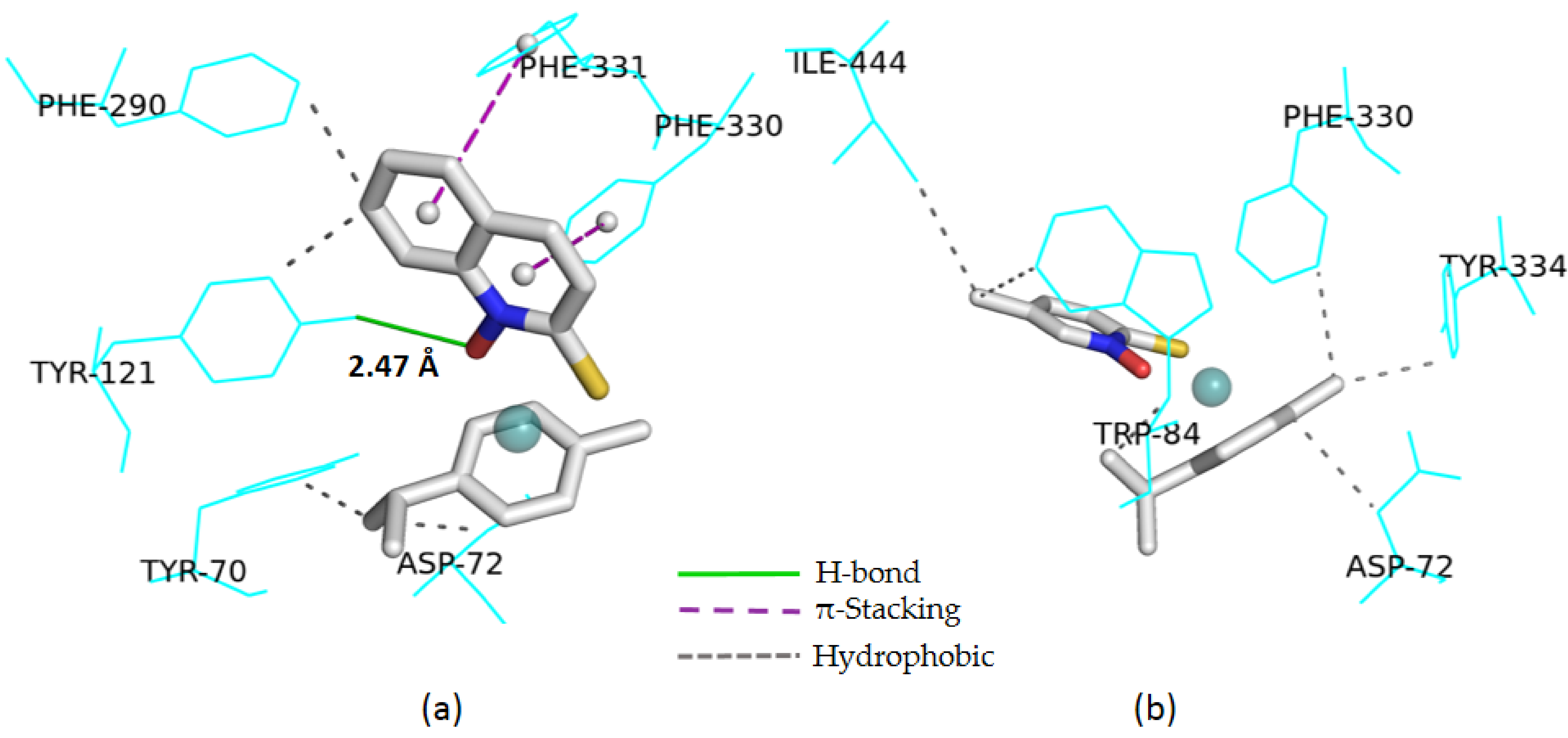
2.4. Computational Study
3. Materials and Methods
3.1. Chemicals
3.2. Characterization
3.3. Syntheses
3.4. Cholinesterase Inhibition Assay
3.5. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACh | acetylcholine |
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| Aβ | amyloid-β |
| BuChE | butyrylcholinesterase |
| ChE | cholinesterase |
| DCM | dichloromethane |
| DMSO | dimethyl sulfoxide |
| ee | electric eel |
| ESI-MS | electrospray ionization mass spectrometry |
| ESI-HRMS | high-resolution electrospray ionization mass spectrometry |
| hs | horse serum |
| MeOH | methanol |
| NMR | nuclear magnetic resonance |
| RuCym | ruthenium precursor [Ru(p-cymene)Cl2]2 |
| Tc | Torpedo californica |
| TLC | thin layer chromatography |
References
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Tarawneh, R.; Holtzman, D.M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Gulisano, W.; Arancio, O.; Palmeri, A. The keystone of Alzheimer pathogenesis might be sought in Aβ physiology. Neuroscience 2015, 307, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, C.; Antollini, S.S. Alzheimer’s disease as a membrane disorder: Spatial cross-talk among beta-amyloid peptides, nicotinic acetylcholine receptors and lipid rafts. Front. Cell. Neurosci. 2019, 13, 28. [Google Scholar] [CrossRef]
- Talesa, V.N. Acetylcholinesterase in Alzheimer’s disease. Mech. Ageing Dev. 2001, 122, 1961–1969. [Google Scholar] [CrossRef]
- Greig, N.H.; Lahiri, D.K.; Sambamurti, K. Butyrylcholinesterase: An important new target in Alzheimer’s disease therapy. Int. Psychogeriatr. 2002, 14, 77–91. [Google Scholar] [CrossRef]
- Ekholm, M. Predicting relative binding free energies of substrates and inhibitors of acetylcholin- and butyrylcholinesterases. J. Mol. Struct. 2001, 572, 25–34. [Google Scholar] [CrossRef]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef]
- Wilkinson, D.G.; Francis, P.T.; Schwam, E.; Payne-Parrish, J. Cholinesterase inhibitors used in the treatment of Alzheimer’s disease the relationship between pharmacological effects and clinical efficacy. Drugs Aging 2004, 21, 453–478. [Google Scholar] [CrossRef]
