Optimized Protocol for Isolation of Small Extracellular Vesicles from Human and Murine Lymphoid Tissues

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
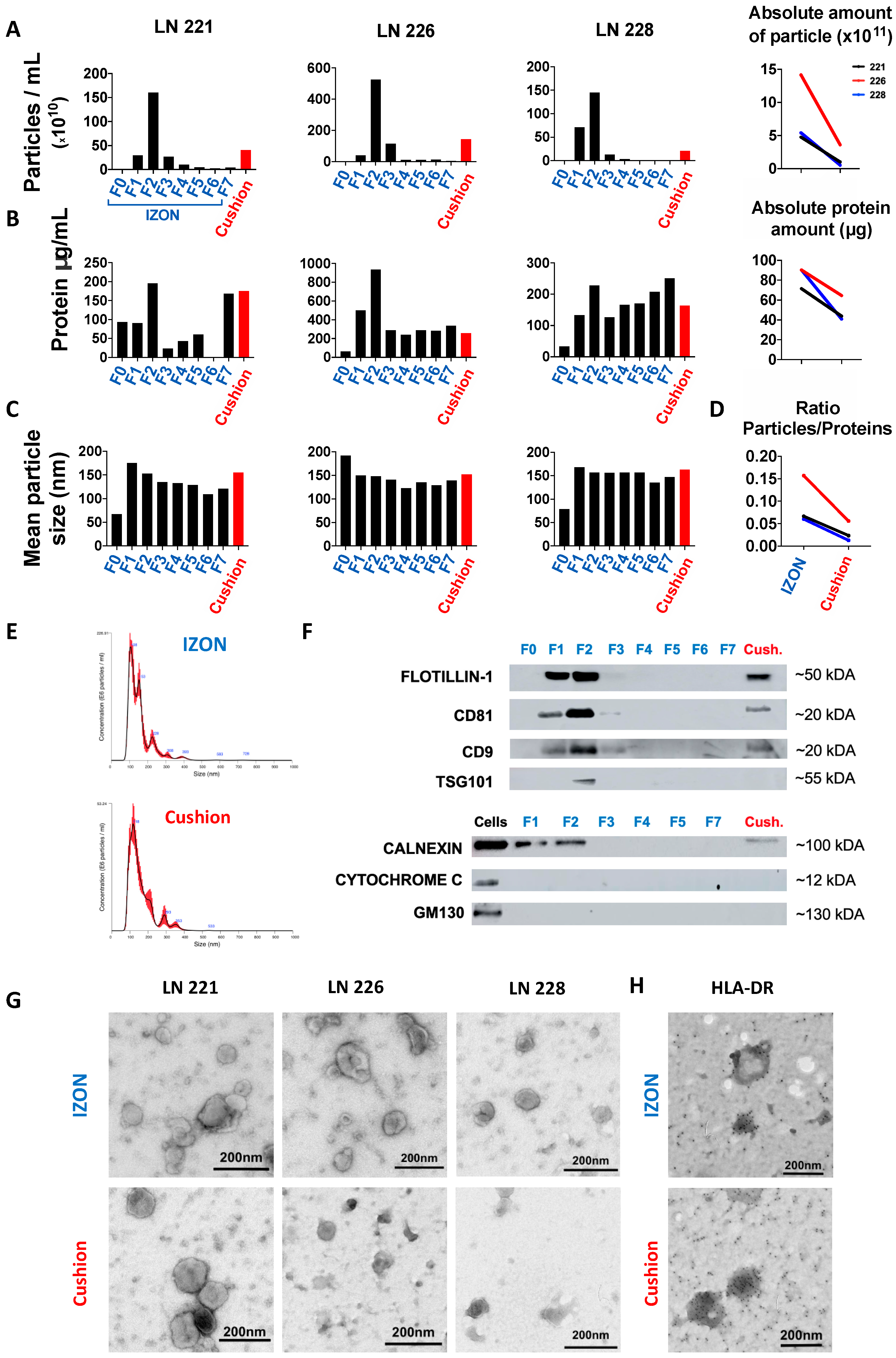
2.1. Isolation and Purification of sEVs from Human Lymph Nodes
2.2. Isolation and Purification of sEVs from Murine Spleen
2.3. Functional Analysis of sEVs Isolated by the Two Different Protocols
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. SEV-Free RPMI Medium
4.3. Isolation of Lymph Node Supernatants
4.4. Isolation of Murine Spleen Supernatants
4.5. Differential Centrifugation
4.6. SEV Isolation on Sucrose Density Cushion
4.7. SEV Isolation on Single qEV 35nm Columns, IZON
4.8. Bicinchonic Acid (BCA) Assay and Nanoparticle Tracking Analyzis (NTA)
4.9. Immunoblotting
4.10. Electron Microscopy (EM)
4.11. Functional Assay
4.12. Statistical Analysis
4.13. EV Track
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BCA | Bicinchoninic acid |
| ER | Endoplasmatic reticulum |
| EVs | Extracellular vesicles |
| FCS | Fetal calf serum |
| i.p. | Intraperitoneal |
| LN | Lymph node |
| MHC | Major histocompatibility complex |
| MVs | Microvesicles |
| NTA | Nanoparticle Tracking Analysis |
| SEC | Size-exclusion chromatography |
| sEVs | Small extracellular vesicles |
| TEM | Transmission electron microscopy |
| TEX | Tumor-derived sEVs |
| WT | Wild-type |
References
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Rajasingh, S.; Drosos, N.; Zhou, Z.; Dawn, B.; Rajasingh, J. Exosomes: New molecular targets of diseases. Acta Pharmacol. Sin. 2018, 39, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Campos-Silva, C.; Suárez, H.; Jara-Acevedo, R.; Linares-Espinós, E.; Martinez-Piñeiro, L.; Yáñez-Mó, M.; Valés-Gómez, M. High sensitivity detection of extracellular vesicles immune-captured from urine by conventional flow cytometry. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yan, J.; Shen, X.; Sun, Y.; Thulin, M.; Cai, Y.; Wik, L.; Shen, Q.; Oelrich, J.; Qian, X.; et al. Profiling surface proteins on individual exosomes using a proximity barcoding assay. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef]
- Raposo, G.; Stahl, P.D. Extracellular vesicles: A new communication paradigm? Nat. Rev. Mol. Cell Biol. 2019, 20, 509–510. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; Van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Palma, J.; Yaddanapudi, S.C.; Pigati, L.; Havens, M.A.; Jeong, S.; Weiner, G.A.; Weimer, K.M.E.; Stern, B.; Hastings, M.L.; Duelli, D.M. MicroRNAs are exported from malignant cells in customized particles. Nucleic Acids Res. 2012, 40, 9125–9138. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Baixauli, F.; Gutiérrez-Vázquez, C.; Sánchez-Madrid, F.; Mittelbrunn, M. Sorting it out: Regulation of exosome loading. Semin. Cancer Biol. 2014, 28, 3–13. [Google Scholar] [CrossRef]
