A Small Molecule, 4-Phenylbutyric Acid, Suppresses HCV Replication via Epigenetically Induced Hepatic Hepcidin

 and
and
Abstract
:1. Introduction
2. Results
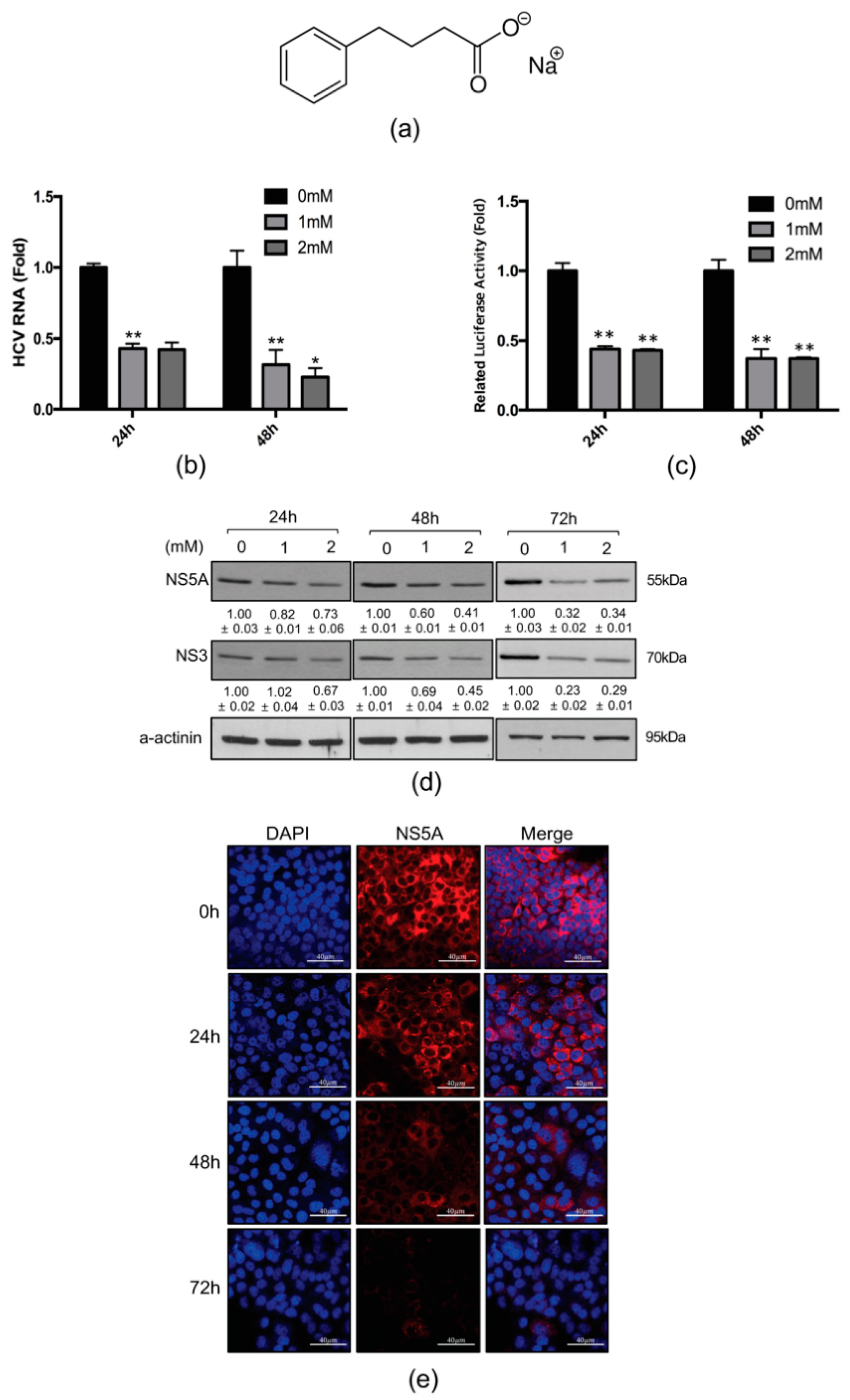
2.1. 4-PBA Inhibited HCV RNA Replication
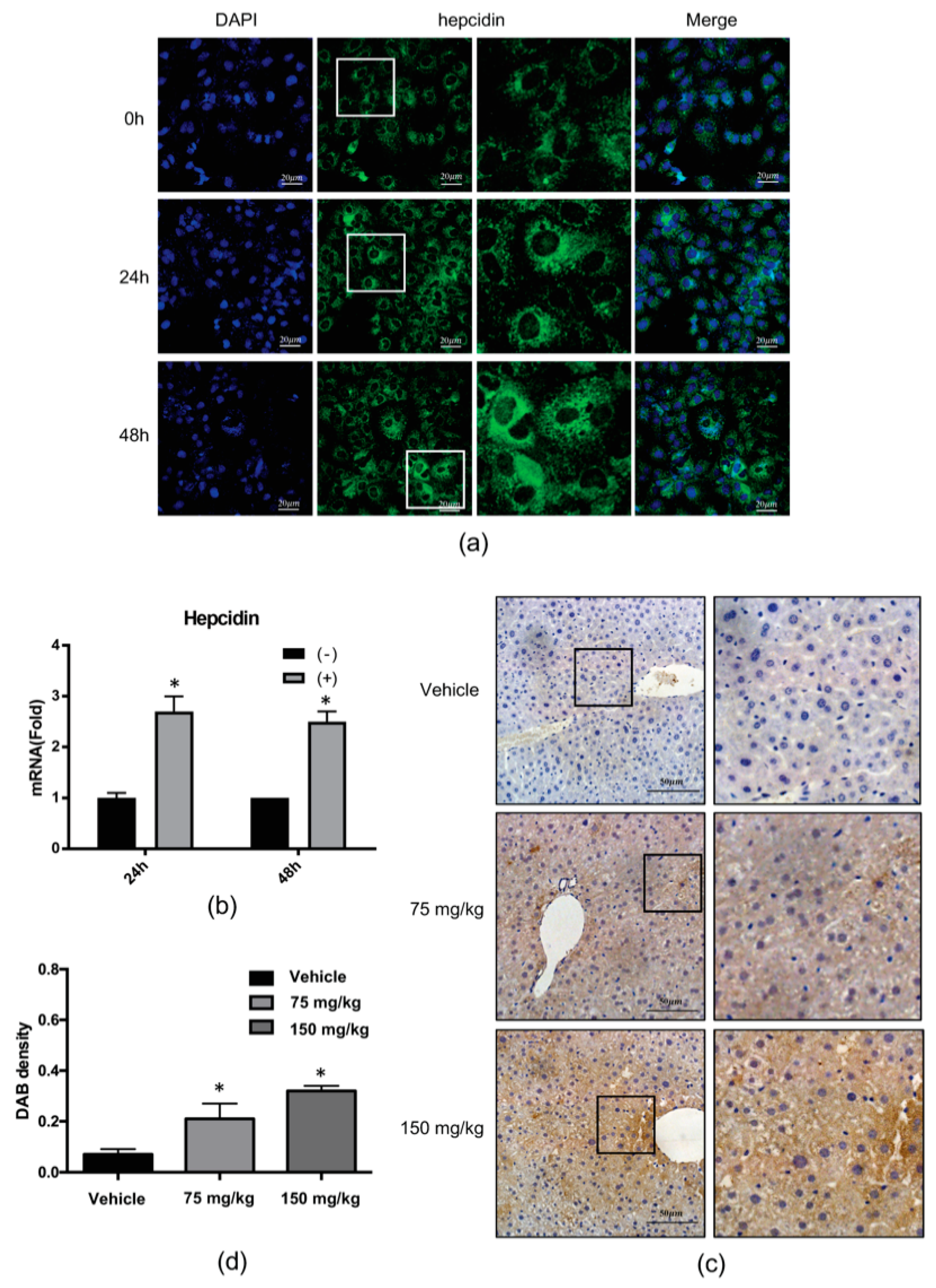
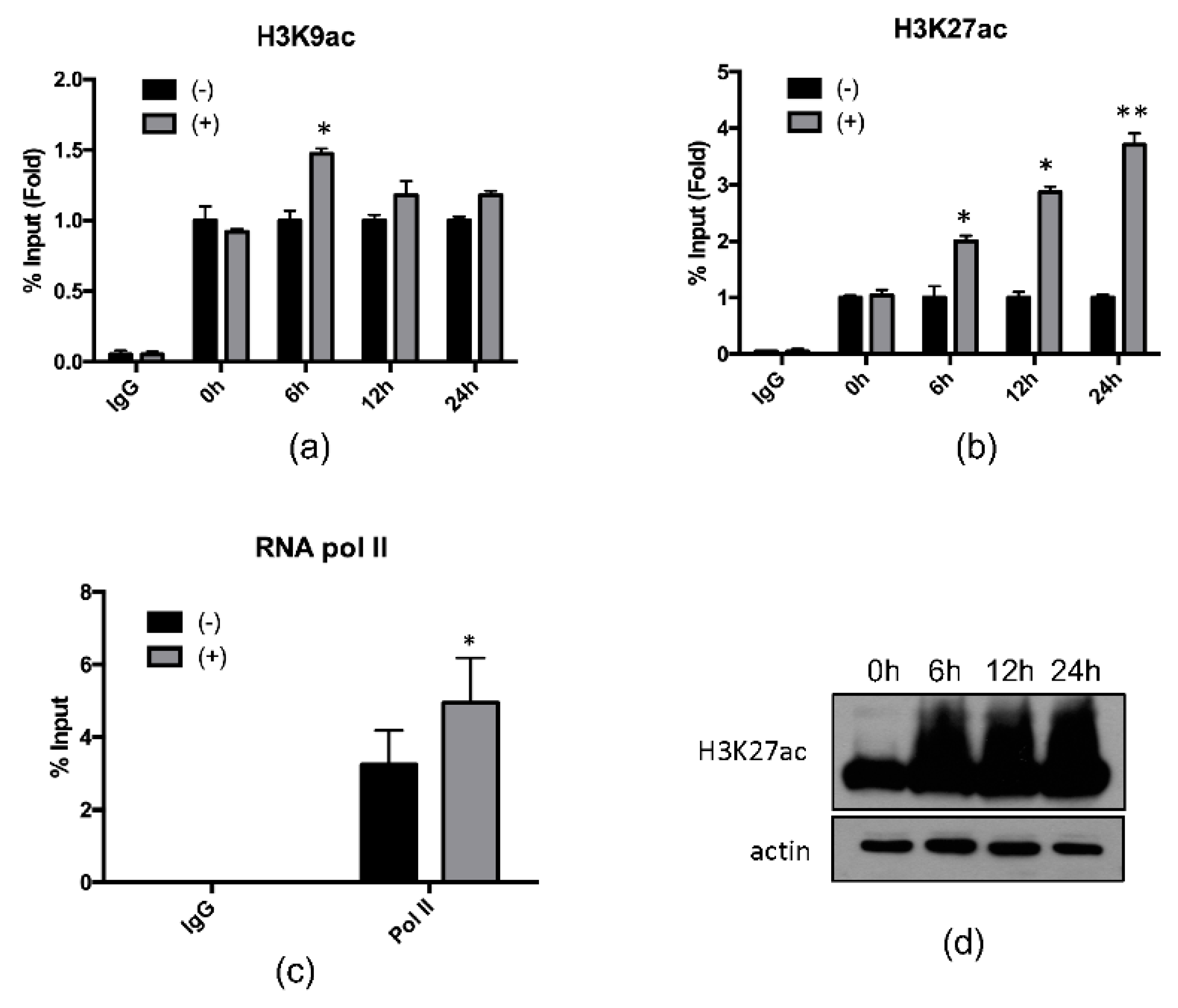
2.2. 4-PBA Induced the Expression of the Antimicrobial Peptide (AMP), Hepcidin
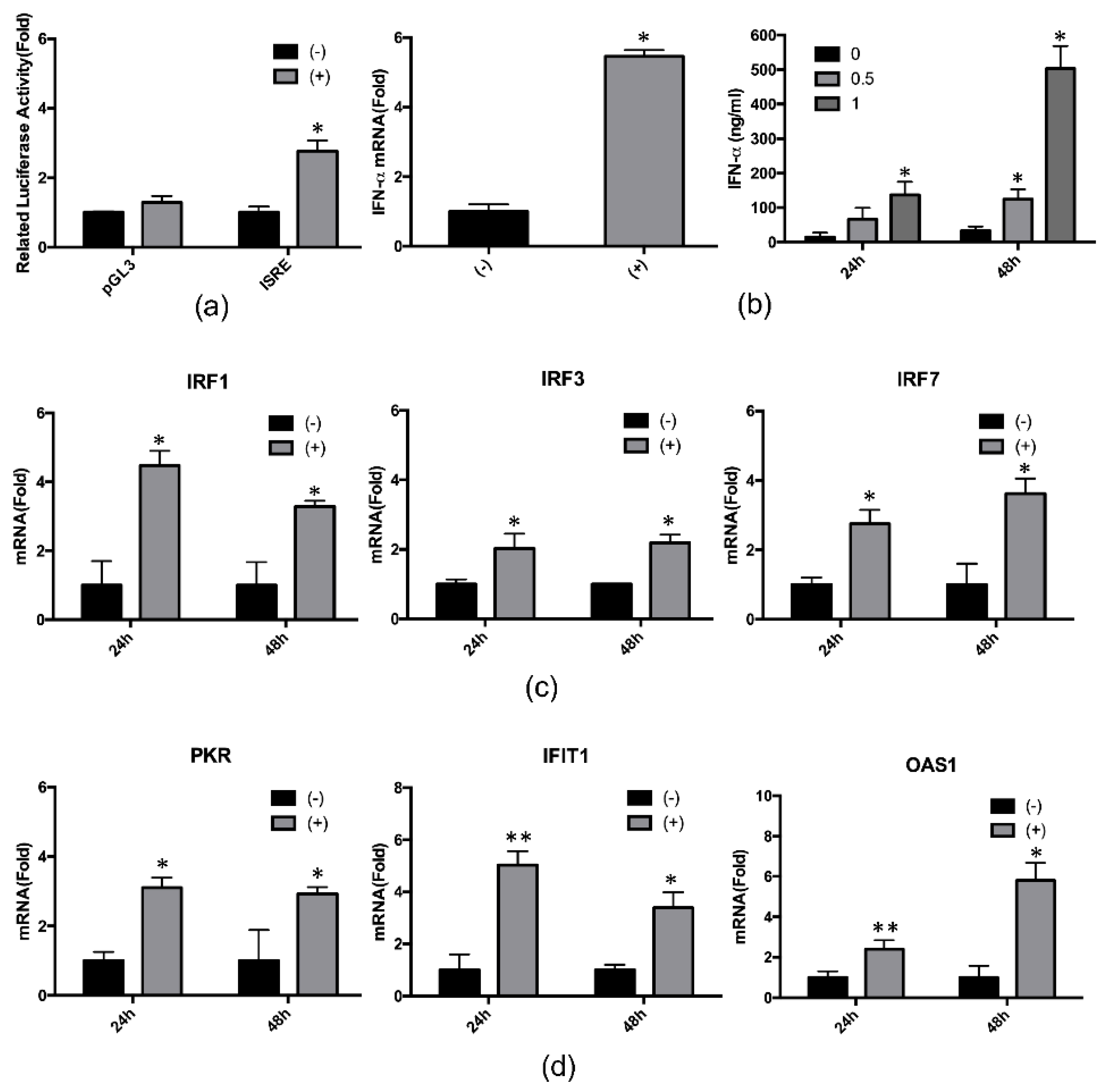
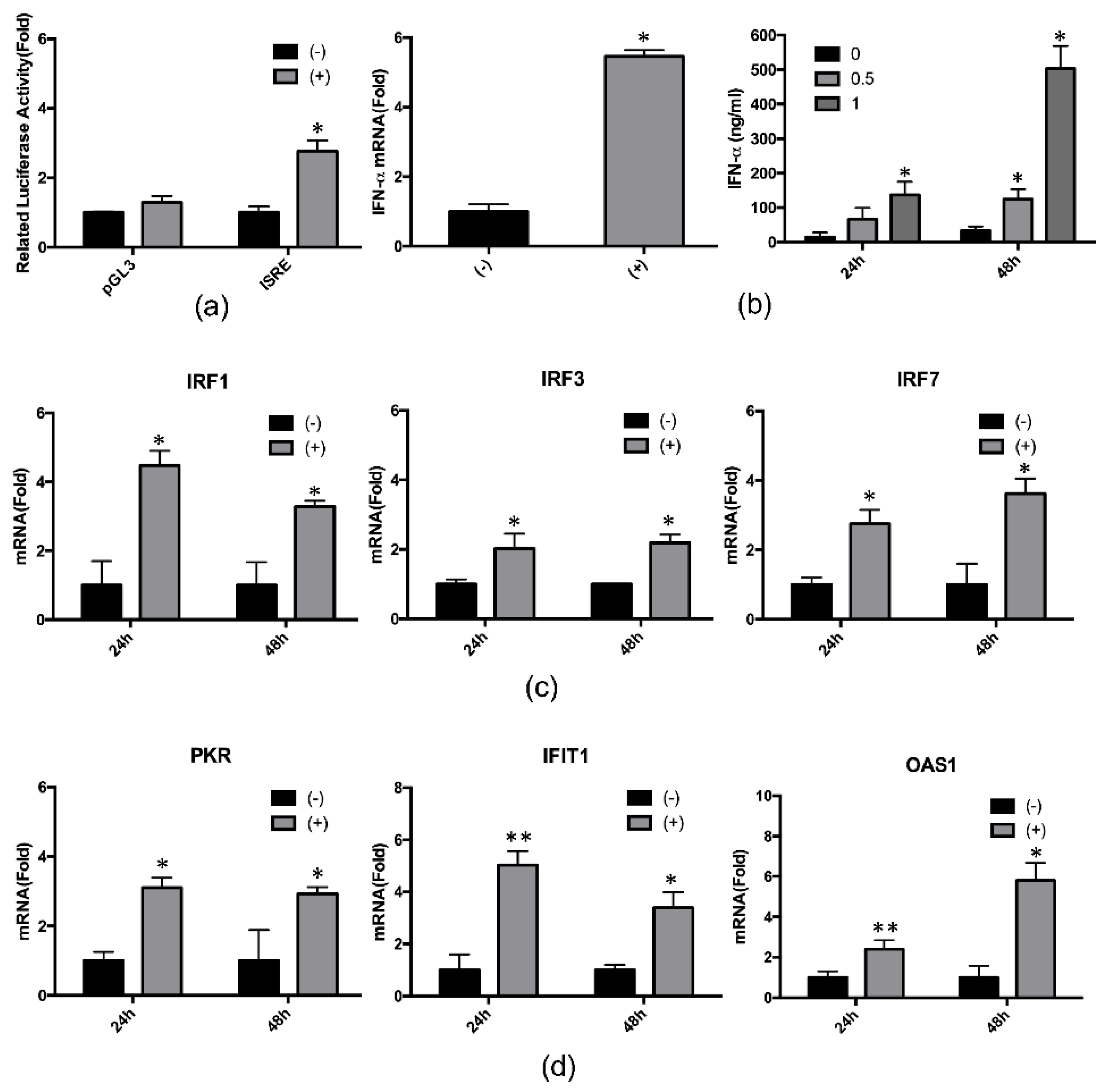
2.3. 4-PBA Enhanced the IFN-α Response
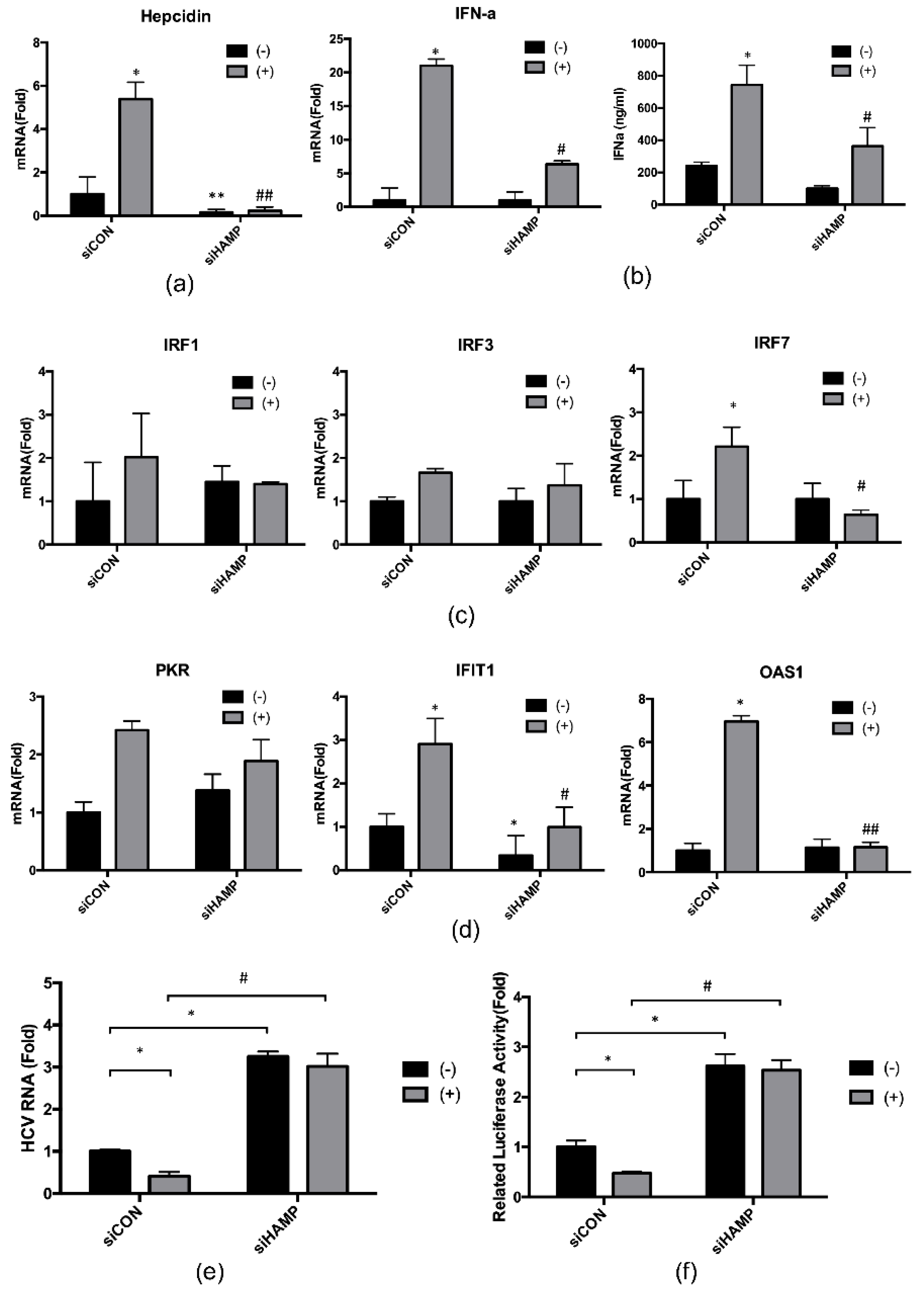
2.4. Hepcidin Mediated 4-PBA-Induced IFN-α Signaling
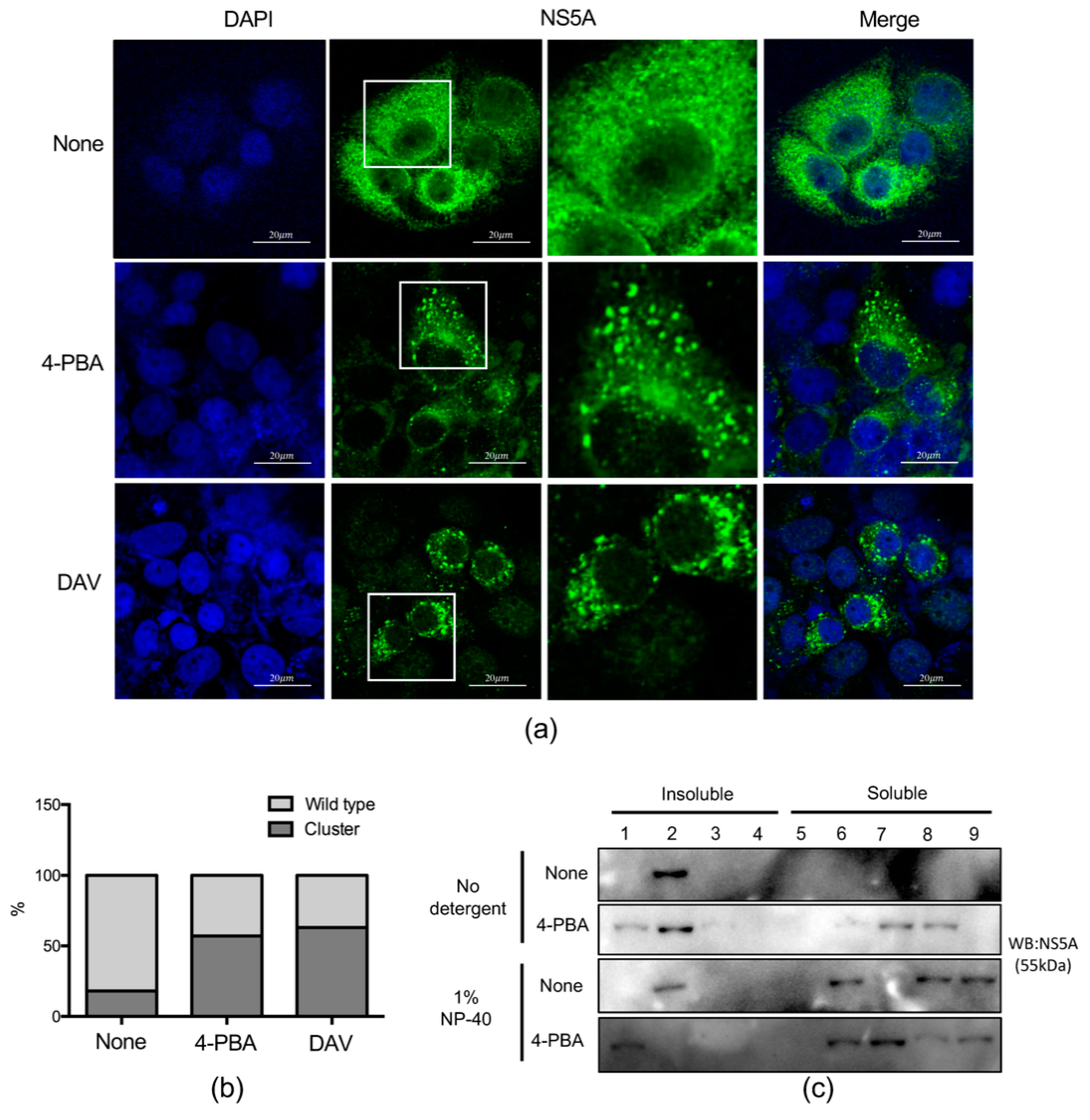
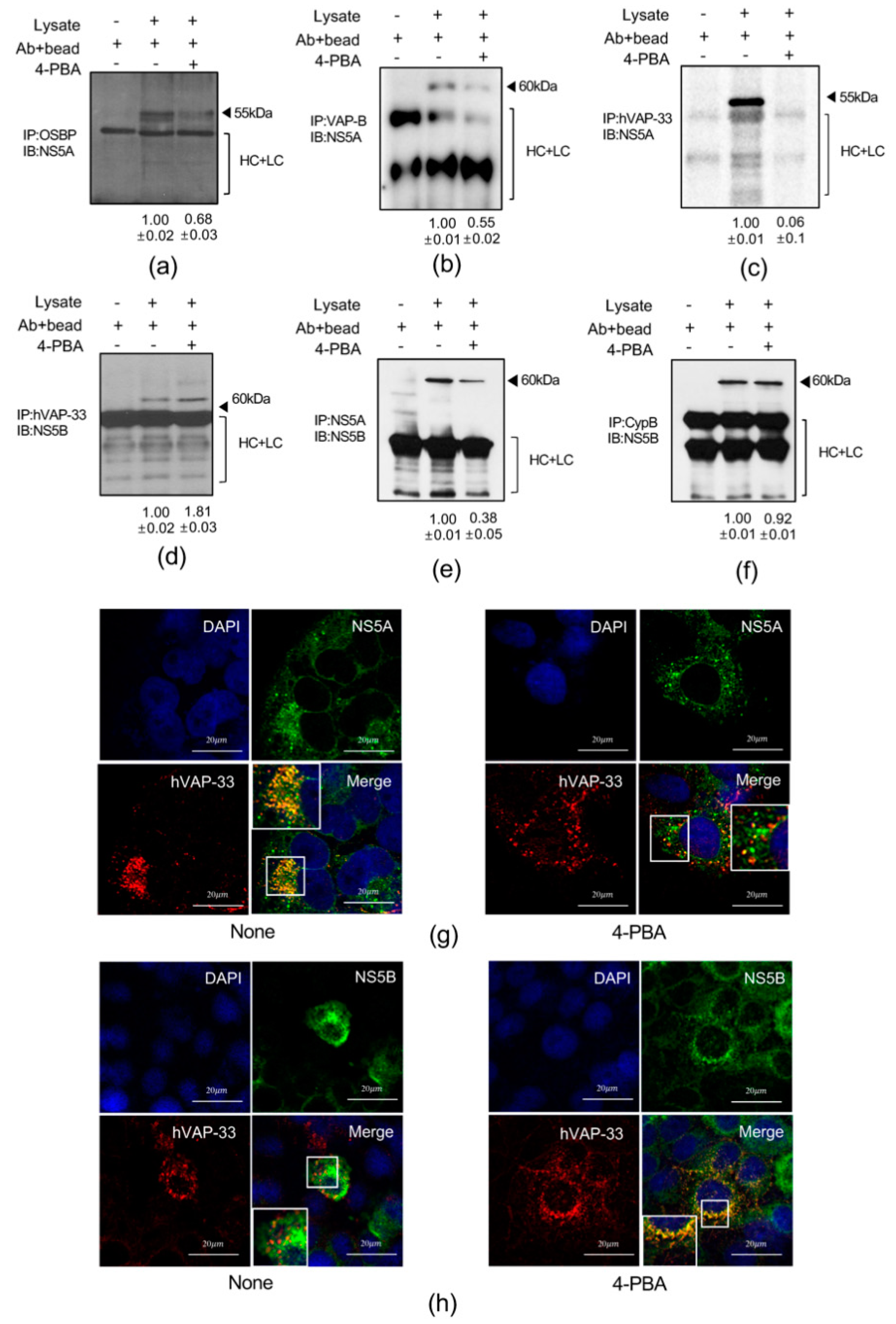
2.5. 4-PBA Disrupted MW Formation
2.6. 4-PBA Inhibited the Formation of Double Membranous Vesicles (DMVs)
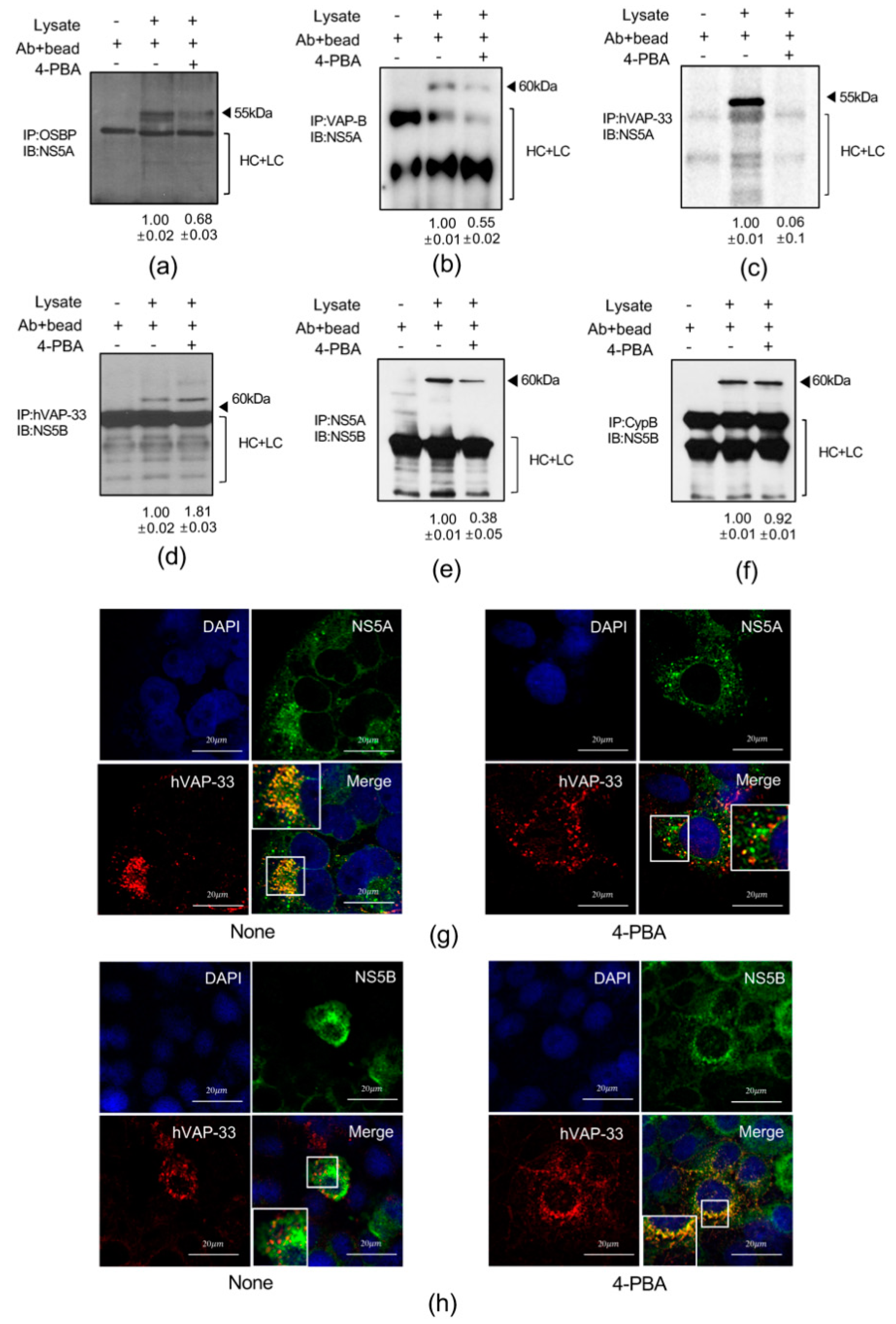
2.7. 4-PBA Interfered with Molecular Interactions in the HCV RNA Replication Complex
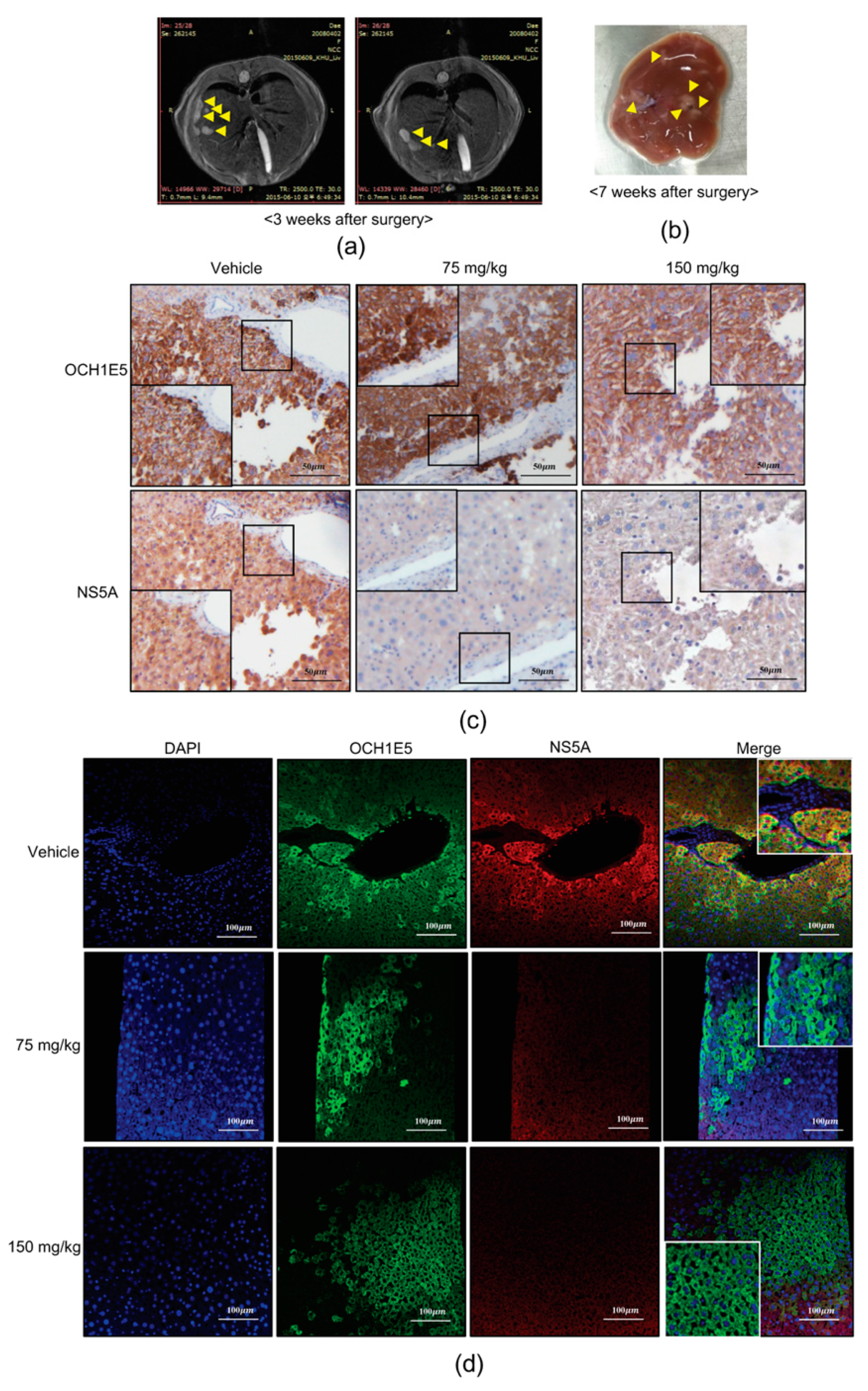
2.8. The Anti-HCV Effects of 4-PBA in a Mouse Model
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.4. Determination of Cell Viability
4.5. Chromatin Immunoprecipitation Assay
4.6. Membrane Flotation Assay
4.7. Luciferase Assay
4.8. Immunoblotting
4.9. Immunofluorescence Analysis
4.10. Immunoprecipitation
4.11. Immunohistochemistry
4.12. DNAs and siRNAs Transfection
4.13. Electron Microscopy
4.14. Animal Procedures