- Pohanka, M. Inhibitors of acetylcholinesterase and butyrylcholinesterase meet immunity. Int. J. Mol. Sci. 2014, 15, 9809–9825. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Yu, J.T.; Wang, H.F.; Tan, M.S.; Meng, X.F.; Wang, C.; Jiang, T.; Zhu, X.C.; Tan, L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimers Dis. 2014, 41, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.E. Improving anti-neurodegenerative benefits of acetylcholinesterase inhibitors in Alzheimer’s disease: Are irreversible inhibitors the future? Int. J. Mol. Sci. 2020, 21, 3438. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal ions in Alzheimer’s disease: A key role or not? Acc. Chem. Res. 2019, 52, 2026–2035. [Google Scholar] [CrossRef]
- Tomljenovic, L. Aluminum and Alzheimer’s disease: After a century of controversy, is there a plausible link? J. Alzheimer’s Dis. 2011, 23, 567–598. [Google Scholar] [CrossRef]
- Alatrash, N.; Narh, E.S.; Yadav, A.; Kim, M.J.; Janaratne, T.; Gabriel, J.; MacDonnell, F.M. Synthesis, DNA cleavage activity, cytotoxicity, acetylcholinesterase inhibition, and acute murine toxicity of redox-active ruthenium(II) polypyridyl complexes. ChemMedChem 2017, 12, 1055–1069. [Google Scholar] [CrossRef]
- Cardoso, C.R.; de Aguiar, I.; Camilo, M.R.; Lima, M.V.S.; Ito, A.S.; Baptista, M.S.; Pavani, C.; Venâncio, T.; Carlos, R.M. Synthesis, spectroscopic characterization, photochemical and photophysical properties and biological activities of ruthenium complexes with mono- and bi-dentate histamine ligand. Dalton Trans. 2012, 41, 6726–6734. [Google Scholar] [CrossRef]
- Mulcahy, S.P.; Li, S.; Korn, R.; Xie, X.; Meggers, E. Solid-phase synthesis of tris-heteroleptic ruthenium(II) complexes and application to acetylcholinesterase inhibition. Inorg. Chem. 2008, 47, 5030–5032. [Google Scholar] [CrossRef]
- Vyas, N.A.; Singh, S.B.; Kumbhar, A.S.; Ranade, D.S.; Walke, G.R.; Kulkarni, P.P.; Jani, V.; Sonavane, U.B.; Joshi, R.R.; Rapole, S. Acetylcholinesterase and Aβ aggregation inhibition by heterometallic ruthenium(II)-platinum(II) polypyridyl complexes. Inorg. Chem. 2018, 57, 7524–7535. [Google Scholar] [CrossRef]
