Abstract
Calcium ions regulate a wide array of physiological functions including cell differentiation, proliferation, muscle contraction, neurotransmission, and fertilization. The endoplasmic reticulum (ER) is the major intracellular Ca2+ store and cellular events that induce ER store depletion (e.g., activation of inositol 1,4,5-triphosphate (IP3) receptors) trigger a refilling process known as store-operated calcium entry (SOCE). It requires the intricate interaction between the Ca2+ sensing stromal interaction molecules (STIM) located in the ER membrane and the channel forming Orai proteins in the plasma membrane (PM). The resulting active STIM/Orai complexes form highly selective Ca2+ channels that facilitate a measurable Ca2+ influx into the cytosol followed by successive refilling of the ER by the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA). STIM and Orai have attracted significant therapeutic interest, as enhanced SOCE has been associated with several cancers, and mutations in STIM and Orai have been linked to immunodeficiency, autoimmune, and muscular diseases. 2-Aminoethyl diphenylborinate (2-APB) is a known modulator and depending on its concentration can inhibit or enhance SOCE. We have synthesized several novel derivatives of 2-APB, introducing halogen and other small substituents systematically on each position of one of the phenyl rings. Using a fluorometric imaging plate reader (FLIPR) Tetra-based calcium imaging assay we have studied how these structural changes of 2-APB affect the SOCE modulation activity at different compound concentrations in MDA-MB-231 breast cancer cells. We have discovered 2-APB derivatives that block SOCE at low concentrations, at which 2-APB usually enhances SOCE.
1. Introduction
Store-operated calcium entry (SOCE) is a ubiquitous pathway by which cells regulate basal calcium (Ca2+), refill intracellular Ca2+ stores, and maintain Ca2+ signaling to control diverse functions such as gene expression, cell proliferation, migration, and differentiation [1]. The stimulation of cell surface receptors coupled to phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) hydrolysis produces inositol 1,4,5-trisphosphate (IP3), a second messenger which upon binding to IP3 receptors (IP3Rs) on the endoplasmic reticulum (ER) membrane, releases Ca2+ from the ER. The depletion of the ER Ca2+ stores, triggers the activation of ER Ca2+-sensing stromal interaction molecules (STIM1 and STIM2) that are embedded in the ER membrane which in turn activate Orai channels (Orai1, Orai2, and Orai3) at the plasma membrane (PM). STIM1 and Orai1 were identified as the predominant isoforms of STIMs and Orais that mediate SOCE in the majority of cell types [2,3,4,5]. Upon activation, structural rearrangements in STIM expose the cytosolic STIM Orai-activating region (SOAR). SOAR physically opens the PM Orai channels, resulting in Ca2+ entry into the cytosol [1]. The STIM/Orai-mediated Ca2+ entry, electrophysiologically characterized as Ca2+ release-activated Ca2+ current (ICRAC) [6], thus allows the acute refilling of the ER Ca2+ stores [7]. Experimentally, sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) blocker thapsigargin (Tg) [8] is widely used for passive depletion of ER Ca2+ stores and subsequent activation of SOCE.
Gain-of-function or loss-of-function mutations in STIM1 or Orai1, or alterations in expression ratios of STIM and Orai isoforms as well as aberrations in other regulatory mechanisms of SOCE pathway lead to its dysfunction [9,10,11]. The disruption of SOCE has various pathophysiological implications such as muscle, immune, and neurodegenerative disorders as well as cancer [12,13,14,15,16]. Breast cancer metastasis has been linked to STIM1/Orai1-mediated Ca2+ influx [17] and several studies also highlighted the upregulation of STIM1 and Orai1 in basal breast cancers [18,19]. Our understanding on the remodeling of SOCE in breast cancer cells has increased over the years and has recently been reviewed [20]. In addition, SOCE has been shown to control the proliferation, migration, invasion, and metastasis in different cancer cell lines [21,22]. Given the role of SOCE in tumor progression [16], developing potent and selective SOCE modulators is of high therapeutic interest.
Several chemical inhibitors [23,24,25,26,27] as well as potentiators of SOCE [28,29,30] have been reported. However, 2-aminoethyl diphenylborinate (2-APB, Figure 1) remains the best characterized compound used to modulate store-operated CRAC channels, despite of its effects on other known targets [31,32,33,34,35,36,37,38,39,40,41,42,43,44]. Although originally described as an inhibitor of IP3 receptors [45,46], it was later shown that inhibition of SOCE and ICRAC by 2-APB is independent of its action on IP3 receptors [36,47,48,49,50,51]. 2-APB is known to exert a very interesting dose-dependent bimodal effect on SOCE. 2-APB when applied in ≤10 µM concentration potentiates whereas ≥30 µM concentration blocks SOCE and STIM1/Orai1-mediated ICRAC in various cell lines such as HEK293, human Jurkat T cells, chicken DT40 B cells, and rat basophilic leukemia cells [42,47,49,50,52,53,54,55,56]. Recent studies have shed some light on the underlying mechanisms of action of potentiation and inhibition of SOCE/ICRAC by 5 µM and 50 µM 2-APB, respectively [57,58,59]. 2-APB not only acts directly on the Orai1 channel for the inhibition of SOCE, but also enhances the intramolecular interactions within STIM1, which leads to the disruption of the functional coupling between STIM1 and Orai1 [59]. On the other hand, 5 µM 2-APB potentiates CRAC channels by a direct dilation of the open pore of Orai1 [57]. The other STIM/Orai isoforms as well as heteromeric channels formed by Orai1/Orai3 have been shown to respond differently to 2-APB [53,55,60,61,62,63,64,65].
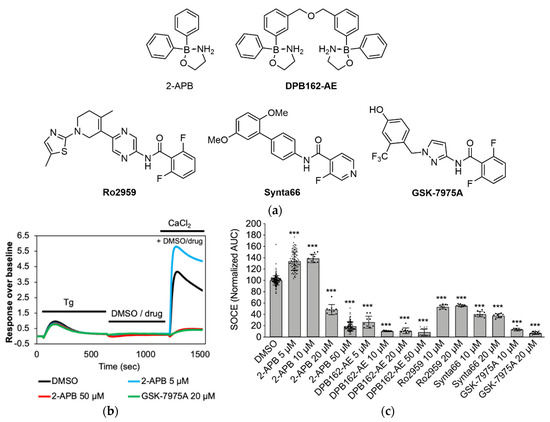
Figure 1.
Pharmacological characterization of store-operated calcium entry (SOCE) in MDA-MB-231 cells by fluorometric imaging plate reader (FLIPR) assay. (a) Structures of known SOCE inhibitors used in the validation of the assay. (b) Recorded intracellular calcium signal over the course of the assay. Black bars above traces indicate duration of applications. MDA-MB-231 cells were incubated with thapsigargin (Tg, 1 μM, 10 min) in nominal calcium-free (NCF) buffer, followed by DMSO (negative control) or drug application (in NCF) for 10 min and finally, addition of 2 mM extracellular CaCl2. Note how low concentration (5 μM) of 2-APB enhanced SOCE (blue trace) whereas at high concentration (50 μM) SOCE is blocked (red trace). (c) Quantification of calcium influx after CaCl2 application (area under the curve (AUC)), normalized to DMSO control (n ≥ 9 for all data points). Data shown are mean ± SD. Statistical significance compared to DMSO control is indicated as “***” for p ≤ 0.001.
Several derivatives of 2-APB that modulate SOCE have been reported previously [28,52,54,58,66,67,68]. Among these derivatives, DPB162-AE (Figure 1), a dimeric, more specific as well as more potent variant of 2-APB, did not exert the typical dose-dependent potentiating effect on SOCE [54,58]. Interestingly, in addition to SOCE inhibition, DPB162-AE (1–10 µM) also released Ca2+ from the ER stores of pancreatic acinar cells as well as HEK293, HeLa and B-cell lymphoma SU-DHL-4 cells [69,70]. In HEK293 and HeLa cells, IP3Rs were partly responsible for this DPB162-AE elicited ER Ca2+ release, with a likely additional role of other ER Ca2+ release channels [69]. A similar IP3R-dependent release of ER Ca2+ stores was reported in DT40 cells by 2-APB at concentrations ranging from 10 to 75 µM [56]. The release of ER Ca2+ by 2-APB is most likely cell type-dependent, as pretreatment of Chinese hamster ovary-K1 cells with 100 µM 2-APB did not affect the thapsigargin-mediated store-depletion [71].
In this study, we used the MDA-MB-231 breast cancer cell line to screen our newly developed 2-APB analogues for their action on SOCE, using a fluorometric imaging plate reader (FLIPR)-based Ca2+ imaging assay. FLIPR assays with MDA-MB-231 cells have been used in the past for discovery of SOCE modulators [26,30,72]. Earlier reports using RNA silencing have shown that SOCE in MDA-MB-231 cells is conducted by STIM1 and Orai1 [18,26,73]. In addition, it was shown that 2-APB at concentrations ≥30 µM blocks SOCE in MDA-MB-231 cell line [73,74,75]. Herein, we report new derivatives of 2-APB, that are more potent than 2-APB in blocking SOCE in MDA-MB-231 cells. We also report the effect of our new 2-APB derivatives on store-depletion as well as proliferation of MDA-MB-231 cells.
2. Results and Discussion
2.1. Synthesis of 2-APB Analogues
The mono-halogenated 2-APB analogues were synthesized by following our previously optimized route [67]. More specifically, mono-halogenated aryl bromides or iodides were transformed into their corresponding aryl lithium species by halogen-lithium exchange at low temperature and then reacted with phenylboronic acid pinacol ester (Scheme 1). This gave the crude aryl phenyl borinic acids after work up and quick flash chromatography purification, which were subsequently esterified with 2-aminoethanol to yield the stable, mono-halogenated 2-APB analogues. This protocol allowed the synthesis of almost all possible mono-halogenated 2-APB analogues (Scheme 1a) in isolated yields ranging from 6 to 65%. For the synthesis of o-Br-2APB it was important to use a mixture of toluene/THF 4:1 [76] in the lithiation and subsequent borination step, in order to significantly reduce the formation of a biphenyl side product that is very difficult to separate from the desired product (Appendix A). We were unable to isolate the ortho-iodo 2-APB derivative o-I-2APB. All our conditions gave inseparable mixtures of ortho-iodophenyl borinic acid and significant amounts of the side product (Appendix A).
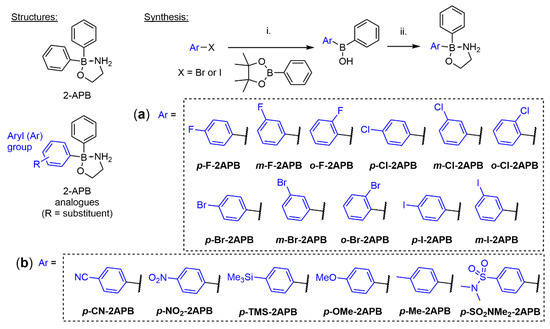
Scheme 1.
Synthesis of (a) mono-halogenated and (b) para-substituted 2-APB analogues. Reagents and conditions: (i) n-BuLi or t-BuLi, Et2O, or THF, −78 °C; then phenylboronic acid pinacol ester, Et2O or THF, −78 °C to rt; then 1 M HCl, rt. For the synthesis of o-Br-2APB toluene/THF 4:1 was used as solvent. (ii) 2-Aminoethanol, EtOH, MeCN, or THF, rt or reflux. Isolated yields over two steps: 6–66%. p-NO2-2APB was synthesized via different route (see text and Materials and Methods).
We were also interested in accessing 2-APB congeners with small substituents other than halogens in order to probe the influence of size and electron-withdrawing or electron-donating properties at the para-position on SOCE activity. These compounds were generated using the same route as above, except for p-NO2-2APB, and yielded 2-APB analogues shown in Scheme 1b in 8–66%. The para-nitro 2-APB analogue was obtained by reacting 4-nitrophenylborinic acid ester with phenyllithium, followed by borinate formation of the isolated borinic acid addition product.
For comparison, we also synthesized the dimeric 2-APB compound DPB162-AE, reported by the Mikoshiba group [54,77], but slightly adapted their procedure (Scheme S1). Recently reported heterocyclic SOCE blockers GSK-7975A and Synta66 were also synthesized (Schemes S2 and S3) applying procedures in the corresponding patents [78,79]. Notably, we have obtained a crystal structure for GSK-7975A which was not described before (Figure S1).
Others and we have shown previously that amino acid adducts of 2-APB, forming acyloxyboranes, are less dynamic in solution than the corresponding amino alcohol esters [67,80]. Furthermore, our studies indicated that amino acid 2-APB adducts with large side chain might block SOCE in HEK293 cells efficiently. In order to study this more systematically, we expanded our 2-APB library in this direction and synthesized several amino acid complexes shown in Scheme 2. Conveniently, such derivatives can be synthesized in one step from diphenylborinic anhydride.
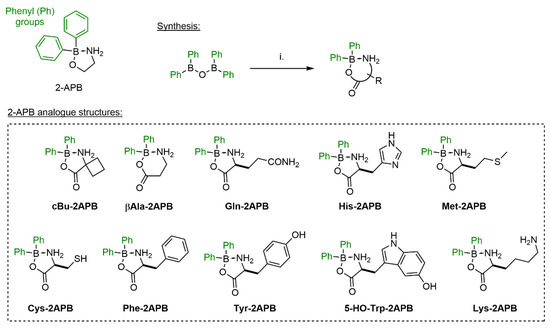
Scheme 2.