- Leidal, A.M.; Huang, H.H.; Marsh, T.; Solvik, T.; Zhang, D.; Ye, J.; Kai, F.B.; Goldsmith, J.; Liu, J.Y.; Huang, Y.H.; et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat. Cell Biol. 2020, 22, 187–199. [Google Scholar] [CrossRef]
- Koppers-Lalic, D.; Hackenberg, M.; Bijnsdorp, I.V.; van Eijndhoven, M.A.J.; Sadek, P.; Sie, D.; Zini, N.; Middeldorp, J.M.; Ylstra, B.; de Menezes, R.X.; et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 2014, 8, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, X.; Shi, H.; Wu, L.; Qian, H.; Xu, W. Exosomes in cancer: Small particle, big player. J. Hematol. Oncol. 2015, 8, 83. [Google Scholar] [CrossRef]
- Reclusa, P.; Taverna, S.; Pucci, M.; Durendez, E.; Calabuig, S.; Manca, P.; Serrano, M.J.; Sober, L.; Pauwels, P.; Russo, A.; et al. Exosomes as diagnostic and predictive biomarkers in lung cancer. J. Thorac. Dis. 2017, 9, S1373–S1382. [Google Scholar] [CrossRef]
- Liu, Y.; Li, D.; Liu, Z.; Zhou, Y.; Chu, D.; Li, X.; Jiang, X.; Hou, D.; Chen, X.; Chen, Y.; et al. Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse. Sci. Rep. 2015, 5, 17543. [Google Scholar] [CrossRef]
- Yong, T.; Zhang, X.; Bie, N.; Zhang, H.; Zhang, X.; Li, F.; Hakeem, A.; Hu, J.; Gan, L.; Santos, H.A.; et al. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat. Commun. 2019, 10, 3838. [Google Scholar] [CrossRef]
- Haderk, F.; Hanna, B.; Richter, K.; Schnölzer, M.; Zenz, T.; Stilgenbauer, S.; Lichter, P.; Seiffert, M. Extracellular vesicles in chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 1826–1830. [Google Scholar] [CrossRef]
- Böing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.W.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.F.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef] [PubMed]
- Stranska, R.; Gysbrechts, L.; Wouters, J.; Vermeersch, P.; Bloch, K.; Dierickx, D.; Andrei, G.; Snoeck, R. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J. Transl. Med. 2018, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef] [PubMed]