- Vyas, N.A.; Bhat, S.S.; Kumbhar, A.S.; Sonawane, U.B.; Jani, V.; Joshi, R.R.; Ramteke, S.N.; Kulkarni, P.P.; Joshi, B. Ruthenium(II) polypyridyl complex as inhibitor of acetylcholinesterase and Aβ aggregation. Eur. J. Med. Chem. 2014, 75, 375–381. [Google Scholar] [CrossRef]
- Vyas, N.A.; Ramteke, S.N.; Kumbhar, A.S.; Kulkarni, P.P.; Jani, V.; Sonawane, U.B.; Joshi, R.R.; Joshi, B.; Erxleben, A. Ruthenium(II) polypyridyl complexes with hydrophobic ancillary ligand as Aβ aggregation inhibitors. Eur. J. Med. Chem. 2016, 121, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Kljun, J.; Anko, M.; Traven, K.; Sinreih, M.; Pavlič, R.; Peršič, Š.; Ude, Ž.; Codina, E.E.; Stojan, J.; Lanišnik Rižner, T.; et al. Pyrithione-based ruthenium complexes as inhibitors of aldo-keto reductase 1C enzymes and anticancer agents. Dalton Trans. 2016, 45, 11791–11800. [Google Scholar] [CrossRef] [PubMed]
- Ristovski, S.; Uzelac, M.; Kljun, J.; Lipec, T.; Uršič, M.; Zemljič Jokhadar, Š.; Žužek, M.C.; Trobec, T.; Frangež, R.; Sepčić, K.; et al. Organoruthenium prodrugs as a new class of cholinesterase and glutathione-S-transferase inhibitors. ChemMedChem 2018, 13, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Marković, K.; Milačič, R.; Marković, S.; Kladnik, J.; Turel, I.; Ščančar, J. Binding kinetics of ruthenium pyrithione chemotherapeutic candidates to human serum proteins studied by HPLC-ICP-MS. Molecules 2020, 25, 1512. [Google Scholar] [CrossRef] [PubMed]
- Lazarević-Pašti, T.; Leskovac, A.; Momić, T.; Petrović, S.; Vasić, V. Modulators of acetylcholinesterase activity: From Alzheimer’s disease to anti-cancer drugs. Curr. Med. Chem. 2017, 24, 3283–3309. [Google Scholar] [CrossRef]
- Jin, X.; Wang, M.; Shentu, J.; Huang, C.; Bai, Y.; Pan, H.; Zhang, D.; Yuan, Z.; Zhang, H.; Xiao, X.; et al. Inhibition of acetylcholinesterase activity and β-amyloid oligomer formation by 6-bromotryptamine A, a multi-target anti-Alzheimer’s molecule. Oncol. Lett. 2020, 19, 1593–1601. [Google Scholar] [CrossRef]
- Hyatt, J.L.; Tsurkan, L.; Morton, C.L.; Yoon, K.J.P.; Harel, M.; Brumshtein, B.; Silman, I.; Sussman, J.L.; Wadkins, R.M.; Potter, P.M. Inhibition of acetylcholinesterase by the anticancer prodrug CPT-11. Chem. Biol. Interact. 2005, 157, 247–252. [Google Scholar] [CrossRef]