Synthesis of 2-APB amino acid analogues. Reagents and conditions: (i) amino acid, EtOH, reflux. Isolated yields: 16%-quantitative.
The cyclic nature of the synthesized 2-APB derivatives in solution is apparent from their 1H and 11B NMR spectra (in DMSO-d6). Donation of electron density of the amino alcohol or amino acid nitrogen to the electron deficient boron atom leads to deshielding of the N-H protons and thus they are low field shifted to 6.1–7.2 ppm. Likewise, the 11B chemical shift of the central boron atom is recorded at 3.7–4.7 ppm for all isolated 2-APB derivatives, except for βAla-2APB which is at 2.05 ppm. For comparison, the “uncoordinated” boron atom in diphenyl borinic acid (Ph2B(OH)) shows a 11B chemical shift of 45 ppm [67]. The cyclic form of the 2-APB derivatives is also visible in the crystal structures of cBu-2APB, p-SO2NMe2-2APB, o-F-2APB, and o-Cl-2APB (Figures S2–S5). For the latter two derivatives the phenyl and ortho-substituted phenyl groups are mutually disordered in the structure (Figures S4 and S5).
2.2. Influence of 2-APB Analogues on Store-Operated Ca2+ Entry in MDA-MB-231 Breast Cancer Cells
For evaluating the activity of the synthetic 2-APB analogues on SOCE, we used a fluorescence-based calcium influx assay into the triple negative breast cancer cells MDA-MB-231. In our assay protocol, 1 μM thapsigargin in nominal calcium-free (NCF) buffer was first applied to the cells for several minutes, which irreversibly blocks SERCA, passively depletes the intracellular ER-stores and initiates the SOCE cascade. This was followed by compound application in NCF buffer for several minutes and a subsequent application of 2 mM extracellular CaCl2 to measure external calcium entry through CRAC channels (Figure 1b). We validated our assay using known SOCE borinic acid inhibitors 2-APB, DPB162-AE and heterocyclic compounds GSK-7975A, Synta66, and Ro2959 [81] (Figure 1a) at concentrations between 5 and 20 μM (Figure 1c). As reported previously in other cell lines, 2-APB showed a similar bimodal effect on SOCE in MDA-MB-231 cells wherein it enhanced Ca2+ entry at low concentrations (5 and 10 μM) and blocked at higher concentrations (partial block at 20 and maximum block at 50 μM) [49,56]. The dimeric 2-APB derivative DPB162-AE exerted a comparable inhibition of SOCE at lower concentration, as 2-APB at 50 µM, confirming its higher activity than 2-APB [77]. The inhibition of SOCE at 10 μM by DPB162-AE was even stronger than by heterocyclic Ro2959 and Synta66. Even doubling the concentration of the latter two compounds did not increase the SOCE block. In our assay, GSK-7975A was the most potent heterocyclic SOCE blocker. We were surprised at the somewhat low activity of Ro2959 and Synta66. The inhibition activity of these heterocyclic compounds has been previously characterized by measuring the influence on the calcium release-activated calcium current (ICRAC) using electrophysiology.
The reported values for Synta66 (IC50 1.4 μM, ICRAC in RBL cells) [82,83], GSK-7975A (IC50 4 μM, ICRAC in HEK293 cells) [84], and Ro2959 (IC50 0.4 μM, ICRAC in RBL-2H3 cells) [85] suggest that they should fully block calcium entry at the concentrations applied in our SOCE assay in MDA-MB-231 cells. The electrophysiology studies have also revealed that all three heterocyclic compounds have a slow onset of inhibition, requiring cells to be preincubated for long periods (e.g., >1 h for Synta66). Indeed, when we preincubated 0.3–0.5 μM GSK-7975A or 1.25–10 µM Synta66 for 20 min before the thapsigargin application (30 min total incubation time), SOCE in MDA-MB-231 cells was inhibited by 50% or 80%, respectively, which is in the range of the reported IC50 values (Figure S6).
Next, we investigated whether the SOCE observed in MDA-MB-231 cells that we could block by inhibitors shown in Figure 1 is conducted by STIM1 and Orai1. Indeed, knock-out of either STIM1 or Orai1 in MDA-MB-231 cells using CRISPR/Cas9 gene editing technique, led to significantly diminished SOCE (Figure S7). This suggests that SOCE in these cells is mainly facilitated by Orai1/STIM1, confirming the RNA interference studies by Motiani et al. [73].
After having validated the SOCE assay, we first tested all our synthetic 2-APB analogues at a single concentration (50 µM, Figure 2). Our data show that all halogenated compounds efficiently blocked SOCE, some even exerted a more pronounced reduction of the Ca2+ entry than the parent compound (e.g., m-Cl-2APB, o-Cl-2APB, o-Br-2APB, and m-I-2APB). Compared to the para-halogenated analogues, most of the other para-substituted congeners did not block SOCE significantly. In fact, 2-APB derivatives with large, polarized substituents in the para-position rather enhanced SOCE (e.g., p-CN-2APB, p-NO2-2APB, and p-SO2NMe2-2APB). The amino acid 2-APB derivatives βAla-2APB, His-2APB, and Tyr-2APB could not be tested because of very poor solubility in the assay buffer. Only few derivatives with polar side chains (e.g., Gln-2APB, Lys-2APB, and Cys-2APB) were able to inhibit SOCE in MDA-MB-231 cells (Figure 2).
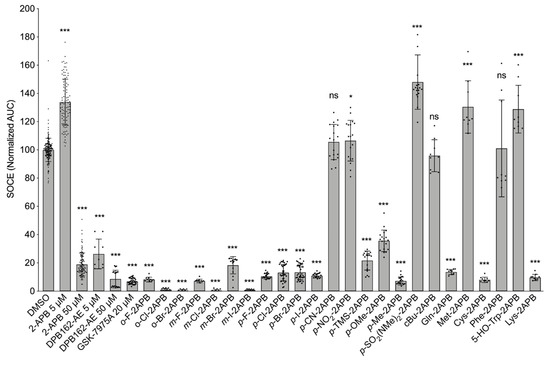
Figure 2.
Screening of synthetic 2-APB analogues on SOCE in MDA-MB-231 cells using the FLIPR-based SOCE assay. Quantification of calcium influx after CaCl2 application (AUC), normalized to DMSO control (n ≥ 9 for all data points). Data shown are mean ± SD. All compounds were tested at 50 µM concentration, unless otherwise noted. Statistical significance compared to DMSO control is indicated as “***” for p ≤ 0.001, “*” for 0.01 < p ≤ 0.05 and “ns” not significant for p > 0.05.
Given the encouraging results, we turned our attention to the mono-halogenated 2-APB analogues. In order to test if any of them have higher SOCE blocking potency than 2-APB, we applied them at lower concentrations (10 and 20 µM, Figure 3). At 10 µM compound concentration all fluorinated 2-APB derivatives enhanced SOCE, similar to 2-APB at the same concentration. This effect was also observed for some of the chloro-compounds, depending on the substitution position, whereas the bromo- and iodo-analogues were overall rather inhibiting SOCE at this low concentration. In terms of the halogen substitution position on one of the phenyl rings of 2-APB, the chloro, bromo, and iodo 2-APB derivatives affect SOCE with the following trend: ortho enhances SOCE, meta blocks efficiently, and para shows only partial or no block of SOCE. This efficient block of SOCE by the meta-substituted derivatives is also apparent at 20 µM compound concentration, at which 2-APB only partially blocks (Figure 3).
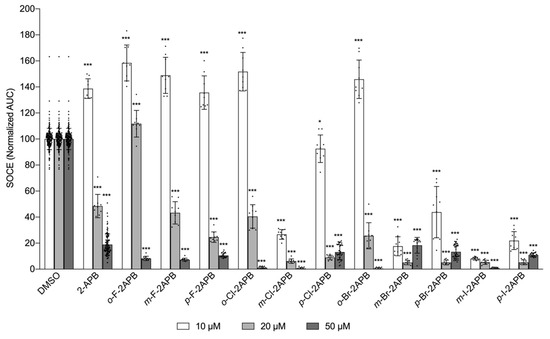
Figure 3.
Select halogenated 2-APB analogues block SOCE more potently at lower concentration than 2-APB. Influence on SOCE in MDA-MB-231 cells after prior depletion of the intracellular Ca2+ store, using the FLIPR-based SOCE assay. Data show quantification of Ca2+ entry (AUC, normalized to DMSO control) after extracellular application of 2 mM CaCl2. Compound concentrations: 10 µM (white bars), 20 µM (grey bars), 50 µM (dark grey bars). All data points from n ≥ 9 independent experiments. Data shown are mean ± SD. Statistical significance is compared to the respective DMSO control and indicated as “***” for p ≤ 0.001 and “*” for 0.01 < p ≤ 0.05.
When comparing the SOCE blocking effect at 10 µM compound concentration with the different halogen substituents at the same position, all ortho-halogenated derivatives did not block, but rather potentiated SOCE. For the meta- and para-positions, inhibiting potency increased with increasing size and decreasing electronegativity (m-F-2APB << m-Cl-2APB < m-Br-2APB < m-I-2APB and p-F-2APB << p-Cl-2APB < p-Br-2APB < p-I-2APB).
2.3. Store Depletion in MDA-MB-231 Breast Cancer Cells by 2-APB Analogues
We also investigated if the synthesized 2-APB derivatives have an effect on intracellular Ca2+ stores, as it has been observed in some cell lines with DPB-162AE [69] and non-boron based SOCE inhibitors [72]. To this end, we treated MDA-MB-231 cells with compounds at 50 µM in NCF buffer first and then added thapsigargin to deplete the stores (Figure 4). The FLIPR recordings of intracellular [Ca2+] show that 2-APB, for instance, does not lead to any detectable mobilization of Ca2+ from intracellular stores of MDA-MB-231 cells. Conversely, p-Br-2APB produced a substantial Ca2+ release signal during application and subsequent store depletion by thapsigargin was unable to mobilize more calcium. Quantification of this effect revealed that dimeric 2-APB compound DPB162-AE mobilizes stored calcium significantly at high concentration compared to 2-APB. From the halogenated compounds, p-Br-2APB and p-I-2APB were as strong or even stronger Ca2+ mobilizers than DPB162-AE.
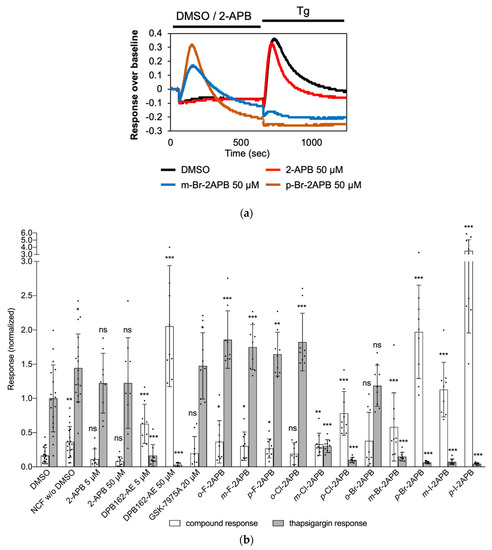
Figure 4.
Store-depletion by selected 2-APB analogues in MDA-MB-231 cells. (a) Examples of recorded intracellular calcium responses for m-Br-2APB and p-Br-2APB in FLIPR assay. Black bars above traces indicate duration of compound applications. MDA-MB-231 cells were incubated with compounds (50 µM, unless otherwise noted) in NCF for 10 min, followed by thapsigargin (Tg, 2.5 µM) application in NCF buffer. (b) Quantifications of Ca2+ mobilization effect from intracellular stores, using the slopes of the curves, by compounds (white bars) and by subsequent thapsigargin (dark bars) application. Normalized to post-thapsigargin response by DMSO, n ≥ 9 for all data points. Data shown are mean ± SD. Statistical significance is compared to the respective DMSO control and indicated as “***” for p ≤ 0.001, “**” for 0.001 < p ≤ 0.01 and “*” for 0.01 < p ≤ 0.05. ns, not significant for p > 0.05.
The compound-induced (pre-thapsigargin) store depletion increases with increasing size and decreasing electronegativity of the halogen substituent (F < Cl < Br < I), in particular at the para-position. Concerning the halogen substitution position, the Ca2+ mobilization effect was strongest for the para-compounds, less for the meta-, and not substantial for the ortho-analogues.
In addition, we checked the ability of m-Br-2APB and p-Br-2APB to block ICRAC in HEK293 cells overexpressing STIM1/Orai1 (Figure 5). Both compounds rapidly inhibited ICRAC in a similar fashion as the parent compound 2-APB, which suggests that in addition to store-depleting activity (Figure 4) SOCE was blocked as well.
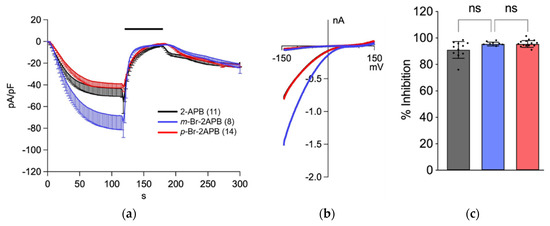
Figure 5.
