Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma


{kind=link}
{kind=link}
{kind=link}
Abstract
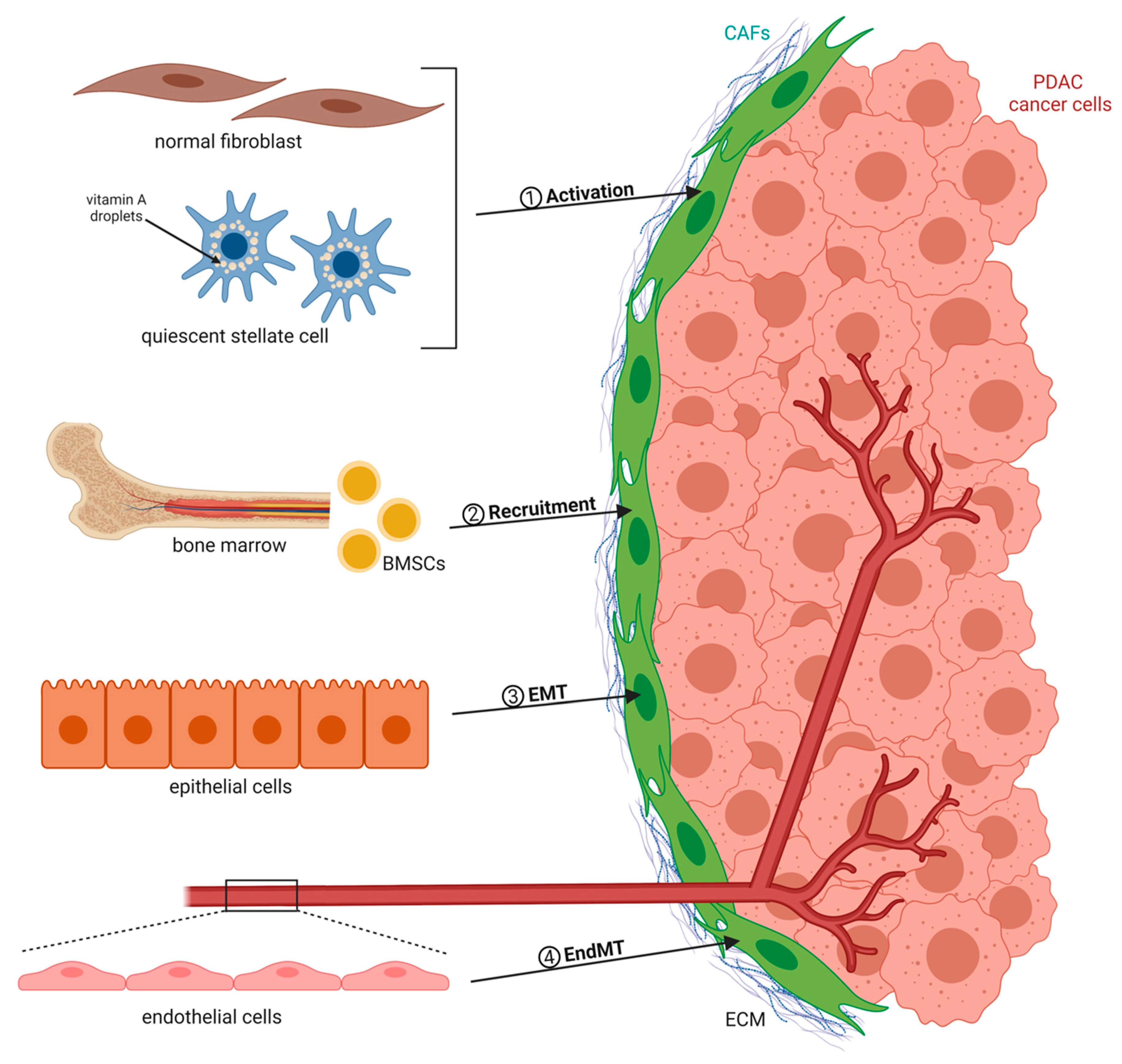
1. Cancer-Associated Fibroblasts Are Key Players in PDAC
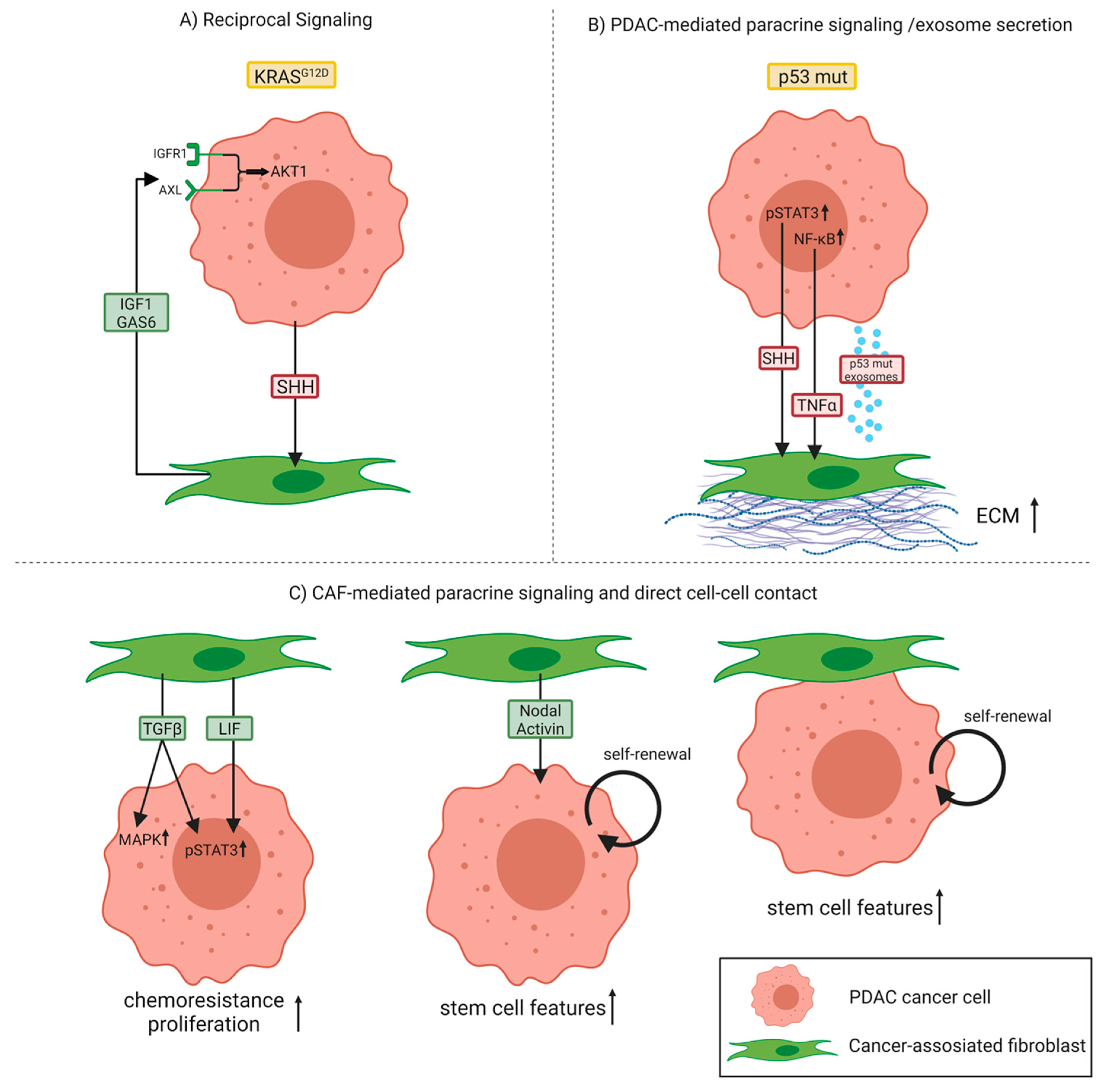
2. Bidirectional Cancer Cell–CAF Crosstalk Promotes Tumor Progression
3. Metabolic Reprograming Mediated by CAFs
4. CAFs Modulate the Immune Microenvironment and Crosstalk with Additional Cell Types in the Desmoplastic Stroma
5. CAF Ablation Studies—Functional Evidence for a Tumor-Suppressive Role of CAFs
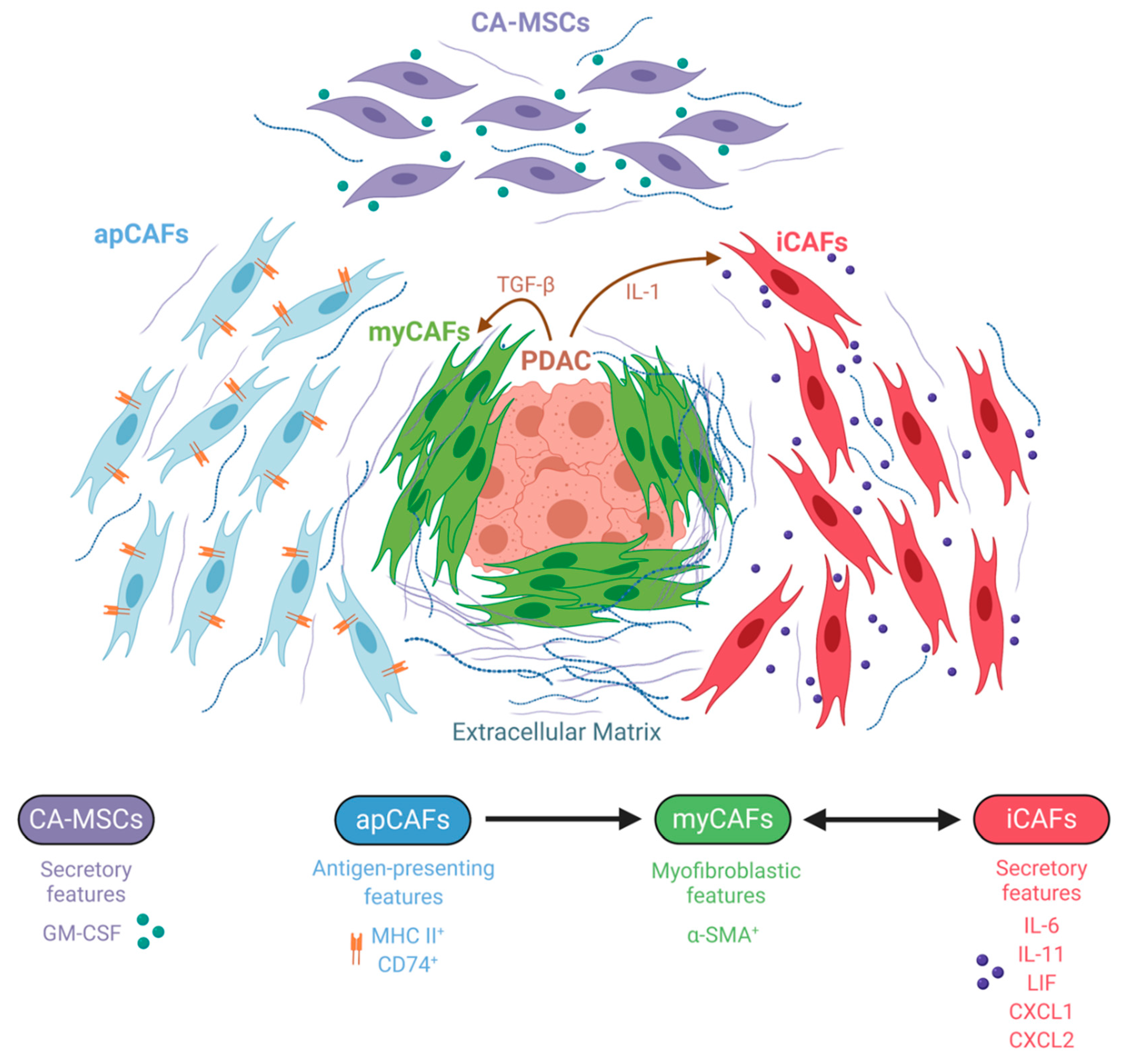
6. The Emerging Field of CAF Heterogeneity
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADM | Acinar-to-ductal metaplasia |
| apCAFs | Antigen-presenting CAFs |
| APCs | Antigen presenting cells |
| α-SMA | alpha-Smooth muscle actin |
| α-SMA-tk | alpha-Smooth muscle actin-thymidine kinase |
| CA-MSCs | Cancer-associated mesenchymal stem cells |
| CAFs | Cancer-associated fibroblasts |
| Cre | Cre recombinase |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| BMSCs | Bone marrow-derived mesenchymal stem cells |
| CCL2 | C-C motif ligand 2 |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| CXCL2 | C-X-C motif chemokine ligand 2 |
| CXCL8 | C-X-C motif chemokine ligand 8 |
| CXCL12 | C-X-C motif chemokine ligand 12 |
| CXCR4 | C-X-C chemokine receptor 4 |
| DC | Dendritic cells |
| DRG | Dorsal root ganglia |
| ECM | Extracellular matrix |
| EndMT | Endothelial-to-mesenchymal transition |
| EMT | Epithelial-to-mesenchymal transition |
| FAK | Focal adhesion kinase |
| FAP | Fibroblast activation protein |
| FoxP3 | Forkhead box P3 |
| FSP1 | Fibroblast-specific protein 1 |
| GAS6 | Growth arrest-specific gene 6 |
| GM-CSF | Granulocytic-macrophage colony-stimulating factor |
| HGF | Hepatocyte growth factor |