- Aljafari, A.A.; Duhaiman, A.S.; Kamal, M.A. Inhibition of human acetylcholinesterase by cyclophosphamide. Toxicology 1995, 96, 1–6. [Google Scholar] [CrossRef]
- Huang, L.; Lin, J.; Xiang, S.; Zhao, K.; Yu, J.; Zheng, J.; Xu, D.; Mak, S.; Hu, S.; Nirasha, S.; et al. Sunitinib, a clinically used anticancer drug, is a potent AChE inhibitor and attenuates cognitive impairments in mice. ACS Chem. Neurosci. 2016, 7, 1047–1056. [Google Scholar] [CrossRef]
- Kamal, M.A.; Nasim, F.H.; Al-Jafari, A.A. Human erythrocyte acetylcholinesterase inhibition by cis-diamminediaquaplatinum (II): A novel kinetic approach. Cancer Lett. 1999, 138, 115–119. [Google Scholar] [CrossRef]
- Jończyk, J.; Godyń, J.; Stawarska, E.; Morak-Młodawska, B.; Jeleń, M.; Pluta, K.; Malawska, B. Dual action of dipyridothiazine and quinobenzothiazine derivatives—Anticancer and cholinesterase-inhibiting activity. Molecules 2020, 25, 2604. [Google Scholar] [CrossRef] [PubMed]
- Kladnik, J.; Kljun, J.; Burmeister, H.; Ott, I.; Romero-Canelon, I.; Turel, I. Towards identification of essential structural elements of organoruthenium(II)-pyrithionato complexes for anticancer activity. Chem. Eur. J. 2019, 25, 14169–14182. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Blachly, P.G.; McCammon, J.A.; Cohen, S.M. Exploring the influence of the protein environment on metal-binding pharmacophores. J. Med. Chem. 2014, 57, 7126–7135. [Google Scholar] [CrossRef] [PubMed]
- Adamek, R.N.; Credille, C.V.; Dick, B.L.; Cohen, S.M. Isosteres of hydroxypyridinethione as drug-like pharmacophores for metalloenzyme inhibition. J. Biol. Inorg. Chem. 2018, 23, 1129–1138. [Google Scholar] [CrossRef]
- Pizarro, A.M.; Habtemariam, A.; Sadler, P.J. Activation mechanisms for organometallic anticancer complexes. In Medicinal Organometallic Chemistry; Jaouen, G., Metzler-Nolte, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 32, pp. 21–56. [Google Scholar]