Inhibition of ICRAC in HEK293 cells overexpressing human STIM1/Orai1 by 2-APB (black traces) and analogues m-Br-2APB (blue) and p-Br-2APB (red). (a) ICRAC was evoked with 20 mM BAPTA and 50 µM IP3 in the patch pipette, currents at −130 mV were normalized to cell capacity and plotted as current density (CD) versus time. Upon ICRAC development, 2-APB (black), m-Br-2APB (blue) or p-Br-2APB (red) were applied from 120–180 s (black bar above traces). (b) I-V curves extracted from recordings in (a) when ICRAC had developed at t = 118 s. (c) Inhibition of ICRAC was calculated from CD at 180 s normalized to the point in time before compounds were applied (t = 118 s) and plotted as inhibition in %. All compounds applied at 50 µM, number of experiments (n) in brackets in panel (a). Data shown are mean ± SD. Statistical significance is indicated as ns for not significant (p > 0.05).
2.4. Synthetic 2-APB Analogues Effect MDA-MB-231 Cell Viability
We wondered if the propensity of the 2-APB analogues to mobilize calcium from internal stores is affecting cell viability. MDA-MB-231 cells as well as MCF-10A, a non-tumorigenic mammary epithelial cell line (control), were incubated with 2-APB or halogenated derivatives thereof at 50 µM concentration for different time points ranging from 24 to 120 h. The XTT cell viability assay showed that 2-APB did not have any substantial effect on proliferation of any of the cell lines (Figure 6a). While the more specific SOCE blocker GSK-7975A had only minor cytotoxic effect in both cell lines, 50 µM DPB162-AE strongly affected the proliferation of MDA-MB-231 cells in comparison to MCF-10A (Figure 6a). On the other hand, the ortho- and para-halogenated analogues did not show contrasting effects on proliferation of MDA-MB-231 cells compared to MCF-10A cells (Figure 6b,d). Conversely, m-Cl-2APB, m-Br-2APB, and m-I-2APB exhibited a stronger cytotoxic effect similar to that of 50 µM DPB162-AE on MDA-MB-231 cells compared to MCF-10A cells (Figure 6c). These compounds also induced changes in morphology of MDA-MB-231 cells (Figure 7). The difference between the ortho- and para-halogenated regioisomers for example, p-Br-2APB and m-Br-2APB is particularly striking considering that the bromine atom is moved by only one carbon position (Figure 6 and Figure 7). In any case, the data suggest that compounds which empty internal calcium stores (e.g., p-Br-2APB) do not influence MDA-MB-231 cell viability much stronger than compounds which empty internal stores very little (e.g., p-F-2APB). Furthermore, we do not see a direct relation between SOCE inhibitory activity of 2-APB analogues or control compounds (e.g., GSK-7975A) and MDA-MB-231 cell proliferation. We cannot rule out the possible off-target effects of our 2-APB analogues in the observed cytotoxicity.
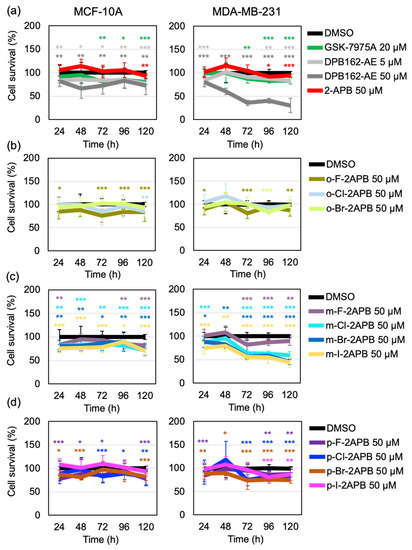
Figure 6.
Dose-response curves of 20 µM GSK-7975A, 5 µM and 50 µM DPB162-AE, and 50 µM 2-APB (a), 50 µM o-F-2APB, o-Cl-2APB, and o-Br-2APB (b) 50 µM m-F-2APB, m-Cl-2APB, m-Br-2APB, and m-I-2APB (c), and 50 µM p-F-2APB, p-Cl-2APB, p-Br-2APB, and p-I-2APB (d) in non-tumorigenic breast epithelial cell line MCF-10A and triple negative breast cancer cell line MDA-MB-231. Relative cell survival in case of each compound treatment was calculated as a percentage of DMSO control. Data shown are mean ± SD (n = 12/compound for each time point) of two independent experiments. Significance levels compared to the DMSO control are indicated as “*” for 0.01 < p ≤ 0.05, “**” for 0.001 < p ≤ 0.01, or “***” for p ≤ 0.001 and not indicated if non-significant (p > 0.05).
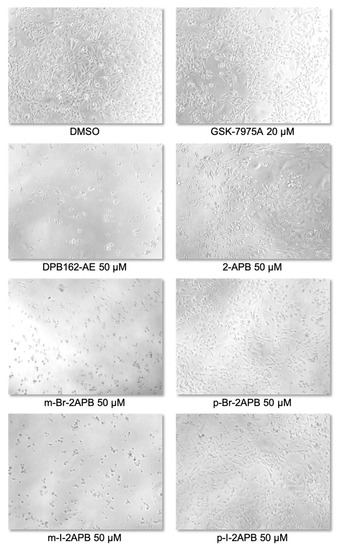
Figure 7.
Photomicrographs show the morphological aspects of the cultures of MDA-MB-231 cells after 72 h treatment with control (DMSO) or the indicated compounds. The shown bright-field microscopy images were captured on Nikon Eclipse TiU inverted microscope.
3. Materials and Methods
3.1. Chemical Synthesis
Commercial solvents and reagents were used without further purification. Dry solvents were obtained by filtration over columns of dried alumina under a positive pressure of argon. The glassware was dried by heating while flushing with argon. In general, reactions were run under argon atmosphere. Flash chromatography was performed on a TeledyneISCO CombiFlash Rf+ (Lincoln, NE, USA) using RediSepRf (Lincoln, NE, USA) pre-packed silica gel columns and cyclohexane/EtOAc gradients to elute compounds, unless otherwise noted. 1H, 11B, 13C, and 19F NMR spectra were measured on a Bruker Avance 300 III HD or Avance II 400 spectrometer. Chemical shifts given in ppm, are referenced to residual solvent peaks (1H and 13C) or internal standard (11B and 19F), and coupling constants J in Hz. (high resolution) mass spectra were recorded on a Thermo Fisher Scientific (Waltham, MA, USA) LTQ Orbitrap XL spectrometer consisting of a linear ion trap (LTQ) featuring a HCD collision cell, coupled to the Orbitrap mass analyzer, equipped with a nanospray ion source (NSI). All X-ray structure determination measurements were made on an Oxford Diffraction SuperNova area-detector diffractometer using mirror optics monochromated Mo Kα radiation (λ = 0.71073 Å) and Al filtered.
Reference compound Ro2925 was purchased from AK Scientific and DPB162-AE, Synta66 and GSK-7975A were resynthesized (Supplementary Materials) by adapting literature [54] and patent protocols [78,79]. The synthesis and spectroscopic characterization of 2-APB derivatives p-Me-2APB and p-I-2APB was reported previously [67].
3.1.1. General Procedure A for the Synthesis of Mono-substituted Phenyl 2-APB Analogues
Dry Et2O or THF (60 mL) was cooled to −78 °C and, unless otherwise noted, t-BuLi solution (10.2 mmol, 1.7 M in pentane) was added dropwise. The resulting mixture was stirred at −78 °C for 5 min. Then, a solution of iodide or bromide (4.96 mmol) in dry Et2O or THF (10 mL) was added to the reaction mixture at a drop rate so that the internal temperature remained below −75 °C. The mixture was stirred for another hour at −78 °C. A solution of phenylboronic acid pinacol ester (4.96 mmol) in dry Et2O or THF (10 mL) was then added, again taking care that the internal temperature did not raise above −75 °C. The reaction mixture was allowed to warm up to r.t. overnight and was then quenched by the addition of aq. 1 M HCl (100 mL). The two resulting phases were separated, and the aqueous phase extracted with Et2O or EtOAc (2 × 20 mL). The combined organic layers were dried over Na2SO4, filtered and evaporated. The crude, oily borinic acid was purified by flash chromatography (silica gel, cyclohexane/EtOAc gradient). The resulting purified borinic acid (oil or solid) was dissolved in MeCN or abs. EtOH (5 mL) and 2-aminoethanol (3.48 mmol, 1 equiv. with respect to purified borinic acid) was added to the solution. The resulting solution was stirred at r.t. for 30 min–1 h to produce the corresponding borinic acid 2-aminoethyl ester, which either precipitated or was isolated after removal of the solvent (for details see individual compounds below).
3.1.2. General Procedure B for the Synthesis of 2-APB Amino Acid Analogues
A mixture of amino acid (0.70 mmol) and diphenylborinic anhydride (0.58 mmol) in abs. EtOH (10 mL) was heated at reflux for 30 min–3 h. The clear solution was allowed to cool to r.t. during which the product often precipitated. For some derivatives, further cooling to 0 °C or −20 °C and concentration of the solution was necessary to initiate precipitation or crystallization. The solid products were washed (e.g., with cold EtOH, n-hexane or Et2O) or recrystallized if necessary (for details see individual compounds below).
3.1.3. 2-(4-Fluorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-F-2APB)
Following general procedure A, 4-fluoroiodobenzene (572 µL, 4.96 mmol) in dry Et2O was lithiated with t-BuLi (6 mL, 1.7 M in pentane, 10.2 mmol) and reacted with phenylboronic acid pinacol ester (1.01 g, 4.96 mmol). Purification of the crude borinic acid by flash chromatography afforded a brownish oil (663 mg), which was dissolved in MeCN and treated with 2-aminoethanol (210 µL, 3.48 mmol) at r.t. for 30 min. Removing the solvents under reduced pressure and drying the residue under high vacuum afforded the title compound as a colorless powder (742 mg, 62%). 1H NMR (300 MHz, DMSO-d6) δ 7.43–7.34 (m, 4H), 7.20–7.09 (m, 2H), 7.08–6.99 (m, 1H), 6.97–6.88 (m, 2H), 6.07 (s, 2H), 3.76 (t, J = 6.5, 2H), 2.82 (p, J = 6.3, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.27. 13C NMR (75 MHz, DMSO-d6) δ 133.20, 133.11, 131.46, 126.66, 124.98, 113.23, 112.98, 62.43, 41.35. 19F NMR (282 MHz, DMSO-d6) δ -118.69 (p, J = 9.1). MS (ESI+) m/z 305.18 [M+C2H8NO]+, 266.11 [M+Na]+, 244.13 [M+H]+, 166.08 [M-C6H5]+, 148.09 [M-C6H4F]+. HRMS (ESI+) m/z calc. for C14H16ONBF [M+H]+: 244.1303; found 244.1303.
3.1.4. 2-(3-Fluorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (m-F-2APB)
According to general procedure A, 1-bromo-3-fluorobenzene (392 µL, 5 mmol) in dry THF was lithiated with t-BuLi (6 mL, 1.7 M in pentane, 10.2 mmol) and condensed with phenylboronic acid pinacol ester (1.02 g, 5 mmol). After aqueous work up, the crude borinic acid was purified by flash chromatography and gave a pale yellow oil (188 mg). This was dissolved in abs. EtOH and treated with 2-aminoethanol (70 µL, 1.16 mmol). After solvent removal and drying the residual oil in a Kugelrohr apparatus under high vacuum at 80 °C, the title compound was obtained as off-white powder (223 mg, 17%). 1H NMR (300 MHz, DMSO-d6) δ 7.43–7.35 (m, 2H), 7.23–6.99 (m, 6H), 6.86–6.75 (m, 1H), 6.13 (s, 2H), 3.76 (t, J = 6.5, 2H), 2.83 (p, J = 6.2, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.01. 13C NMR (75 MHz, DMSO-d6) δ 163.59, 160.38, 131.41, 128.45, 128.36, 127.32, 127.30, 126.70, 125.08, 117.32, 117.10, 111.52, 111.25, 62.44, 41.35. 19F NMR (282 MHz, DMSO-d6) δ -115.64 (m). MS (ESI+) m/z 244.13 [M+H]+, 266.11 [M+Na]+, 166.08 [M-C6H5]+. HRMS (ESI+) m/z calc. for C14H16ONBF [M+H]+: 244.1303; found 244.1300.
3.1.5. 2-(2-Fluorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (o-F-2APB)
1-Bromo-2-fluorobenzene (547 µL, 5 mmol) was lithiated in dry THF using t-BuLi, following general procedure A, and reacted with phenylboronic acid pinacol ester (1.02 g, 5 mmol). After work up and purification of the crude borinic acid by flash chromatography an almost colorless oil was isolated (841 mg) that was mixed with abs. EtOH and 2-aminoethanol (275 µL, 4.56 mmol) to generate the borinate. Drying the crude product in the Kugelrohr under high vacuum at 80 °C gave a slightly brown solid (997 mg), which was recrystallized from MeCN (7 mL), yielding off-white crystals (837 mg, 65%) suitable for X-ray structure determination (Figure S4). 1H NMR (300 MHz, DMSO-d6) δ 7.47–7.33 (m, 3H), 7.17–6.84 (m, 6H), 6.33–6.06 (m, 2H), 3.87–3.75 (m, 1H), 3.73–3.61 (m, 1H), 2.95–2.73 (m, 2H). 11B NMR (96 MHz, DMSO-d6) δ 3.70. 13C NMR (75 MHz, DMSO-d6) δ 133.60, 133.43, 131.58, 131.55, 127.26, 127.15, 126.57, 125.13, 123.15, 123.12, 114.00, 113.66, 62.36, 41.69. 19F NMR (376 MHz, DMSO-d6) δ -105.62 (m). MS (ESI+) m/z 244.13 [M+H]+, 266.11 [M+Na]+, 305.18 [M+C2H8NO]+. HRMS (ESI+) m/z calc. for C14H16ONBF [M+H]+: 244.1303; found 244.1305.