4.15. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HCV | Hepatitis C Virus |
| 4-PBA | 4-Phenyl butyrate |
| HDACi | Histone Deacetylase Inhibitor |
| HCC | Hepatocellcular Carcinoma |
| DAAs | Direct Acting antiviral reagents |
| FDA | Food and Drug Administration |
| ER | Endoplasmic Reticulum |
| AMPs | Antimicrobial Peptides |
References
- Sarrazin, C.; Hezode, C.; Zeuzem, S.; Pawlotsky, J.M. Antiviral strategies in hepatitis C virus infection. J. Hepatol. 2012, 56 (Suppl. 1), S88–S100. [Google Scholar] [CrossRef]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.; Vutien, P.; Hoang, J.; Trinh, S.; Le, A.; Yasukawa, L.A.; Weber, S.; Henry, L.; Nguyen, M.H. Barriers to care for chronic hepatitis C in the direct-acting antiviral era: A single-centre experience. BMJ Open Gastroenterol. 2017, 4, e000181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozbial, K.; Moser, S.; Schwarzer, R.; Laferl, H.; Al-Zoairy, R.; Stauber, R.; Stattermayer, A.F.; Beinhardt, S.; Graziadei, I.; Freissmuth, C.; et al. Unexpected high incidence of hepatocellular carcinoma in cirrhotic patients with sustained virologic response following interferon-free direct-acting antiviral treatment. J. Hepatol. 