- Turel, I.; Kljun, J.; Perdih, F.; Morozova, E.; Bakulev, V.; Kasyanenko, N.; Byl, J.A.W.; Osheroff, N. First ruthenium organometallic complex of antibacterial agent ofloxacin. Crystal structure and interactions with DNA. Inorg. Chem. 2010, 49, 10750–10752. [Google Scholar] [CrossRef]
- Peacock, A.F.A.; Melchart, M.; Deeth, R.J.; Habtemariam, A.; Parsons, S.; Sadler, P.J. Osmium(II) and ruthenium(II) arene maltolato complexes: Rapid hydrolysis and nucleobase binding. Chem. Eur. J. 2007, 13, 2601–2613. [Google Scholar] [CrossRef]
- Grguric-Sipka, S.; Stepanenko, I.N.; Lazic, J.M.; Bartel, C.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. Synthesis, X-ray diffraction structure, spectroscopic properties and antiproliferative activity of a novel ruthenium complex with constitutional similarity to cisplatin. Dalton Trans. 2009, 3334–3339. [Google Scholar] [CrossRef]
- Diaz-Torres, R.; Alvarez, S. Coordinating ability of anions and solvents towards transition metals and lanthanides. Dalton Trans. 2011, 40, 10742–10750. [Google Scholar] [CrossRef]
- Seršen, S.; Kljun, J.; Kryeziu, K.; Panchuk, R.; Alte, B.; Körner, W.; Heffeter, P.; Berger, W.; Turel, I. Structure-related mode-of-action differences of anticancer organoruthenium complexes with β-diketonates. J. Med. Chem. 2015, 58, 3984–3996. [Google Scholar] [CrossRef]
- Briš, A.; Jašik, J.; Turel, I.; Roithova, J. Anti-cancer organoruthenium(II) complexes and their interactions with cysteine and its analogues. A mass-spectrometric study. Dalton Trans. 2019, 48, 2626–2634. [Google Scholar] [CrossRef]
- Sheng, Y.; Hou, Z.; Cui, S.; Cao, K.; Yuan, S.; Sun, M.; Kljun, J.; Huang, G.; Turel, I.; Liu, Y. Covalent versus noncovalent binding of ruthenium η6-p-cymene complexes to zinc-finger protein NCp7. Chem. Eur. J. 2019, 25, 12789–12794. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J. Lipophilicity in drug discovery. Expert. Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Botić, T.; Defant, A.; Zanini, P.; Žužek, M.C.; Frangež, R.; Janussen, D.; Kersken, D.; Knez, Ž.; Mancini, I.; Sepčić, K. Discorhabdin alkaloids from Antarctic Latrunculia spp. sponges as a new class of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 136, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Jankowiak, A.; Kaszynski, P. 4-substituted 1-acyloxypyridine-2(1H)-thiones: Experimental and computational studies of the substituent effect on electronic absorption spectra. J. Org. Chem. 2009, 74, 7441–7448. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian, Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Waller, M.P.; Bühl, M. Vibrational corrections to geometries of transition metal complexes from density functional theory. J. Comput. Chem. 2007, 28, 1531–1537. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Galdeano, C.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Bartolini, M.; Perez, B.; Clos, M.V.; Silman, I.; Jean, L.; Colletier, J.P.; Renard, P.Y.; et al. Increasing polarity in tacrine and huprine derivatives: Potent anticholinesterase agents for the treatment of myasthenia gravis. Molecules 2018, 23, 634. [Google Scholar] [CrossRef] [PubMed]
- Protein-Ligand Interaction Profiler (PLIP)! Available online: Biotec.tu-dresden.de (accessed on 8 May 2020).
- Swiss ADME. Available online: http://www.swissadme.ch/ (accessed on 8 May 2020).





{kind=link}
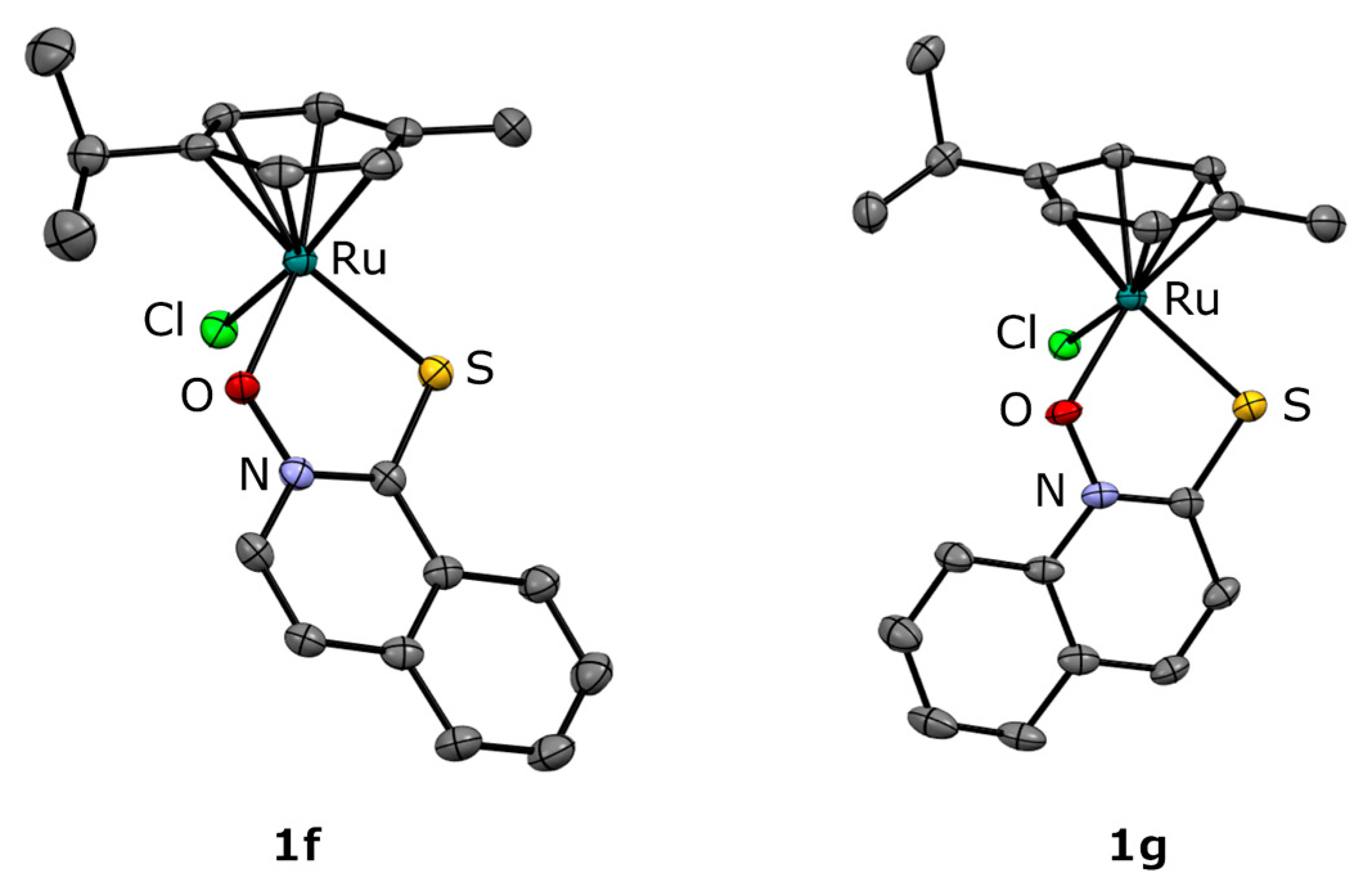{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | eeAChE | hsBuChE | ||
|---|---|---|---|---|
| IC50 (µM) 1 | Ki (µM) 2 | IC50 (µM) 1 | Ki (µM) 2 | |
| a | / | / | / | / |
| b | / | / | / | / |
| c | / | / | / | / |
| d | / | / | / | / |
| e | / | / | >100 | / |
| f | / | / | / | / |
| g | / | / | / | / |
| h | >100 | / | 53 | n.d. |
| 1a | 7.8 ± 0.8 | 15.1 | 2.3 ± 0.1 | 0.5 |
| 1b | 10.5 ± 0.7 | 9.7 | 1.2 ± 0.02 | 0.4 |
| 1c | 9.5 ± 1.1 | 8.5 | 2.7 ± 0.8 | 0.9 |
| 1d | 6.6 ± 0.5 | 9.7 | 1.9 ± 0.4 | 0.4 |
| 1e | 5.1 ± 0.4 | 4.9 | 0.7 ± 0.1 | 3.4 |
| 1f | 8.1 ± 0.5 | 12.3 | 1.3 ± 0.1 | 0.4 |
| 1g | 4.9 ± 0.1 | 5.6 | 0.2 ± 0.05 | 0.2 |
| 1h | 14.3 ± 2.0 | 20.1 | 0.7 ± 0.1 | 0.2 |
| Neostigmine bromide | 4.3 ± 0.8 | / | 37.3 ± 4.2 | / |
| Compound 1 | Interactions | ||||
|---|---|---|---|---|---|
| ΔE (Kcal/mol) | H-Bond 2 | Hydrophobic 2 | π-Stacking 2 | Salt Bridge 2 | |
| 1a | −7.55 | - | Y70 (3.43); Y121(3.47) F330(3.57; 3.14) | W84 (4.38; 3.73) | - |
| 1b | −7.16 | S122 (3.09) | D72 (3.82); F330 (3.34) | W84 (4.04) | - |