3.1.6. 2-(4-Chlorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-Cl-2APB)
Following general procedure A, the title compound was synthesized from 1-chloro-4-iodobenzene (1.1 g, 4.61 mmol) using dry Et2O as reaction solvent. Work up and purification by flash chromatography yielded a colorless solid (620 mg) which was treated with 2-aminoethanol (200 µL, 3.32 mmol) in MeCN. The mixture was heated to reflux for 1 h in order to dissolve all the solid. Removing the solvent under reduced pressure afforded a foam, which was dried in vacuo. The solidified foam was crushed and washed with n-hexane. Further drying under high vacuum gave the title compound as a colorless powder (743 mg, 62%). 1H NMR (300 MHz, DMSO-d6) δ 7.43–7.33 (m, 4H), 7.20–7.10 (m, 4H), 7.08–7.00 (m, 1H), 6.11 (s, 2H), 3.75 (t, J = 6.5, 2H), 2.82 (p, J = 6.2, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.14. 13C NMR (75 MHz, DMSO-d6) δ 133.38, 131.43, 129.85, 126.69, 126.44, 125.05, 62.45, 41.36. MS (ESI+) m/z 260.10 [M+H]+, 282.08 [M+Na]+, 300.13 [M+C2H3N]+, 321.15 [M+C2H8NO]+, 182.05 [M-C6H5]+, 148.09 [M-C6H4Cl]+. HRMS (ESI+) m/z calc. for C14H16ONBCl [M+H]+: 260.1008; found 260.1011.
3.1.7. 2-(3-Chlorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (m-Cl-2APB)
1-Bromo-3-chlorobenzene (587 µL, 5 mmol) was lithiated in dry THF and reacted with phenylboronic acid pinacol ester according to general procedure A. Work up and purification by flash chromatography gave a pale yellow oil (741 mg) that was treated with 2-aminoethanol in abs. EtOH for 1 h at r.t. After solvent removal and drying of the residual oil in a Kugelrohr under high vacuum at 80 °C the title compounds was isolated as a colorless powder (746 mg, 58%). 1H NMR (300 MHz, DMSO-d6) δ 7.42–7.29 (m, 4H), 7.20–7.10 (m, 3H), 7.10–7.01 (m, 2H), 6.16 (s, 2H), 3.76 (t, J = 6.5, 2H), 2.83 (p, J = 6.2, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.17. 13C NMR (75 MHz, DMSO-d6) δ 132.07, 131.37, 130.99, 129.93, 128.62, 126.74, 125.12, 124.72, 62.45, 41.37. MS (ESI+) m/z 260.10 [M+H]+, 282.08 [M+Na]+, 321.15 [M+C2H8NO]+, 182.05 [M-C6H5]+, 148.09 [M-C6H4Cl]+. HRMS (ESI+) m/z calc. for C14H16ONBCl [M+H]+: 260.1008; found 260.1006.
3.1.8. 2-(2-Chlorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (o-Cl-2APB)
The title compound was synthesized from 1-bromo-2-chlorobenzene (584 µL, 5 mmol) following general procedure A using THF as the reaction solvent. Work up and flash chromatography purification of the borinic acid afforded a colorless oil (450 mg) that was subjected to 2-aminoethanol treatment in abs. EtOH. After removing the solvent and drying of the oily product (Kugelrohr, 80 °C, high vacuum) a colorless solid was isolated, which was suspended in Et2O (10 mL) using an ultrasonic bath. After the precipitate settled, the supernatant was removed. Washing with Et2O was repeated two more times and the remaining solid (490 mg) was recrystallized from MeCN (3 mL), yielding colorless crystals (235 mg, 18%) suitable for X-ray diffraction analysis (Figure S5). 1H NMR (300 MHz, DMSO-d6) δ 7.70–7.59 (m, 1H), 7.37–7.27 (m, 2H), 7.24–6.93 (m, 6H), 6.39 (s, 1H), 6.14 (s, 1H), 3.84–3.68 (m, 1H), 3.68–3.57 (m, 1H), 2.88 (dd, J = 15.4, 8.2, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.38. 13C NMR (75 MHz, DMSO-d6) δ 137.62, 133.77, 131.94, 131.52, 128.20, 127.27, 126.34, 125.55, 125.00, 62.09, 42.25. MS (ESI+) m/z 260.10 [M+H]+, 282.08 [M+Na]+, 336.13 [M+C6H5]+, 182.05 [M-C6H5]+. HRMS (ESI+) m/z calc. for C14H16ONBCl [M+H]+: 260.1008; found 260.1005.
3.1.9. 2-(4-Bromophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-Br-2APB)
The starting material 1,4-dibromobenzene (1 g, 4.24 mmol) was purified by flash chromatography (silica gel, cyclohexane/EtOAc gradient) prior to use in the reaction. General procedure A was followed using dry Et2O as the reaction solvent and n-BuLi solution (2.5 M in hexanes) for the lithiation. The thus obtained pale yellow, oily borinic acid (502 mg) after work up and flash chromatography was transformed into its borinate by 2-aminoethanol treatment in abs. EtOH. The oily product was dried (Kugelrohr, 80 °C, high vacuum), which yielded the title compound as colorless powder (472 mg, 37%). 1H NMR (300 MHz, DMSO-d6) δ 7.39–7.27 (m, 6H), 7.17–7.09 (m, 2H), 7.08–7.00 (m, 1H), 6.11 (s, 2H), 3.75 (t, J = 6.5, 2H), 2.82 (t, J = 6.3, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.11. 13C NMR (75 MHz, DMSO-d6) δ 133.83, 131.41, 129.33, 126.69, 125.05, 118.56, 62.44, 41.36. MS (ESI+) m/z 304.05 [M+H]+, 326.03 [M+Na]+, 226.00 [M-C6H5]+, 148.09 [M-C6H4Br]+. HRMS (ESI+) m/z calc. for C14H16ONBBr [M+H]+: 304.0503; found 304.0500.
3.1.10. 2-(3-Bromophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (m-Br-2APB)
Following general procedure A, the title compound was synthesized from 1,3-dibromobenzene (550 µL, 4.55 mmol) using dry Et2O as reaction solvent. Work up and purification by flash chromatography gave the borinic acid as a yellow oil (1.06 g), which was dissolved in abs. EtOH and esterified with 2-aminoethanol. Solvent removal and drying the oily product in the Kugelrohr at 80 °C in high vacuum yielded the final borinate as a colorless powder (887 mg, 64%). 1H NMR (400 MHz, DMSO-d6) δ 7.54–7.49 (m, 1H), 7.39–7.35 (m, 3H), 7.24–7.19 (m, 1H), 7.17–7.02 (m, 4H), 6.17 (s, 2H), 3.76 (t, J = 6.5, 2H), 2.88–2.78 (m, 2H). 11B NMR (96 MHz, DMSO-d6) δ 3.84. 13C NMR (75 MHz, DMSO-d6) δ 133.91, 131.37, 130.30, 129.07, 127.61, 126.75, 125.14, 121.46, 62.45, 41.38. MS (ESI+) m/z 304.05 [M+H]+, 326.03 [M+Na]+, 365.10 [M+C2H8NO]+. HRMS (ESI+) m/z calc. for C14H16ONBBr [M+H]+: 304.0503; found 304.0504.
3.1.11. 2-(2-Bromophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (o-Br-2APB)
A mixture of THF (10 mL), toluene (40 mL), 1,2-dibromobenzene (602 μL, 5 mmol), and phenylboronic acid pinacol ester (2.04 g, 10 mmol) was cooled down to −78 °C. n-BuLi solution (2 mL, 2.5 M in hexanes, 5 mmol) was added dropwise over a period of 3 h. The cloudy white mixture was allowed to warm up to r.t. overnight. The mixture was quenched by the addition of aq. 1 M HCl (50 mL) and EtOAc (100 mL) was added. The resulting two phases were separated and the organic phase was washed with aq. 1 M HCl (3 × 50 mL). The organic phase was dried over Na2SO4 and the solvents were removed under reduced pressure. The residue was dried in vacuo, yielding a viscous sticky solid. The solid was purified by flash chromatography (silica gel, with cyclohexane/EtOAc gradient) affording an oil (446 mg). The oil was dissolved in abs. EtOH (5 mL) and 2-aminoethanol (120 μL, 2.00 mmol) was added to the solution. The resulting mixture was stirred at r.t. for 1 h. The solvent was removed under reduced pressure and the residue was dried in the Kugelrohr under high vacuum at 80 °C. The resulting solid was recrystallized from MeCN yielding the title compound as colorless crystals (189 mg, 12%). 1H NMR (300 MHz, DMSO-d6) δ 7.68 (dd, J = 7.4, 1.9, 1H), 7.38–7.27 (m, 3H), 7.22 (td, J = 7.3, 1.2, 1H), 7.12–6.97 (m, 4H), 6.40 (s, 1H), 6.12 (s, 1H), 3.80–3.69 (m, 1H), 3.66–3.56 (m, 1H), 3.02–2.80 (m, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.53. 13C NMR (75 MHz, DMSO-d6) δ 134.25, 132.14, 131.61, 128.57, 127.64, 126.28, 126.00, 124.96, 62.10, 42.51. MS (ESI+) m/z 304.05 [M+H]+. HRMS (ESI+) m/z calc. for C14H16ONBBr [M+H]+: 304.0503; found 304.0496.
3.1.12. 2-(3-Iodophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (m-I-2APB)
1,3-Diiodobenzene (1.65 g, 5 mmol) was lithiated with n-BuLi (2 mL, 2.5 M in hexanes, 5 mmol) in dry THF, following general procedure A, and reacted with phenylboronic acid pinacol ester to yield the corresponding borinic acid (604 mg) after work up and flash chromatography purification as a pale yellow oil. Treatment with 2-aminoethanol in abs. EtOH yielded, after solvent removal and drying (Kugelrohr, 80 °C, high vacuum), a sticky solid. This was reprecipitated with MeCN, filtered and dried again in the Kugelrohr (80 °C, high vacuum). The title compound was thus obtained as a colorless powder (434 mg, 25%). 1H NMR (300 MHz, DMSO-d6) δ 7.76–7.69 (m, 1H), 7.44–7.31 (m, 4H), 7.20–6.92 (m, 4H), 6.15 (s, 2H), 3.75 (t, J = 6.5, 2H), 2.82 (p, J = 6.2, 4H). 11B NMR (96 MHz, DMSO-d6) δ 3.74. 13C NMR (75 MHz, DMSO-d6) δ 139.99, 133.49, 131.36, 130.71, 129.30, 126.74, 125.12, 95.30, 62.44, 41.38. MS (ESI+) m/z 352.04 [M+H]+, 374.02 [M+Na]+, 413.09 [M+C2H8NO]+, 273.99 [M-C6H5]+, 148.09 [M-C6H4I]+. HRMS (ESI+) m/z calc. for C14H16ONBI [M+H]+: 352.0364; found 352.0355.
3.1.13. 4-(2-Phenyl-1,3,2λ4-oxazaborolidin-2-yl)benzonitrile (p-CN-2APB)
Following general procedure A, the title compound was synthesized from 4-bromobenzonitrile (900 mg, 4.95 mmol) using dry THF as reaction solvent. Work up and purification by flash chromatography yielded the borinic acid (712 mg) as a beige powder. It was dissolved in MeCN and treated with 2-aminoethanol, which gave the borinate as a sticky solid after solvent removal. This was redissolved in CH2Cl2, the solvent evaporated and the residual solid was dried under high vacuum. This afforded the title compound as a colorless powder (816 mg, 66%). 1H NMR (300 MHz, DMSO-d6) δ 7.66–7.51 (m, 4H), 7.41–7.33 (m, 2H), 7.19–7.08 (m, 2H), 7.09–7.00 (m, 1H), 6.26 (s, 2H), 3.88–3.68 (m, 2H), 2.94–2.76 (m, 2H). 11B NMR (96 MHz, DMSO-d6) δ 3.94. 13C NMR (75 MHz, DMSO-d6) δ 132.19, 131.34, 130.14, 126.81, 125.25, 119.89, 107.53, 62.48, 41.40, 39.52. MS (ESI+) m/z 251.13 [M+H]+, 273.12 [M+Na]+, 312.19 [M+C2H8NO]+, 173.09 [M-C6H5]+. HRMS (ESI+) m/z calc. for C15H16ON2B [M+H]+: 251.1350; found 251.1349.