2016, 65, 856–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrenson, B.; Suetani, R.J.; Williams, M.J.; Bickley, V.M.; George, P.M.; Jones, G.T.; McCormick, S.P. Functional rescue of mutant ABCA1 proteins by sodium 4-phenylbutyrate. J. Lipid Res. 2013, 54, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannitti, T.; Palmieri, B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D 2011, 11, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, J.; Halldorsson, S.; Agerberth, B.; Gudmundsson, G.H. Phenylbutyrate induces antimicrobial peptide expression. Antimicrob. Agents Chemother. 2009, 53, 5127–5133. [Google Scholar] [CrossRef] [Green Version]
- Chriett, S.; Dabek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent action of butyrate over beta-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef] [Green Version]
- Correa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef]
- Lauth, X.; Babon, J.J.; Stannard, J.A.; Singh, S.; Nizet, V.; Carlberg, J.M.; Ostland, V.E.; Pennington, M.W.; Norton, R.S.; Westerman, M.E. Bass hepcidin synthesis, solution structure, antimicrobial activities and synergism, and in vivo hepatic response to bacterial infections. J. Biol. Chem. 2005, 280, 9272–9282. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Trinh, T.L.; Dong, H.; Keith, R.; Nelson, D.; Liu, C. Iron regulator hepcidin exhibits antiviral activity against hepatitis C virus. PLoS ONE 2012, 7, e46631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girelli, D.; Pasino, M.; Goodnough, J.B.; Nemeth, E.; Guido, M.; Castagna, A.; Busti, F.; Campostrini, N.; Martinelli, N.; Vantini, I.; et al. Reduced serum hepcidin levels in patients with chronic hepatitis C. J. Hepatol. 