| HH | Hedgehog |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| iCAFs | inflammatory CAFs |
| IFN-γ | Interferon-gamma |
| IGF1 | Insulin-like growth factor 1 |
| IGF1R | Insulin-like growth factor 1 receptor |
| IHC | Immunohistochemistry |
| IL-1 | Interleukin-1 |
| IL-1R | Interleukin-1 receptor |
| IL-6 | Interleukin-6 |
| IL-11 | Interleukin-11 |
| IL-33 | Interleukin-33 |
| JAK | Janus kinase |
| KC | Pdx1-Cre;LSL-KrasG12D/+ |
| KPC | Pdx1-Cre;LSL-KrasG12D/+;LSL-p53R172H |
| KPCY | Pdx1-Cre;LSL-KrasG12D/+;p53flox/+;LSL-Rosa26YFP/+ |
| KPflC | Pdx1-Cre;LSL-KrasG12D/+;p53flox/+ |
| KPP | Pdx1-Cre;LSL-KrasG12D/+;p16/p19flox/flox |
| KRAS | Kirsten rat sarcoma virus |
| LCK | Lymphocyte-specific protein tyrosine kinase |
| LGD IPMN | Low-grade intraductal papillary mucinous neoplasm |
| HGD IPMN | High-grade intraductal papillary mucinous neoplasm |
| LIF | Leukemia inhibitory factor |
| LIFR | Leukemia inhibitory factor receptor |
| LPC | Lysophosphatidylcholine |
| LRRC15 | Leucine-rich repeat containing 15 |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| M-CSF | Macrophage-colony stimulating factor |
| MCT1 | Monocarboxylate transporter 1 |
| MCT4 | Monocarboxylate transporter 4 |
| MDSCs | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| miRNA | microRNA |
| MMP9 | Matrix metallopeptidase 9 |
| MSCs | Mesenchymal stem cells |
| myCAFs | myofibroblastic CAFs |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGF | Nerve growth factor |
| NI | Neural invasion |
| NK cells | Natural killer cells |
| OTI | MHC class I-restricted ovalbumin-specific T cell receptor |
| OTII | MHC class II-restricted ovalbumin-specific T cell receptor |
| OVA | Ovalbumin |
| PanIN | Pancreatic intraepithelial neoplasia |
| PBMCs | Peripheral blood mononuclear cells |
| PD-1 | Programmed death-1 |