3.1.14. 2-(4-Nitrophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-NO2-2APB)
A solution of 4-nitrophenylboronic acid pinacol ester (500 mg, 2.01 mmol) in dry THF (20 mL) was cooled to −78 °C. Phenyllithium solution (1.1 mL, 1.8 M in Bu2O, 1.98 mmol) was added dropwise to the mixture. During the addition the drop rate was adjusted so that the temperature of the mixture remained below −75 °C. The mixture was stirred at −78 °C for 4 h. While still stirring at −78 °C, the mixture was quenched by the addition of aq. 1 M HCl (10 mL) over a period of 2 min. More aq. 1 M HCl (40 mL) and CH2Cl2 (100 mL) were added to the mixture. The resulting two phases were separated. The clear yellow organic phase was dried over Na2SO4 and the solvent was removed under reduced pressure. The remaining brown oil was purified by flash chromatography (silica gel, gradient with CH2Cl2 and MeOH/CH2Cl2 1:19, (v/v)) affording a black oil (146 mg). The oil was dissolved in abs. EtOH (4 mL) and 2-aminoethanol (40 μL, 0.66 mmol) were added to the solution. The resulting mixture was stirred at r.t. for 30 min. The solvent was removed under reduced pressure and the residue was dried in vacuo affording a sticky yellow solid with a slight greenish tint. The solid was suspended in Et2O (20 mL) using an ultrasonic bath. After the solid settled, the supernatant was removed. This washing step was repeated once. The residue was dried in the Kugelrohr at 80 °C under high vacuum affording the title compound as a beige powder (47 mg, 9%). 1H NMR (300 MHz, DMSO-d6) δ 8.06–7.96 (m, 2H), 7.71–7.62 (m, 2H), 7.45–7.34 (m, 2H), 7.20–7.11 (m, 2H), 7.11–7.01 (m, 1H), 6.30 (s, 2H), 3.87–3.69 (m, 2H), 2.92–2.79 (m, 1H). 11B NMR (96 MHz, DMSO-d6) δ 4.25. 13C NMR (75 MHz, DMSO-d6) δ 145.69, 132.35, 131.38, 126.86, 125.34, 121.39, 117.03, 62.53, 41.47. MS (ESI+) m/z 271.12 [M+H]+, 293.11 [M+Na]+, 311.16 [M+C2H3N]+, 332.18 [M+C2H8NO]+, 193.08 [M-C6H5]+. HRMS (ESI+) m/z calc. for C14H16O3N2B [M+H]+: 271.1248; found 271.1249.
3.1.15. 2-Phenyl-2-(4-(trimethylsilyl)phenyl)-1,3,2λ4-oxazaborolidine (p-TMS-2APB)
1-Bromo-4-(trimethylsilyl)benzene (400 µL, 2.05 mmol) was reacted according to general procedure A in dry Et2O as reaction solvent. After work up and flash chromatography purification the borinic acid was obtained as pale yellow oil (170 mg), which was subjected to 2-aminoethanol treatment in abs. EtOH. The solid, isolated after solvent removal, was suspended in Et2O (10 mL) using an ultrasonic bath. After the solid settled, the supernatant was removed. This washing procedure was repeated two times with Et2O (10 mL) and once with n-hexane (5 mL). After drying the solid residue in high vacuum the title compound was isolated as colorless powder (49 mg, 8%). 1H NMR (300 MHz, DMSO-d6) δ 7.44–7.35 (m, 4H), 7.33–7.24 (m, 2H), 7.17–7.07 (m, 2H), 7.06–6.98 (m, 1H), 6.06 (s, 2H), 3.75 (t, J = 6.5, 2H), 2.81 (p, J = 6.4, 2H), 0.18 (s, 9H). 11B NMR (96 MHz, DMSO-d6) δ 4.42. 13C NMR (75 MHz, DMSO-d6) δ 134.96, 131.55, 131.44, 131.01, 126.58, 124.87, 62.41, 41.37, -0.92. MS (ESI+) m/z 298.18 [M+H]+, 320.16 [M+Na]+, 359.23 [M+C2H8NO]+, 220.13 [M-C6H5]+. HRMS (ESI+) m/z calc. for C17H25ONBSi [M+H]+: 298.1793; found 298.1793.
3.1.16. 2-(4-Methoxyphenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-OMe-2APB)
Starting from 4-bromoanisole (669 µL, 5.35 mmol) the title compound was synthesized according to general procedure A. The borinic acid (763 mg), obtained as pale yellow oil after work up and flash chromatography purification, was treated with 2-aminoethanol in MeCN at r.t. Removal of solvent gave a residual sticky solid that was redissolved in CH2Cl2. The solvent was evaporated under reduced pressure yielding the borinate as a colorless powder (825 mg, 60%). 1H NMR (300 MHz, DMSO-d6) δ 7.41–7.33 (m, 2H), 7.31–7.24 (m, 2H), 7.17–7.08 (m, 2H), 7.06–6.98 (m, 1H), 6.74–6.68 (m, 2H), 5.96 (s, 2H), 3.75 (t, J = 6.3, 2H), 2.81 (p, J = 6.3, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.40. 13C NMR (75 MHz, DMSO-d6) δ 157.34, 132.63, 131.56, 126.58, 124.83, 112.34, 62.42, 54.65, 41.33, 39.52. MS (ESI+) m/z 256.15 [M+H]+, 278.13 [M+Na]+, 178.10 [M-C6H5]+, 148.09 [M-C7H5O]+. HRMS (ESI+) m/z calc. for C15H19O2NB [M+H]+: 256.1503; found 256.1500.
3.1.17. N,N-Dimethyl-4-(2-phenyl-1,3,2λ4-oxazaborolidin-2-yl)benzenesulfonamide (p-SO2NMe2-2APB)
4-Bromo-N,N-diemthylbenzenesulfonamide (1 g, 3.79 mmol), which was freshly purified by flash chromatography (silica gel, cyclohexane/EtOAc gradient), was used as starting material. Dry THF was used as the reaction solvent and n-BuLi solution (2.5 M in hexanes) for the lithiation to synthesize the corresponding borinic acid according to general procedure A. The borinic acid was obtained as brown oil (930 mg) after work up and flash chromatography purification (silica gel, gradient with CH2Cl2 and MeOH/CH2Cl2 1:19 (v/v)). The oil was dissolved in abs. EtOH and 2-aminoethanol added to the solution. Removal of solvent yielded a sticky solid that was dried (Kugelrohr, 80 °C, high vacuum), affording an off-white solid. Recrystallization from MeCN gave the title compound (630 mg, 52%) as off-white crystals, suitable for X-ray diffraction analysis (Figure S3). 1H NMR (300 MHz, DMSO-d6) δ 7.68–7.63 (m, 2H), 7.53–7.49 (m, 2H), 7.43–7.37 (m, 2H), 7.20–7.12 (m, 2H), 7.09–7.02 (m, 1H), 6.24 (s, 2H), 3.78 (t, J = 6.5, 2H), 2.84 (p, J = 6.1, 2H), 2.55 (s, 6H). 11B NMR (96 MHz, DMSO-d6) δ 4.17. 13C NMR (75 MHz, DMSO-d6) δ 131.96, 131.36, 126.82, 125.76, 125.21, 117.00, 62.51, 41.44, 37.59. MS (ESI+) m/z 333.14 [M+H]+, 355.13 [M+Na]+, 394.20 [M+C2H8NO]+, 255.10 [M-C6H5]+; HRMS (ESI+) m/z calc. for C16H22O3N2BS [M+H]+: 333.1439; found 333.1433.
3.1.18. 6,6-Diphenyl-7-oxa-5-aza-6λ4-boraspiro[3.4]octan-8-one (cBu-2APB)
Following general procedure B (reflux for 55 min), 1-amino-1-cyclobutanecarboxylic acid (80 mg, 0.70 mmol) was used as starting material. The crude precipitate was washed with Et2O and n-hexane, reprecipitated in a mixture of EtOH and n-hexane and washed several times with EtOH and n-hexane. Drying the solid product in high vacuum afforded the title compound (32 mg, 16%), containing crystals suitable for X-ray structure determination (Figure S2). 1H NMR (300 MHz, DMSO-d6) δ 7.43–7.29 (m, 6H), 7.24–7.14 (m, 4H), 7.14–7.06 (m, 2H), 2.20–2.05 (m, 4H), 1.99–1.75 (m, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.27. 13C NMR (75 MHz, DMSO-d6) δ 176.39, 130.69, 126.97, 125.79, 59.16, 30.71, 14.72. MS (ESI+) m/z 280.15 [M+H]+, 302.13 [M+Na]+. HRMS (ESI+) m/z calc. for C17H19O2NB [M+H]+: 280.1503; found 280.1502.
3.1.19. 2,2-Diphenyl-1,3,2λ4-oxazaborinan-6-one (βAla-2APB)
β-Alanine (60 mg, 0.67 mmol) was reacted as described in general procedure B (reflux for 1.5 h). The product crystallized from the reaction solution upon cooling. The colorless crystals were washed with n-hexane and dried in high vacuum. This gave the title compound as colorless crystals (88 mg, 52%). 1H NMR (300 MHz, DMSO-d6) δ 7.41 (dd, J = 8.0, 1.3, 4H), 7.25–7.16 (m, 4H), 7.14–7.05 (m, 2H), 6.76 (s, 2H), 2.90–2.80 (m, 2H), 2.54–2.41 (m, 2H). 11B NMR (96 MHz, DMSO-d6) δ 2.05. 13C NMR (75 MHz, DMSO-d6) δ 169.23, 130.78, 127.02, 125.61, 35.48, 30.01. MS (ESI+) m/z 254.13 [M+H]+, 276.12 [M+Na]+, 176.09 [M-C6H5]+. HRMS (ESI+) m/z calc. for C15H17O2NB [M+H]+: 254.1347; found 254.1345.
3.1.20. (S)-3-(5-Oxo-2,2-diphenyl-1,3,2λ4-oxazaborolidin-4-yl)propanamide (Gln-2APB)
L-Glutamine (95 mg, 0.65 mmol) was treated as described in general procedure B (reflux for 3.75 h) and the product precipitated from the reaction mixture upon cooling. The solid was washed with EtOH, Et2O, and n-hexane and dried in high vacuum. The residue was recrystallized from EtOH affording the title compound as colorless crystals (96 mg, 48%). 1H NMR (300 MHz, DMSO-d6) δ 7.43–7.28 (m, 6H), 7.21 (td, J = 7.1, 1.6, 4H), 7.18–7.07 (m, 2H), 7.03–6.90 (m, 1H), 6.86 (s, 1H), 3.56 (p, J = 7.5, 1H), 2.30 (td, J = 7.2, 2.7, 2H), 1.89 (ddt, J = 39.3, 14.7, 7.4, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.39. 13C NMR (75 MHz, DMSO-d6) δ 174.07, 173.86, 131.00, 130.93, 127.08, 127.03, 126.01, 125.91, 54.44, 30.88, 25.31. MS (ESI+) m/z 311.16 [M+H]+, 333.14 [M+Na]+. HRMS (ESI+) m/z calc. for C17H20O3N2B [M+H]+: 311.1567; found 311.1561.
3.1.21. (S)-4-((1H-Imidazol-5-yl)methyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (His-2APB)
L-Histidine (100 mg, 0.65 mmol) was used as starting material and general procedure B followed to synthesize the title compound (reflux for 1 h). The crude product precipitated from the reaction mixture upon cooling. It was washed with Et2O and n-hexane and dried in high vacuum. This gave the title compound as colorless solid (92 mg, 45%). 1H NMR (300 MHz, DMSO-d6) δ 12.04 (s, 1H), 7.62–7.54 (m, 1H), 7.48–7.07 (m, 12H), 6.91 (s, 1H), 3.84 (t, J = 7.2, 1H), 2.95 (d, J = 6.0, 2H). 11B NMR (96 MHz, DMSO-d6) δ 4.40. 13C NMR (75 MHz, DMSO-d6) δ 173.32, 135.19, 131.18, 131.07, 127.04, 127.01, 126.01, 55.18. MS (ESI+) m/z 320.16 [M+H]+, 342.14 [M+Na]+, 242.11 [M-C6H5]+. HRMS (ESI+) m/z calc. for C18H19O2N3B [M+H]+: 320.1565; found 320.1562.
3.1.22. (S)-4-(2-(Methylthio)ethyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Met-2APB)
Following general procedure B, L-methionine (100 mg, 0.67 mmol) was used as the starting material. After refluxing for 45 min the clear solution was cooled to r.t. and then stored at −20 °C to initiate precipitation of the crude product. The obtained crystals were washed with Et2O and n-hexane, and dried in high vacuum. This afforded the desired product as colorless crystals (212 mg, quantitative). 1H NMR (300 MHz, DMSO-d6) δ 7.45–7.09 (m, 12H), 6.78 (t, J = 10.4, 1H), 3.69–3.53 (m, 1H), 2.72–2.52 (m, 2H), 2.14–1.78 (m, 4H). 11B NMR (96 MHz, DMSO-d6) δ 4.92. 13C NMR (75 MHz, DMSO-d6) δ 174.04, 130.99, 130.92, 127.14, 127.08, 126.08, 125.98, 53.60, 29.51, 28.95, 14.15. MS (ESI+) m/z 314.14 [M+H]+, 336.12 [M+Na]+, 236.09 [M-C6H5]+. HRMS (ESI+) m/z calc. for C17H21O2NBS [M+H]+: 314.1381; found 314.1380.
3.1.23. (R)-4-(Mercaptomethyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Cys-2APB)
L-Cysteine (75 mg, 0.62 mmol) was reacted according to general procedure B (reflux for 1 h). The crude product, which precipitated from the reaction mixture upon cooling, was washed with n-hexane. This gave the title compound as colorless solid (125 mg, 71%) after drying in high vacuum. 1H NMR (300 MHz, DMSO-d6) δ 7.58–7.36 (m, 5H), 7.29–7.09 (m, 6H), 6.63 (t, J = 10.8, 1H), 3.79–3.63 (m, 1H), 3.04–2.65 (m, 3H). 11B NMR (96 MHz, DMSO-d6) δ 4.58. 13C NMR (75 MHz, DMSO-d6) δ 172.29, 131.14, 131.08, 127.08, 127.06, 126.12, 126.07, 57.81, 23.83. MS (ESI+) m/z 286.11 [M+H]+, 308.09 [M+Na]+. HRMS (ESI+) m/z calc. for C15H17O2NBS [M+H]+: 286.1068; found 286.1066.