2009, 51, 845–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriishi, K.; Matsuura, Y. Exploitation of lipid components by viral and host proteins for hepatitis C virus infection. Front. Microbiol. 2012, 3, 54. [Google Scholar] [CrossRef] [Green Version]
- Neufeldt, C.J.; Joyce, M.A.; Van Buuren, N.; Levin, A.; Kirkegaard, K.; Gale, M., Jr.; Tyrrell, D.L.; Wozniak, R.W. The Hepatitis C Virus-Induced Membranous Web and Associated Nuclear Transport Machinery Limit Access of Pattern Recognition Receptors to Viral Replication Sites. PLoS Pathog. 2016, 12, e1005428. [Google Scholar] [CrossRef] [Green Version]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Aizaki, H.; He, J.W.; Lai, M.M. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 2004, 78, 3480–3488. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Perry, J.W.; Lauring, A.S.; Neddermann, P.; De Francesco, R.; Tai, A.W. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 2014, 146, 1373–1385.e11. [Google Scholar] [CrossRef]
- Tu, H.; Gao, L.; Shi, S.T.; Taylor, D.R.; Yang, T.; Mircheff, A.K.; Wen, Y.; Gorbalenya, A.E.; Hwang, S.B.; Lai, M.M. Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology 1999, 263, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Watashi, K.; Ishii, N.; Hijikata, M.; Inoue, D.; Murata, T.; Miyanari, Y.; Shimotohno, K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 2005, 19, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Ayala, P.; Montenegro, J.; Vivar, R.; Letelier, A.; Urroz, P.A.; Copaja, M.; Pivet, D.; Humeres, C.; Troncoso, R.; Vicencio, J.M.; et al. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp. Mol. Pathol. 2012, 92, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Kanto, T.; Hayashi, N. Innate immunity in hepatitis C virus infection: Interplay among dendritic cells, natural killer cells and natural killer T cells. Hepatol. Res. Off. J. Japan Soc. Hepatol. 2007, 37 (Suppl. 3), S319–S326. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Masaki, T.; Williford, S.; Ingravallo, P.; Feng, Z.; Lahser, F.; Asante-Appiah, E.; Neddermann, P.; De Francesco, R.; Howe, A.Y.; et al. Kinetic analyses reveal potent and early blockade of hepatitis C virus assembly by NS5A inhibitors. Gastroenterology 2014, 147, 453–462.e7. [Google Scholar] [CrossRef]
- Berger, C.; Romero-Brey, I.; Radujkovic, D.; Terreux, R.; Zayas, M.; Paul, D.; Harak, C.; Hoppe, S.; Gao, M.; Penin, F.; et al. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology 2014, 147, 1094–1105.e25. [Google Scholar] [CrossRef]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Kusaczuk, M.; Kretowski, R.; Bartoszewicz, M.; Cechowska-Pasko, M. Phenylbutyrate-a pan-HDAC inhibitor-suppresses proliferation of glioblastoma LN-229 cell line. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 931–942. [Google Scholar] [CrossRef] [Green Version]