- Gorgun, G.; Ramsay, A.G.; Holderried, T.A.W.; Zahrieh, D.; Dieu, R.L.; Liu, F.; Quackenbush, J.; Croce, C.M.; Gribben, J.G. Eμ-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 6250–6255. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Valero, A.; Monguió-Tortajada, M.; Carreras-Planella, L.; Franquesa, M.; Beyer, K.; Borràs, F.E. Size-Exclusion Chromatography-based isolation minimally alters Extracellular Vesicles’ characteristics compared to precipitating agents. Sci. Rep. 2016, 6, 33641. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Sana, B.; Johnson, E.; Sheah, K.; Poh, C.L.; Lim, S. Iron-based ferritin nanocore as a contrast agent. Biointerphases 2010, 5, FA48–FA52. [Google Scholar] [CrossRef]
- Falvo, E.; Tremante, E.; Arcovito, A.; Papi, M.; Elad, N.; Boffi, A.; Morea, V.; Conti, G.; Toffoli, G.; Fracasso, G.; et al. Improved Doxorubicin Encapsulation and Pharmacokinetics of Ferritin−Fusion Protein Nanocarriers Bearing Proline, Serine, and Alanine Elements. Biomacromolecules 2015, 17, 514–522. [Google Scholar] [CrossRef]
- Fleming, V.; Hu, X.; Weller, C.; Weber, R.; Groth, C.; Riester, Z.; Hüser, L.; Sun, Q.; Nagibin, V.; Kirschning, C.; et al. Melanoma extracellular vesicles generate immunosuppressive myeloid cells by upregulating PD-L1 via TLR4 signaling. Cancer Res. 2019, 79, 4715–4728. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. OncoImmunology 2018, 7, e1412909. [Google Scholar] [CrossRef]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.; Di Vizio, D.; Sahoo, S.; Théry, C.; Witwer, K.M.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5, 32945. [Google Scholar] [CrossRef] [PubMed]
- Yuana, Y.; Levels, J.; Grootemaat, A.; Sturk, A.; Nieuwland, R. Co-isolation of extracellular vesicles and high-density lipoproteins using density gradient ultracentrifugation. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Karimi, N.; Cvjetkovic, A.; Jang, C.J.; Crescitelli, S.; Feizi, M.A.H.; Nieuwland, R.; Lötvall, J.; Lässer, C. Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cell. Mol. Life Sci. 2018, 75, 2873–2886. [Google Scholar] [CrossRef]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Takov, K.; Yellon, D.M.; Davidson, S. Comparison of small extracellular vesicles isolated from plasma by ultracentrifugation or size-exclusion chromatography: Yield, purity and functional potential. J. Extracell. Vesicles 2018, 8, 1560809. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.K.; Geuze, H.J. B Lymphocytes Secrete Antigen-Presenting Vesicles. J. Exp. Med. 1996, 1, 1161–1172. [Google Scholar] [CrossRef]
- André, F.; Chaput, N.; Schartz, N.E.C.; Flament, C.; Aubert, N.; Bernard, J.; Lemonnier, F.; Raposo, G.; Escudier, B.; Hsu, D.-H.; et al. Exosomes as Potent Cell-Free Peptide-Based Vaccine. I. Dendritic Cell-Derived Exosomes Transfer Functional MHC Class I/Peptide Complexes to Dendritic Cells. J. Immunol. 2004, 172, 2126–2136. [Google Scholar]