| PD-L1 | Programmed death-ligand 1 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDGF | Platelet-derived growth factor |
| PDGFRα | Platelet-derived growth factor receptor-alpha |
| PDGFRβ | Platelet-derived growth factor receptor-beta |
| PDPN | Podoplanin |
| Pdx1 | Pancreatic and duodenal homeobox 1 |
| PKT | Ptf1a-Cre;LSL-KrasG12D/+;Tgfbr2flox/flox |
| PODXL | Podocalyxin |
| PSCs | Pancreatic stellate cells |
| PTCH | Patched |
| PTF1α | Pancreas associated transcription factor 1-alpha |
| RCP | Rab-coupling protein |
| ROS | Reactive oxygen species |
| SAA3 | Serum amyloid A3 |
| scRNA-seq | Single cell RNA-sequencing |
| SHH | Sonic hedgehog |
| SMAD4 | Mothers against decapentaplegic homolog 4 |
| SMO | Smoothened |
| SOCS1 | Suppressor of cytokine signaling 1 |
| ST2 | Suppression of tumorigenicity 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAMs | Tumor-associated macrophages |
| TCA | Tricarboxylic acid |
| TCR | T-cell receptor |
| TGF-β | Transforming growth factor-beta |
| TGFBI | Transforming growth factor-beta-induced |
| TGFBR2 | Transforming growth factor-beta receptor 2 |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor-alpha |
| Tregs | Regulatory T cells |
| VEGF | Vascular endothelial growth factor |
| YAP1 | Yes-associated protein 1 |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Maitra, A. Subtyping Pancreatic Cancer. Cancer Cell 2015, 28, 411–413. [Google Scholar] [CrossRef]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef]
- Gao, Z.; Wang, X.; Wu, K.; Zhao, Y.; Hu, G. Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/CXCR4 axis. Pancreatology 2010, 10, 186–193. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Lieubeau, B.; Garrigue, L.; Barbieux, I.; Meflah, K.; Gregoire, M. The role of transforming growth factor beta 1 in the fibroblastic reaction associated with rat colorectal tumor development. Cancer Res. 1994, 54, 6526–6532. [Google Scholar] [PubMed]