- Gondcaille, C.; Depreter, M.; Fourcade, S.; Lecca, M.R.; Leclercq, S.; Martin, P.G.; Pineau, T.; Cadepond, F.; ElEtr, M.; Bertrand, N.; et al. Phenylbutyrate up-regulates the adrenoleukodystrophy-related gene as a nonclassical peroxisome proliferator. J. Cell Biol. 2005, 169, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Taura, K.; Kodama, Y.; Schnabl, B.; Brenner, D.A. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 2008, 48, 1420–1429. [Google Scholar] [CrossRef]
- Aizaki, H.; Lee, K.J.; Sung, V.M.; Ishiko, H.; Lai, M.M. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 2004, 324, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Reghellin, V.; Donnici, L.; Fenu, S.; Berno, V.; Calabrese, V.; Pagani, M.; Abrignani, S.; Peri, F.; De Francesco, R.; Neddermann, P. NS5A inhibitors impair NS5A-phosphatidylinositol 4-kinase IIIα complex formation and cause a decrease of phosphatidylinositol 4-phosphate and cholesterol levels in hepatitis C virus-associated membranes. Antimicrob. Agents Chemother. 2014, 58, 7128–7140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Udali, S.; Castagna, A.; Corbella, M.; Ruzzenente, A.; Moruzzi, S.; Mazzi, F.; Campagnaro, T.; De Santis, D.; Franceschi, A.; Pattini, P.; et al. Hepcidin and DNA promoter methylation in hepatocellular carcinoma. Eur. J. Clin. Investig. 2018, 48, e12870. [Google Scholar] [CrossRef] [PubMed]