- Möst, J.; Schwaeble, W.; Drach, J.; Sommerauer, A.; Dierich, M.P. Regulation of the Expression of ICAM-1 on Human Monocytes and Monocytic Tumor Cell Lines. J. Immunol. 1992, 148, 1635–1642. [Google Scholar]
- Fujihara, M.; Ikebuchi, K.; Yamaguchi, M.; Abe, H.; Niwa, K.; Sekiguchi, S. Effects of Liposome-Encapsulated Hemoglobin on Phorbol Ester-Induced Superoxide Production and Expression of Costimulatory Molecules by Monocytes in Vitro. Artif. Cells Blood Substit. Immobil. Biotechnol. 1998, 26, 487–495. [Google Scholar] [CrossRef] [PubMed]
- McClanahan, F.; Riches, J.C.; Miller, S.; Day, W.P.; Kotsiou, E.; Neuberg, D.; Croce, C.M.; Capasso, M.; Gribben, J.G. Mechanisms of PD-L1/PD-1–mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Eµ-TCL1 CLL mouse model. Blood 2015, 126, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Hanna, B.; McClanahan, F.; Yazdanparast, H.; Zaborsky, N.; Kalter, V.; Rössner, P.M.; Benner, A.; Dürr, C.; Egle, A.; Gribben, J.G.; et al. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia 2016, 30, 570–579. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bordas, M.; Genard, G.; Ohl, S.; Nessling, M.; Richter, K.; Roider, T.; Dietrich, S.; Maaß, K.K.; Seiffert, M. Optimized Protocol for Isolation of Small Extracellular Vesicles from Human and Murine Lymphoid Tissues. Int. J. Mol. Sci. 2020, 21, 5586. https://doi.org/10.3390/ijms21155586
Bordas M, Genard G, Ohl S, Nessling M, Richter K, Roider T, Dietrich S, Maaß KK, Seiffert M. Optimized Protocol for Isolation of Small Extracellular Vesicles from Human and Murine Lymphoid Tissues. International Journal of Molecular Sciences. 2020; 21(15):5586. https://doi.org/10.3390/ijms21155586
Chicago/Turabian StyleBordas, Marie, Géraldine Genard, Sibylle Ohl, Michelle Nessling, Karsten Richter, Tobias Roider, Sascha Dietrich, Kendra K. Maaß, and Martina Seiffert. 2020. "Optimized Protocol for Isolation of Small Extracellular Vesicles from Human and Murine Lymphoid Tissues" International Journal of Molecular Sciences 21, no. 15: 5586. https://doi.org/10.3390/ijms21155586
APA StyleBordas, M., Genard, G., Ohl, S., Nessling, M., Richter, K., Roider, T., Dietrich, S., Maaß, K. K., & Seiffert, M. (2020). Optimized Protocol for Isolation of Small Extracellular Vesicles from Human and Murine Lymphoid Tissues. International Journal of Molecular Sciences, 21(15), 5586. https://doi.org/10.3390/ijms21155586