| 1c | −7.02 | - | Y70 (3.60); D72 (3.62) N85 (3.96); F330 (3.92; 3.35); Y334 (3.38) | W84 (4.62) F330 (5.09) | - |
| 1d | −8.60 | - | D72 (3.62); W84 (3.39; 3.78) F330 (3.66); Y334 (3.39); I444 (3.96) W432 (3.77); Y442 (3.67) I444 (372) | - | - |
| 1e | −6.73 | - | D72 (3.99); W 84 (3.37; 3.41) F290 (3.73); F330 (3.13) F331 (3.58) | - | - |
| 1f | −8.31 | - | D72 (3.66); W84 (3.55; 3.87); F330 (3.23; 3.16); L333 (3.94) Y334 (2.95; 3.27); W432 (3.08) I439 (3.52) | W84 (3.81) F330 (3.63) | - |
| 1g | −9.86 | Y121 (2.47) | Y70 (3.02); D72 (3.40) Y121 (3.46); F290 (3.51) | F330 (4.35) F331 (5.34) | - |
| 1h | −8.28 | - | W84 (3.57; 3.94; 3.97) F330 (3.63); W432 (3.80) Y442 (3.51) | W84 (3.61) F330 (3.84) | - |
| neostigmine bromide | −7.03 | Y121 (2.93) | Y70 (3.72); W279 (3.36) F330 (3.57) | - | D72 (3.95) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kladnik, J.; Ristovski, S.; Kljun, J.; Defant, A.; Mancini, I.; Sepčić, K.; Turel, I. Structural Isomerism and Enhanced Lipophilicity of Pyrithione Ligands of Organoruthenium(II) Complexes Increase Inhibition on AChE and BuChE. Int. J. Mol. Sci. 2020, 21, 5628. https://doi.org/10.3390/ijms21165628
Kladnik J, Ristovski S, Kljun J, Defant A, Mancini I, Sepčić K, Turel I. Structural Isomerism and Enhanced Lipophilicity of Pyrithione Ligands of Organoruthenium(II) Complexes Increase Inhibition on AChE and BuChE. International Journal of Molecular Sciences. 2020; 21(16):5628. https://doi.org/10.3390/ijms21165628
Chicago/Turabian StyleKladnik, Jerneja, Samuel Ristovski, Jakob Kljun, Andrea Defant, Ines Mancini, Kristina Sepčić, and Iztok Turel. 2020. "Structural Isomerism and Enhanced Lipophilicity of Pyrithione Ligands of Organoruthenium(II) Complexes Increase Inhibition on AChE and BuChE" International Journal of Molecular Sciences 21, no. 16: 5628. https://doi.org/10.3390/ijms21165628
APA StyleKladnik, J., Ristovski, S., Kljun, J., Defant, A., Mancini, I., Sepčić, K., & Turel, I. (2020). Structural Isomerism and Enhanced Lipophilicity of Pyrithione Ligands of Organoruthenium(II) Complexes Increase Inhibition on AChE and BuChE. International Journal of Molecular Sciences, 21(16), 5628. https://doi.org/10.3390/ijms21165628