- Jyothi, K.R.; Beloor, J.; Jo, A.; Nguyen, M.N.; Choi, T.G.; Kim, J.H.; Akter, S.; Lee, S.K.; Maeng, C.H.; Baik, H.H.; et al. Liver-targeted cyclosporine A-encapsulated poly (lactic-co-glycolic) acid nanoparticles inhibit hepatitis C virus replication. Int. J. Nanomed. 2015, 10, 903–921. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Direction | Sequence |
|---|---|---|
| HCV | forward | CGGGAGAGCCATAGTGGTCTG CG |
| reverse | CTCGCAAGCACCCTATCA GGCAGTA | |
| b-actin | forward | AGGCTGTGCTGTCCCTCT |
| reverse | TCCGGTGAGGAGGATGCG | |
| hepcidin | forward | ACCAGAGCAAGCTCAAGACC |
| reverse | CAGGGCAGGTAGGTTCTACG | |
| IFN-a | forward | TTTCTCCTGCCTGAAGGACAG |
| reverse | GCTCATGATTTCTGCTCTGACA | |
| OAS1 | forward | AGGTGGTAAAGGGTGGCTCC |
| reverse | ACAACCAGGTCAGCGTCAGAT | |
| IFIT1 | forward | TGGCTAAGCAAAACCCTGCA |
| reverse | TCTGGCCTTTCAGGTGTTTCAC | |
| IRF1 | forward | CTCACCAGGAACCAGAGGAA |
| reverse | TGAGTGGTGTAACTGCTGTGG | |
| IRF3 | forward | ACCAGCCGTGGACCAAGAG |
| reverse | TACCAAGGCCCTGAGGCAC | |
| IRF7 | forward | TGGTCCTGGTGAAGCTGGAA |
| reverse | GATGTCGTCATAGAGGCTGTTGG | |
| PKR | forward | TCTCTGGCGGTCTTCAGAAT |
| reverse | ACTCCCTGCTTCTGACGGTA | |
| hVAP-33 | forward | GAAGACTACAGCACCTCGCC |
| reverse | GCCTCTTTCCACACAGCTTC | |