3.1.24. (S)-4-Benzyl-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Phe-2APB)
The title compound was synthesized from L-phenylalanine (100 mg, 0.60 mmol) according to general procedure B (reflux for 30 min). The precipitated crude product was washed with n-hexane and dried in high vacuum. This yielded the final compound as colorless solid (128 mg, 64%). 1H NMR (300 MHz, DMSO-d6) δ 7.47–7.01 (m, 16H), 6.76 (t, J = 10.5, 1H), 3.77–3.61 (m, 1H), 3.17 (dd, J = 14.5, 4.0, 1H), 2.92 (dd, J = 14.5, 10.3, 1H). 11B NMR (96 MHz, DMSO-d6) δ 4.61. 13C NMR (75 MHz, DMSO-d6) δ 173.48, 137.26, 131.15, 130.81, 129.27, 128.47, 127.02, 126.95, 126.65, 125.95, 125.86, 56.68, 35.04. MS (ESI+) m/z 330.17 [M+H]+, 352.15 [M+Na]+. HRMS (ESI+) m/z calc. for C21H21O2NB [M+H]+: 330.1660; found 330.1658.
3.1.25. (S)-4-(4-Hydroxybenzyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Tyr-2APB)
Following general procedure B, L-tyrosine (120 mg, 0.68 mmol) was reacted (reflux for 40 min) and the reaction mixture cooled to r.t. and then −20 °C. The formed precipitate was washed with Et2O and n-hexane. The isolated solid was suspended in EtOH, layered with n-hexane, and the mixture stored at 5 °C. The obtained precipitate was filtered, washed with n-hexane, and dried in high vacuum. This gave the title compound as shimmery beige solid (39 mg, 17%). 1H NMR (300 MHz, DMSO-d6) δ 9.26 (s, 1H), 7.39 (td, J = 7.9, 1.6, 4H), 7.30–6.99 (m, 9H), 6.74–6.55 (m, 3H), 3.64–3.53 (m, 1H), 3.04 (dd, J = 14.5, 4.0, 1H), 2.79 (dd, J = 14.5, 10.3, 1H). 11B NMR (96 MHz, DMSO-d6) δ 4.73. 13C NMR (75 MHz, DMSO-d6) δ 173.56, 156.18, 131.15, 130.81, 130.22, 127.10, 127.00, 126.94, 125.93, 125.85, 115.26, 57.04, 34.18. MS (ESI+) m/z 346.16 [M+H]+, 368.14 [M+Na]+. HRMS (ESI+) m/z calc. for C21H21O3NB [M+H]+: 346.1609; found 346.1605.
3.1.26. (S)-4-((5-Hydroxy-1H-indol-3-yl)methyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (5-HO-Trp-2APB)
5-Hydroxytryptophan (191 mg, 0.87 mmol) was used as the starting material to synthesize the title compound according to general procedure B (reflux until all the solids were dissolved). Precipitation of the crude product was initiated by first cooling to r.t. and then to −20 °C. The precipitate was washed with Et2O and n-hexane and the obtained solid recrystallized from EtOH. This gave the desired compound as a colorless solid (220 mg, 66%) after drying in high vacuum. 1H NMR (300 MHz, DMSO-d6) δ 10.67–10.58 (m, 1H), 8.60 (s, 1H), 7.42–7.34 (m, 4H), 7.29 (dd, J = 12.0, 6.2, 1H), 7.23–7.04 (m, 7H), 7.03 (d, J = 2.3, 1H), 6.75 (d, J = 2.2, 1H), 6.59 (dd, J = 8.6, 2.2, 1H), 6.52–6.35 (m, 1H), 3.73–3.58 (m, 1H), 3.14 (dd, J = 15.1, 3.5, 1H), 2.93 (dd, J = 15.2, 10.1, 1H). 11B NMR (96 MHz, DMSO-d6) δ 8.58. 13C NMR (75 MHz, DMSO-d6) δ 174.01, 150.52, 131.19, 130.93, 127.47, 127.12, 127.05, 126.05, 125.99, 124.97, 111.99, 111.58, 107.80, 101.86, 55.71, 25.58. MS (ESI+) m/z 385.1726 [M+H]+, 407.1543 [M+Na]+, 307.1254 [M-C6H5]+. HRMS (ESI+) m/z calc. for C23H22O3N2B [M+H]+: 385.1718; found 385.1726.
3.1.27. (S)-4-(4-Aminobutyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Lys-2APB)
Following general procedure B, L-lysine (90 mg, 0.62 mmol) was reacted (reflux for 50 min) and the solvents evaporated under reduced pressure. The residue was layered with a mixture of Et2O and n-hexane, and the precipitation allowed to settle. The supernatant was removed and the solid washed with Et2O and n-hexane. After drying the solid in high vacuum, it was recrystallized from a mixture of i-PrOH and EtOH, affording the title compound as colorless crystals (96 mg, 50%). 1H NMR (300 MHz, DMSO-d6) δ 7.43–7.36 (m, 5H), 7.25–7.17 (m, 5H), 7.16–7.09 (m, 2H), 3.41 (dd, J = 9.4, 4.6, 1H), 2.48–2.44 (m, 2H), 1.80–1.21 (m, 8H). 11B NMR (96 MHz, DMSO-d6) δ 4.42. 13C NMR (75 MHz, DMSO-d6) δ 174.27, 131.00, 130.92, 127.05, 126.99, 125.95, 125.86, 55.17, 41.21, 32.74, 29.37, 22.97. MS (ESI+) m/z 311.19 [M+H]+, 333.17 [M+Na]+, 233.15 [M-C6H5]+. HRMS (ESI+) m/z calc. for C18H24O2N2B [M+H]+: 311.1925; found 311.1925.
3.2. Cell Culture
Cell culture reagents were purchased from Thermo Fisher Scientific (Waltham, MA, USA). MDA-MB-231 cells (ATCC) were cultured at 37 °C in RPMI cell culture medium supplemented with 10% FBS, 10 mM HEPES, and 1% penicillin/streptomycin. HEK 293 cells stably expressing STIM1 were grown at 37 °C and 5% CO2 in MEM medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FCS and 500 µg/mL G418 (Gibco). Cells were transfected according to manufacturer’s guidelines in a Nucleofector 4D device with Nucleofector Kit SF (Lonza, Basel, Switzerland) with 1 µg DNA per 106 cells.
3.3. Fluorescent Ca2+ Influx Assay Using FLIPR® Tetra
3.3.1. General
The calcium indicator, Calcium-5 was purchased from Molecular Devices LLC and all other chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA). For loading the cells and subsequent administrations during the FLIPR experiments a nominal calcium free (NCF) modified Krebs buffer was used, containing 117 mM NaCl, 4.8 mM KCl, 1 mM MgCl2, 5 mM D-glucose, and 10 mM HEPES. The pH of the NCF buffer was adjusted to 7.4 using HCl and NaOH. Thapsigargin (Tg) solutions were always prepared freshly a few minutes before starting the FLIPR experiment. Cells were excited using a 470–495 nm LED module, and the emitted fluorescence signal was filtered with a 515–575 nm emission filter (manufacturer’s guidelines). Fluorescence signals were analyzed using the ScreenWorks 3.1.1.8 software (Molecular Devices).
3.3.2. SOCE Modulation Assay
The attached MDA-MB-231 cells from T75 flasks were trypsinized and plated onto Corning® 96-well black polystyrene clear bottom microplates (CLS3603 Sigma–Aldrich, St. Louis, MO, USA) at a density 60,000 cells/well in a final volume of 100 µL per well. Phenol-red free RPMI medium supplemented with 10% FBS and 10 mM HEPES was used for seeding the cells. After culturing the cells for 24 h, the medium of the confluent cells was removed and the cells were loaded with 50 μL NCF Krebs buffer containing calcium-5 dye (50 μL per mL). The cells then were incubated with the dye in the dark for 1 h at 37 °C. Right after the dye-incubation period, the FLIPR experiment was started. After establishment of stable baselines for 50 s, first ER Ca2+ stores were depleted by adding and incubating 50 μL of 2 μM thapsigargin (Tg) in NCF Krebs buffer (1 μM final concentration) for 10 min. Next, 100 μL of 2× of the intended final concentration of the drug of interest (synthetic 2-APB analogue or other SOCE inhibitor) in NCF Krebs buffer was applied and incubated for 10 min. Finally, 50 µL of NCF Krebs buffer containing 10 mM CaCl2 (2 mM final concentration) was administered while maintaining the 1× concentration of the drug of interest. The subsequent response was monitored for 5 min after the addition of CaCl2. The modulation of the SOCE signal was quantified by calculating the area under the curve (AUC) of the relative change of the fluorescence intensity over time (trace) from the last 5 min of the experiment. The values were then normalized to the DMSO positive control (DMSO response = 100%).
3.3.3. ER Ca2+ Mobilization Assay (Compound Followed by Thapsigargin)
The medium of the confluent cells was replaced by 50 μL buffer containing 2 mM CaCl2 as well as calcium-5 dye (50 μL per mL). The cells were incubated in the dark for 1 h at 37 °C. After the dye was incubated, the cells were washed twice: first with 250 μL of 500 μM EGTA in NCF Krebs buffer and afterwards with 250 μL NCF Krebs buffer. Finally, the cells were loaded with 50 μL NCF Krebs buffer and subjected to the FLIPR experiment. Total of 50 μL of 2× of the intended concentration of drug of interest in NCF Krebs buffer was applied and incubated for 10 min while recording the Ca2+ response. Afterwards 25 μL of 12.5 μM Tg (2.5 µM final concentration) was applied and the subsequent response was monitored for 10 min. The response after each addition step was quantified by calculating the slope of the curve and normalizing it to the slope of the response after Tg administration.
3.4. Electrophysiology
Currents from whole cell patch clamp recordings were acquired and filtered at 2.9 kHz with a HEKA EPC-10 amplifier (HEKA Elektronik, Lambrecht (Pfalz), Germany). Currents were corrected for a liquid junction potential of 10 mV, recorded and analyzed with HEKA Patchmaster (v2×53) software and Igor Pro 6.37 (Wavemetrics, Lake Oswego, OR, USA). 50 ms voltage ramps spanning, 150 mV to 150 mV were delivered every 2 s from a holding potential of 0. Before each voltage ramp, capacitive currents were corrected. Currents at −130 mV and 130 mV were extracted, normalized to cell capacitance, and plotted versus time. Bath solution contained (in mM): 120 NaCl, 20 CaCl2, 10 TEA-Cl, 10 HEPES, 2 MgCl2. Pipette solution contained (in mM): 120 Cs-glutamate, 20 BAPTA, 10 HEPES, 3 MgCl2, and 0.050 IP3. Osmolarity was adjusted with glucose to 310 mOsm and pH was adjusted to 7.2 with CsOH.
3.5. CRISPR/Cas9-Mediated Generation and Validation of STIM1 and Orai1 Knockout MDA-MB-231 Cells
Earlier described methods were used to generate STIM1 knockout [86] and Orai1 knockout MDA-MB-231 cells [87]. Single-cell derived clones D11 and G10 were functionally confirmed to have undergone knockout of STIM1 and Orai1, respectively by using SOCE functional assay on FLIPR Tetra® (Molecular Devices). The knockouts were further confirmed by Western blotting. STIM1 was detected using the polyclonal hSTIM1 antibody raised in guinea pig [88] in a 1:1000 dilution. Peroxidase-conjugated affinipure goat anti-guinea pig secondary antibody was used in a 1:10,000 dilution (106-035-003, Jackson Immuno Research, West Grove, PA, USA). Orai1 was detected using rabbit polyclonal hOrai1 antibody (O8264, Sigma-Aldrich) in a 1:1000 dilution. Horseradish peroxidase (HRP)-conjugated secondary goat anti-rabbit IgG (W4011, Promega, Madison, WI, USA) was used in a 1:20,000 dilution. Tubulin (loading control) was detected using mouse monoclonal Tubulin antibody (T9028, Sigma-Aldrich) in a 1:2000 dilution. HRP-conjugated goat anti-mouse secondary antibody was used in a 1:3000 dilution (172-1011, Bio-Rad, Hercules, CA, USA).
3.6. Cell Viability Assay
The XTT assay was used to evaluate the effect of 2-APB analogues on cell proliferation. Cells were plated at a density of 5 × 103 cells/well in 96-well plates and were incubated at 37 °C and 5% CO2 for 24 h. On the following day, the medium was carefully aspirated and replaced with 100 μL medium supplemented with DMSO control or different 2-APB compounds and incubated over a range of different time points from 24 to 120 h. At each time point, 25 μL of sodium 3′-[1-[(phenylamino)-carbony]-3,4-tetrazolium]-bis(4-methoxy-6-nitro)benzene-sulfonic acid hydrate (XTT 1mg/mL prepared in Hank’s Balanced Salt Solution) containing 1.25% phenazine methosulphonate (PMS) was added to each well and incubated at 37 °C for 2 h. Subsequently, the absorbance was read at 650 nm and subtracted from the absorbance of 450 nm by spectrophotometry (Vmax Kinetic Microplate Reader, Molecular Devices LLC). The resulting subtracted absorbance for each compound was expressed as a percentage of viable cells relative to the DMSO control. XTT sodium salt and PMS were purchased from Sigma-Aldrich. The MCF-10A cells were kindly provided by Prof. Dr. Olivier Pertz, Institute of Cell Biology, University of Bern and were cultured in growth medium composed by DMEM/F12 supplemented with 5% horse serum, 20 ng/mL recombinant human EGF (Sigma-Aldrich), 10 μg/mL insulin (Sigma-Aldrich), and 0.5 mg/mL hydrocortisone (Sigma-Aldrich).