| Hepcidin_locus | forward | CTGTTTTCCCACAACAGGTG |
| reverse | CTCAGTGCTCGGGTGTCTC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.; Lee, Y.-s.; Jeong, S.; Kim, D.; Chon, S.; Pak, Y.K.; Kim, S.; Ha, J.; Kang, I.; Choe, W. A Small Molecule, 4-Phenylbutyric Acid, Suppresses HCV Replication via Epigenetically Induced Hepatic Hepcidin. Int. J. Mol. Sci. 2020, 21, 5516. https://doi.org/10.3390/ijms21155516
Kim K, Lee Y-s, Jeong S, Kim D, Chon S, Pak YK, Kim S, Ha J, Kang I, Choe W. A Small Molecule, 4-Phenylbutyric Acid, Suppresses HCV Replication via Epigenetically Induced Hepatic Hepcidin. International Journal of Molecular Sciences. 2020; 21(15):5516. https://doi.org/10.3390/ijms21155516
Chicago/Turabian StyleKim, Kiyoon, Young-seok Lee, Suyun Jeong, Daehong Kim, Suk Chon, Youngmi Kim Pak, Sungsoo Kim, Joohun Ha, Insug Kang, and Wonchae Choe. 2020. "A Small Molecule, 4-Phenylbutyric Acid, Suppresses HCV Replication via Epigenetically Induced Hepatic Hepcidin" International Journal of Molecular Sciences 21, no. 15: 5516. https://doi.org/10.3390/ijms21155516
APA StyleKim, K., Lee, Y.-s., Jeong, S., Kim, D., Chon, S., Pak, Y. K., Kim, S., Ha, J., Kang, I., & Choe, W. (2020). A Small Molecule, 4-Phenylbutyric Acid, Suppresses HCV Replication via Epigenetically Induced Hepatic Hepcidin. International Journal of Molecular Sciences, 21(15), 5516. https://doi.org/10.3390/ijms21155516