3.7. Data Analysis
Statistical significance between the two groups was estimated using student’s T-test where both groups were normally distributed and Mann Whitney test in case of data that were not distributed normally. GraphPad Prism 8.4.3 was used to perform the statistical analysis as well as to generate the bar graphs with scattered individual data points. Data are usually shown as mean ± SD.
4. Conclusions
In an effort to develop new potent inhibitors of store-operated calcium entry we have synthesized several mono-substituted and amino acid analogues of 2-APB. We tested these 2-APB derivatives using a FLIPR assay format in MDA-MB-231 cells. This breast cancer cell line already has been established by others [26,72] as a viable tool for screening new SOCE inhibitors. The compound series of mono-halogenated 2-APB derivatives, in particular, showed promising inhibitory activity compared to 2-APB. Some members of this series, e.g., m-Cl-2APB, m-Br-2APB, m-I-2APB, and p-I-2APB, exhibited efficient SOCE block at a five times lower concentration than 2-APB. It is worth noting that at this low concentration 2-APB generally enhances SOCE. Throughout the course of our investigation we have also demonstrated that some of our novel analogues (m-I-2APB, p-Cl-2APB, p-Br-2APB, p-I-2APB), but also known 2-APB-type SOCE inhibitors (DPB162-AE), are able to mobilize calcium from intracellular stores of MDA-MB-231 cells at high concentration. In this regard, m-Cl-2APB is a promising tool compound from our synthetic series as it efficiently blocked SOCE at a low concentration (10 µM) but did not completely deplete internal stores at higher concentrations (50 µM). While structurally very close isomers like m-Br-2APB and p-Br-2APB did not show different behaviour in blocking ICRAC, they had a strikingly different effect on MDA-MB-231 cells viability.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/21/16/5604/s1. Supplementary data (adapted synthesis of DPB162-AE (Scheme S1), GSK-7975A (Scheme S2), and Synta66 (Scheme S3), crystal structures of GSK-7975A (Figure S1), cBu-2APB (Figure S2), p-SO2NMe2-2APB (Figure S3), o-F-2APB (Figure S4), and o-Cl-2APB (Figure S5), SOCE blockade by GSK-7975A and Synta66 with pre-incubation (Figure S6), SOCE in STIM1 KO and Orai1 KO cells (Figure S7) and copies of 1H, 11B, 13C, and 19F NMR spectra of all final compounds).
Author Contributions
Conceptualization, M.L.; methodology, A.S. and R.B. (Rajesh Bhardwaj); validation, A.S., R.B. (Rajesh Bhardwaj) and R.B. (Roland Baur); formal analysis, A.S., R.B. (Rajesh Bhardwaj), and R.B. (Roland Baur); investigation, A.S., R.B. (Rajesh Bhardwaj), N.W., D.T., P.K., J.D., and R.B. (Roland Baur); data curation, A.S. and R.B. (Rajesh Bhardwaj); writing—original draft preparation, A.S., R.B. (Rajesh Bhardwaj), C.P., and M.L.; writing—review and editing, A.S., R.B. (Rajesh Bhardwaj), C.P., and M.L.; visualization, A.S. and R.B. (Rajesh Bhardwaj); supervision, C.P., M.A.H., and M.L.; project administration, M.A.H. and M.L.; funding acquisition, M.A.H. and M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Swiss National Science Foundation (Sinergia grants CRSII3_160782 and CRSII5_180326 to M.A.H. and M.L.).
Acknowledgments
We thank the analytical services from the Department of Chemistry and Biochemistry, University of Bern, Switzerland, for measuring the spectra of synthetic intermediates and final compounds: the group of Chemical Crystallography (Piero Macchi) for the X-ray structure solutions and the Swiss National Science Foundation (R’equip grant 206021_128724) for co-funding the single crystal X-ray diffractometer, the group of Julien Furrer for measuring NMR spectra, and the group of Stefan Schürch for MS and HRMS spectra.
Conflicts of Interest
The authors declare no conflict of interest. The funder had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| 2-APB | 2-Aminoethyl diphenylborinate |
| AUC | Area under the curve |
| CD | Current density |
| CRAC | Ca2+ release-activated Ca2+ current |
| ER | Endoplasmic reticulum |
| FLIPR | Fluorometric imaging plate reader |
| IP3 | Inositol 1,4,5-triphosphate |
| NCF | Nominal calcium-free (buffer) |
| PI(4,5)P2 | Phosphatidylinositol 4,5-bisphosphate |
| PM | Plasma membrane |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| SOCE | Store-operated calcium entry |
| STIM | Stromal interaction molecule |
| Tg | Thapsigargin |
Appendix A
When we used the general lithiation/borination protocol with 1,2-dibromobenzene (Scheme A1) we observed significant attack of the aryl lithium species at the halogenated carbon of the borinic intermediate. This led to the shown biphenyl side product which we were unable to separate from the desired borinic acid by chromatography or crystallization. Changing the solvent to toluene/THF 4:1 to carry out the halogen lithium exchange and subsequent reaction with phenylboronic acid pinacol ester suppressed the formation of the biphenyl side product and enabled us to isolate pure (2-bromophenyl)(phenyl) borinic acid. The solvent change strategy did not work with 1,2-diiodobenzene, however, and gave inseparable mixtures of desired (2-iodophenyl)(phenyl)borinic acid and biphenyl side product.

Scheme A1.
Lithiation of 1,2-dibromo- or 1,2-diiodobenzene, and subsequent borination of the organolithium species led to significant formation of biphenyl side products that were inseparable from the desired (aryl)(phenyl)borinic acids.
Scheme A1.
Lithiation of 1,2-dibromo- or 1,2-diiodobenzene, and subsequent borination of the organolithium species led to significant formation of biphenyl side products that were inseparable from the desired (aryl)(phenyl)borinic acids.

References
- Prakriya, M.; Lewis, R.S. Store-operated calcium channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef]
- Zheng, S.; Zhou, L.; Ma, G.; Zhang, T.; Liu, J.; Li, J.; Nguyen, N.T.; Zhang, X.; Li, W.; Nwokonko, R.; et al. Calcium store refilling and STIM activation in STIM- and Orai-deficient cell lines. Pflugers Arch. Eur. J. Physiol. 2018, 470, 1555–1567. [Google Scholar] [CrossRef]
- Thastrup, O.; Cullen, P.J.; Drobak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef]
- Feske, S. CRAC channels and disease—From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116. [Google Scholar] [CrossRef]
- Kappel, S.; Borgstrom, A.; Stoklosa, P.; Dorr, K.; Peinelt, C. Store-operated calcium entry in disease: Beyond STIM/Orai expression levels. Semin. Cell Dev. Biol. 2019, 94, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Han, W.; Cao, W.M.; Wang, Y.; Wen, S.; Huang, Y.; Li, M.; Du, L.; Zhou, Y. Store-operated calcium entry mediated by ORAI and STIM. Compr. Physiol. 2018, 8, 981–1002. [Google Scholar] [CrossRef] [PubMed]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordonez, A.; Rosado, J.A.; Smani, T. The complex role of store operated calcium entry pathways and related proteins in the function of cardiac, skeletal and vascular smooth muscle cells. Front. Physiol. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Yoo, S.-A.; Kim, M.; Kim, W.-U. The role of calcium-calcineurin-NFAT signaling pathway in health and autoimmune diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Secondo, A.; Bagetta, G.; Amantea, D. On the role of store-operated calcium entry in acute and chronic neurodegenerative diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef]
- Chen, Y.F.; Lin, P.C.; Yeh, Y.M.; Chen, L.H.; Shen, M.R. Store-operated Ca2+ entry in tumor progression: From molecular mechanisms to clinical implications. Cancers 2019, 11, 899. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef]
- Azimi, I.; Milevskiy, M.J.G.; Chalmers, S.B.; Yapa, K.; Robitaille, M.; Henry, C.; Baillie, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Orai1 and Orai3 in breast cancer molecular subtypes and the identification of Orai3 as a hypoxia sensitive gene and a regulator of hypoxia responses. Cancers 2019, 11, 208. [Google Scholar] [CrossRef]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Store-operated Ca2+ entry in breast cancer cells: Remodeling and functional role. Int. J. Mol. Sci. 2018, 19, 4053. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting STIM and Orai proteins as an alternative approach in anticancer therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A review in the theme: Cell and molecular processes in cancer metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C457–C469. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, Z.K.; Minatti, A.; Button, D.C.; Patrick, S. Small-molecule inhibitors of store-operated calcium entry. ChemMedChem 2009, 4, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Jairaman, A.; Prakriya, M. Molecular pharmacology of store-operated CRAC channels. Channels 2013, 7, 402–414. [Google Scholar] [CrossRef]
- Tian, C.; Du, L.; Zhou, Y.; Li, M. Store-operated CRAC channel inhibitors: Opportunities and challenges. Future Med. Chem. 2016, 8, 817–832. [Google Scholar] [CrossRef]
- Azimi, I.; Flanagan, J.U.; Stevenson, R.J.; Inserra, M.; Vetter, I.; Monteith, G.R.; Denny, W.A. Evaluation of known and novel inhibitors of Orai1-mediated store operated Ca2+ entry in MDA-MB-231 breast cancer cells using a Fluorescence Imaging Plate Reader assay. Bioorg. Med. Chem. 2017, 25, 440–449. [Google Scholar] [CrossRef]
- Riva, B.; Griglio, A.; Serafini, M.; Cordero-Sanchez, C.; Aprile, S.; Di Paola, R.; Gugliandolo, E.; Alansary, D.; Biocotino, I.; Lim, D.; et al. Pyrtriazoles, a novel class of store-operated calcium entry modulators: Discovery, biological profiling, and in vivo proof-of-concept efficacy in acute pancreatitis. J. Med. Chem. 2018, 61, 9756–9783. [Google Scholar] [CrossRef]
- Djillani, A.; Doignon, I.; Luyten, T.; Lamkhioued, B.; Gangloff, S.C.; Parys, J.B.; Nusse, O.; Chomienne, C.; Dellis, O. Potentiation of the store-operated calcium entry (SOCE) induces phytohemagglutinin-activated Jurkat T cell apoptosis. Cell Calcium 2015, 58, 171–185. [Google Scholar] [CrossRef]
- Doignon, I.; Fayol, O.; Dellis, O. Improvement of the rituximab-induced cell death by potentiation of the store-operated calcium entry in mantle cell lymphoma cell lines. Oncotarget 2019, 10, 4466–4478. [Google Scholar] [CrossRef][Green Version]
- Azimi, I.; Stevenson, R.J.; Zhang, X.; Meizoso-Huesca, A.; Xin, P.; Johnson, M.; Flanagan, J.U.; Chalmers, S.B.; Yoast, R.E.; Kapure, J.S.; et al. A new selective pharmacological enhancer of the Orai1 Ca2+ channel reveals roles for Orai1 in smooth and skeletal muscle functions. ACS Pharmacol. Transl. Sci. 2020, 3, 135–147. [Google Scholar] [CrossRef]
- Ma, K.T.; Guan, B.C.; Yang, Y.Q.; Nuttall, A.L.; Jiang, Z.G. 2-Aminoethoxydiphenyl borate blocks electrical coupling and inhibits voltage-gated K+ channels in guinea pig arteriole cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H335–H346. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, L.; Prevarskaya, N.; Mazurier, J.; Shuba, Y.; Skryma, R. 2-APB inhibits volume-regulated anion channels independently from intracellular calcium signaling modulation. FEBS Lett. 2004, 556, 121–126. [Google Scholar] [CrossRef]
- Bai, D.; del Corsso, C.; Srinivas, M.; Spray, D.C. Block of specific gap junction channel subtypes by 2-aminoethoxydiphenyl borate (2-APB). J. Pharmacol. Exp. Ther. 2006, 319, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Bilmen, J.G.; Wootton, L.L.; Godfrey, R.E.; Smart, O.S.; Michelangeli, F. Inhibition of SERCA Ca2+ pumps by 2-aminoethoxydiphenyl borate (2-APB). 2-APB reduces both Ca2+ binding and phosphoryl transfer from ATP, by interfering with the pathway leading to the Ca2+ binding sites. Eur. J. Biochem. 2002, 269, 3678–3687. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Starkov, A.A.; Fiskum, G. Cyclosporin a-insensitive permeability transition in brain mitochondria: Inhibition by 2-aminoethoxydiphenyl borate. J. Biol. Chem. 2003, 278, 27382–27389. [Google Scholar] [CrossRef]
- Ma, H.T.; Venkatachalam, K.; Li, H.S.; Montell, C.; Kurosaki, T.; Patterson, R.L.; Gill, D.L. Assessment of the role of the inositol 1,4,5-trisphosphate receptor in the activation of transient receptor potential channels and store-operated Ca2+ entry channels. J. Biol. Chem. 2001, 276, 18888–18896. [Google Scholar] [CrossRef]
- Schindl, R.; Kahr, H.; Graz, I.; Groschner, K.; Romanin, C. Store depletion-activated CaT1 currents in rat basophilic leukemia mast cells are inhibited by 2-aminoethoxydiphenyl borate. Evidence for a regulatory component that controls activation of both CaT1 and CRAC (Ca2+ release-activated Ca2+ channel) channels. J. Biol. Chem. 2002, 277, 26950–26958. [Google Scholar] [CrossRef]
- Xu, S.Z.; Zeng, F.; Boulay, G.; Grimm, C.; Harteneck, C.; Beech, D.J. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: A differential, extracellular and voltage-dependent effect. Br. J. Pharmacol. 2005, 145, 405–414. [Google Scholar] [CrossRef]
- Li, M.; Jiang, J.; Yue, L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef]
- Togashi, K.; Inada, H.; Tominaga, M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br. J. Pharmacol. 2008, 153, 1324–1330. [Google Scholar] [CrossRef]
- Callera, G.E.; He, Y.; Yogi, A.; Montezano, A.C.; Paravicini, T.; Yao, G.; Touyz, R.M. Regulation of the novel Mg2+ transporter transient receptor potential melastatin 7 (TRPM7) cation channel by bradykinin in vascular smooth muscle cells. J. Hypertens. 2009, 27, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Hermosura, M.C.; Monteilh-Zoller, M.K.; Scharenberg, A.M.; Penner, R.; Fleig, A. Dissociation of the store-operated calcium current ICRAC and the Mg-nucleotide-regulated metal ion current MagNuM. J. Physiol. 2002, 539, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Z.; Gu, Q.; Wang, C.; Colton, C.K.; Tang, J.; Kinoshita-Kawada, M.; Lee, L.Y.; Wood, J.D.; Zhu, M.X. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 2004, 279, 35741–35748. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Montalbetti, N.; Simonin, A.; Danko, T.; Balazs, B.; Zsembery, A.; Hediger, M.A. Inhibition of the human epithelial calcium channel TRPV6 by 2-aminoethoxydiphenyl borate (2-APB). Cell Calcium 2012, 52, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kanaji, T.; Nakade, S.; Kanno, T.; Mikoshiba, K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 1997, 122, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.T.; Patterson, R.L.; van Rossum, D.B.; Birnbaumer, L.; Mikoshiba, K.; Gill, D.L. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science 2000, 287, 1647–1651. [Google Scholar] [CrossRef]
- Braun, F.J.; Broad, L.M.; Armstrong, D.L.; Putney, J.W., Jr. Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. J. Biol. Chem. 2001, 276, 1063–1070. [Google Scholar] [CrossRef]
- Broad, L.M.; Braun, F.J.; Lievremont, J.P.; Bird, G.S.; Kurosaki, T.; Putney, J.W., Jr. Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. J. Biol. Chem. 2001, 276, 15945–15952. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. 2001, 536, 3–19. [Google Scholar] [CrossRef]
- Bakowski, D.; Glitsch, M.D.; Parekh, A.B. An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current ICRAC in RBL-1 cells. J. Physiol. 2001, 532, 55–71. [Google Scholar] [CrossRef]
- Iwasaki, H.; Mori, Y.; Hara, Y.; Uchida, K.; Zhou, H.; Mikoshiba, K. 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Recept. Channels 2001, 7, 429–439. [Google Scholar] [PubMed]
- Djillani, A.; Nusse, O.; Dellis, O. Characterization of novel store-operated calcium entry effectors. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2341–2347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Bird, G.S.; Putney, J.W., Jr. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 2008, 283, 19265–19273. [Google Scholar] [CrossRef] [PubMed]
- Goto, J.-I.; Suzuki, A.Z.; Ozaki, S.; Matsumoto, N.; Nakamura, T.; Ebisui, E.; Fleig, A.; Penner, R.; Mikoshiba, K. Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca2+ entry via STIM proteins. Cell Calcium 2010, 47, 1–10. [Google Scholar] [CrossRef]
- Peinelt, C.; Lis, A.; Beck, A.; Fleig, A.; Penner, R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J. Physiol. 2008, 586, 3061–3073. [Google Scholar] [CrossRef]
- Ma, H.-T.; Venkatachalam, K.; Parys, J.B.; Gill, D.L. Modification of store-operated channel coupling and inositol trisphosphate receptor function by 2-aminoethoxydiphenyl borate in DT40 lymphocytes. J. Biol. Chem. 2002, 277, 6915–6922. [Google Scholar] [CrossRef]
- Xu, X.; Ali, S.; Li, Y.; Yu, H.; Zhang, M.; Lu, J.; Xu, T. 2-aminoethoxydiphenyl borate potentiates CRAC current by directly dilating the pore of open Orai1. Sci. Rep. 2016, 6, 29304. [Google Scholar] [CrossRef]
- Hendron, E.; Wang, X.; Zhou, Y.; Cai, X.; Goto, J.I.; Mikoshiba, K.; Baba, Y.; Kurosaki, T.; Wang, Y.; Gill, D.L. Potent functional uncoupling between STIM1 and Orai1 by dimeric 2-aminodiphenyl borinate analogs. Cell Calcium 2014, 56, 482–492. [Google Scholar] [CrossRef]
- Wei, M.; Zhou, Y.; Sun, A.; Ma, G.; He, L.; Zhou, L.; Zhang, S.; Liu, J.; Zhang, S.L.; Gill, D.L.; et al. Molecular mechanisms underlying inhibition of STIM1-Orai1-mediated Ca2+ entry induced by 2-aminoethoxydiphenyl borate. Pflugers Arch. Eur. J. Physiol. 2016, 468, 2061–2074. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, X.; Zhou, Y.; Hendron, E.; Mancarella, S.; Ritchie, M.F.; Tang, X.D.; Baba, Y.; Kurosaki, T.; Mori, Y.; et al. STIM protein coupling in the activation of Orai channels. Proc. Natl. Acad. Sci. USA 2009, 106, 7391–7396. [Google Scholar] [CrossRef]
- Parvez, S.; Beck, A.; Peinelt, C.; Soboloff, J.; Lis, A.; Monteilh-Zoller, M.; Gill, D.L.; Fleig, A.; Penner, R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008, 22, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Zhang, X.; Pathak, T.; Nwokonko, R.; Gueguinou, M.F.; Subedi, K.P.; Zhou, Y.; Ambudkar, I.S.; et al. Cross-talk between N-terminal and C-terminal domains in stromal interaction molecule 2 (STIM2) determines enhanced STIM2 sensitivity. J. Biol. Chem. 2019, 294, 6318–6332. [Google Scholar] [CrossRef] [PubMed]
- Schindl, R.; Bergsmann, J.; Frischauf, I.; Derler, I.; Fahrner, M.; Muik, M.; Fritsch, R.; Groschner, K.; Romanin, C. 2-aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J. Biol. Chem. 2008, 283, 20261–20267. [Google Scholar] [CrossRef] [PubMed]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. 2007, 17, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Kappel, S.; Kilch, T.; Baur, R.; Lochner, M.; Peinelt, C. The number and position of Orai3 units within heteromeric store-operated Ca2+ channels alter the pharmacology of ICRAC. Int. J. Mol. Sci. 2020, 21, 2458. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Iwasaki, H.; Nakamura, T.; Nakamura, K.; Maruyama, T.; Hamano, S.; Ozaki, S.; Mizutani, A.; Mikoshiba, K. 2-Aminoethyl diphenylborinate analogues: Selective inhibition for store-operated Ca2+ entry. Biochem. Biophys. Res. Commun. 2007, 352, 277–282. [Google Scholar] [CrossRef]
- Hofer, A.; Kovacs, G.; Zappatini, A.; Leuenberger, M.; Hediger, M.A.; Lochner, M. Design, synthesis and pharmacological characterization of analogs of 2-aminoethyl diphenylborinate (2-APB), a known store-operated calcium channel blocker, for inhibition of TRPV6-mediated calcium transport. Bioorg. Med. Chem. 2013, 21, 3202–3213. [Google Scholar] [CrossRef]
- Dellis, O.; Mercier, P.; Chomienne, C. The boron-oxygen core of borinate esters is responsible for the store-operated calcium entry potentiation ability. BMC Pharmacol. 2011, 11, 1. [Google Scholar] [CrossRef]
- Bittremieux, M.; Gerasimenko, J.V.; Schuermans, M.; Luyten, T.; Stapleton, E.; Alzayady, K.J.; De Smedt, H.; Yule, D.I.; Mikoshiba, K.; Vangheluwe, P.; et al. DPB162-AE, an inhibitor of store-operated Ca2+ entry, can deplete the endoplasmic reticulum Ca2+ store. Cell Calcium 2017, 62, 60–70. [Google Scholar] [CrossRef]
- Bittremieux, M.; La Rovere, R.M.; Schuermans, M.; Luyten, T.; Mikoshiba, K.; Vangheluwe, P.; Parys, J.B.; Bultynck, G. Extracellular and ER-stored Ca2+ contribute to BIRD-2-induced cell death in diffuse large B-cell lymphoma cells. Cell Death Discov. 2018, 4, 101. [Google Scholar] [CrossRef]
- Kukkonen, J.P.; Lund, P.E.; Akerman, K.E. 2-aminoethoxydiphenyl borate reveals heterogeneity in receptor-activated Ca2+ discharge and store-operated Ca2+ influx. Cell Calcium 2001, 30, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.J.; Azimi, I.; Flanagan, J.U.; Inserra, M.; Vetter, I.; Monteith, G.R.; Denny, W.A. An SAR study of hydroxy-trifluoromethylpyrazolines as inhibitors of Orai1-mediated store operated Ca2+ entry in MDA-MB-231 breast cancer cells using a convenient Fluorescence Imaging Plate Reader assay. Bioorg. Med. Chem. 2018, 26, 3406–3413. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor a-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, X.; Shen, Q.; Yang, X.; Yu, C.; Cai, C.; Cai, G.; Meng, X.; Zou, F. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem. Biophys. Res. Commun. 2015, 458, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Seven, Ö.; Bolte, M.; Lerner, H.-W.; Wagner, M. High-yield syntheses and reactivity studies of 1,2-diborylated and 1,2,4,5-tetraborylated benzenes. Organometallics 2014, 33, 1291–1299. [Google Scholar] [CrossRef]
- Suzuki, A.Z.; Ozaki, S.; Goto, J.-I.; Mikoshiba, K. Synthesis of bisboron compounds and their strong inhibitory activity on store-operated calcium entry. Bioorg. Med. Chem. Lett. 2010, 20, 1395–1398. [Google Scholar] [CrossRef]
- Allen, D.G.; Coe, D.M.; Cooper, A.W.J.; Gore, P.M.; House, D.; Senger, S.; Sollis, S.L.; Vile, S.; Wilson, C. N-Pyrazolyl Carboxamides as CRAC Channel Inhibitors. U.S. Patent 8399436B2, 19 March 2013. [Google Scholar]
- Chen, S.; Jiang, J.; Li, H.; James, D.; Chimmanamada, D.; Borella, C.; Sun, L.; Xie, Y.; Holmqvist, M.; Mahiou, J.; et al. Compounds for Inflammation and Immune-Related Uses. U.S. Patent 7709518B2, 4 May 2010. [Google Scholar]
- Flückiger, R.; Henson, E.; Hess, G.M.; Gallop, P.M. Mass spectral and HPLC analysis of biological compounds with diphenylborinic acid. Biomed. Mass Spectrom. 1984, 11, 611–615. [Google Scholar] [CrossRef]
- Bohnert, G.; Chen, S.; Xie, Y. Vinyl-Phenyl Derivatives for Inflammation and Immune-Related Uses. U.S. Patent 7816535B2, 19 October 2010. [Google Scholar]
- Ng, S.W.; di Capite, J.; Singaravelu, K.; Parekh, A.B. Sustained activation of the tyrosine kinase Syk by antigen in mast cells requires local Ca2+ influx through Ca2+ release-activated Ca2+ channels. J. Biol. Chem. 2008, 283, 31348–31355. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Rovedatti, L.; Kaur, R.; Spencer, J.P.; Brown, J.T.; Morisset, V.D.; Biancheri, P.; Leakey, N.A.B.; Wilde, J.I.; Scott, L.; et al. Targeting gut T cell Ca2+ release-activated Ca2+ channels inhibits T cell cytokine production and T-Box transcription factor T-Bet in inflammatory bowel disease. J. Immunol. 2009, 183, 3454–3462. [Google Scholar] [CrossRef]
- Derler, I.; Schindl, R.; Fritsch, R.; Heftberger, P.; Riedl, M.C.; Begg, M.; House, D.; Romanin, C. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 2013, 53, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Panicker, S.; Lau, K.-Y.; Apparsundaram, S.; Patel, V.A.; Chen, S.-L.; Soto, R.; Jung, J.K.C.; Ravindran, P.; Okuhara, D.; et al. Characterization of a novel CRAC inhibitor that potently blocks human T cell activation and effector functions. Mol. Immunol. 2013, 54, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Augustynek, B.S.; Ercan-Herbst, E.; Kandasamy, P.; Seedorf, M.; Peinelt, C.; Hediger, M.A. Ca2+/Calmodulin binding to STIM1 hydrophobic residues facilitates slow Ca2+-dependent inactivation of the Orai1 channel. Cell. Physiol. Biochem. 2020, 54, 252–270. [Google Scholar] [CrossRef] [PubMed]
- Butorac, C.; Muik, M.; Derler, I.; Stadlbauer, M.; Lunz, V.; Krizova, A.; Lindinger, S.; Schober, R.; Frischauf, I.; Bhardwaj, R.; et al. A novel STIM1-Orai1 gating interface essential for CRAC channel activation. Cell Calcium 2019, 79, 57–67. [Google Scholar] [CrossRef]
- Ercan, E.; Chung, S.-H.; Bhardwaj, R.; Seedorf, M. Di-arginine signals and the K-rich domain retain the Ca2+ sensor STIM1 in the endoplasmic reticulum. Traffic 2012, 13, 992–1003. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).