- Löhr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Müller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar] [PubMed]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.M.; Nguyen, M.; Barsky, S.H. Human breast carcinoma desmoplasia is PDGF initiated. Oncogene 2000, 19, 4337–4345. [Google Scholar] [CrossRef]
- Kahlert, C.; Kalluri, R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. (Berl.) 2013, 91, 431–437. [Google Scholar] [CrossRef]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef]
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Cardon, M.; Lefort, S.; Richardson, M.; Rigaill, G.; Parrini, M.C.; Lucchesi, C.; et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2010, 2, 211–230. [Google Scholar] [CrossRef]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. eLife 2019, 8. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef]
- Scarlett, C.J.; Colvin, E.K.; Pinese, M.; Chang, D.K.; Morey, A.L.; Musgrove, E.A.; Pajic, M.; Apte, M.; Henshall, S.M.; Sutherland, R.L.; et al. Recruitment and activation of pancreatic stellate cells from the bone marrow in pancreatic cancer: A model of tumor-host interaction. PLoS ONE 2011, 6, e26088. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Chen, Y.; LeBleu, V.S.; Carstens, J.L.; Sugimoto, H.; Zheng, X.; Malasi, S.; Saur, D.; Kalluri, R. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Vennin, C.; Melenec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637. [Google Scholar] [CrossRef]
- Djurec, M.; Grana, O.; Lee, A.; Troule, K.; Espinet, E.; Cabras, L.; Navas, C.; Blasco, M.T.; Martin-Diaz, L.; Burdiel, M.; et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E1147–E1156. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Novo, D.; Heath, N.; Mitchell, L.; Caligiuri, G.; MacFarlane, A.; Reijmer, D.; Charlton, L.; Knight, J.; Calka, M.; McGhee, E.; et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat. Commun. 2018, 9, 5069. [Google Scholar] [CrossRef] [PubMed]
- Wormann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Gorgulu, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193.e112. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef]
- Scholz, A.; Heinze, S.; Detjen, K.M.; Peters, M.; Welzel, M.; Hauff, P.; Schirner, M.; Wiedenmann, B.; Rosewicz, S. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003, 125, 891–905. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 2016, 76, 5395–5404. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; McMillan, R.H.; Chang, Y.T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct Interactions With Cancer-Associated Fibroblasts Lead to Enhanced Pancreatic Cancer Stem Cell Function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically "fertilize" the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.H.; Fadare, O.; Pizzo, D.P.; et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef]
- Pavlides, S.; Tsirigos, A.; Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010, 9, 3485–3505. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezene, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; Del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2017, 23, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Vander Heiden, M.G.; et al. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res. 2019, 79, 5723–5733. [Google Scholar] [CrossRef]
- Sandforth, L.; Ammar, N.; Dinges, L.A.; Rocken, C.; Arlt, A.; Sebens, S.; Schafer, H. Impact of the Monocarboxylate Transporter-1 (MCT1)-Mediated Cellular Import of Lactate on Stemness Properties of Human Pancreatic Adenocarcinoma Cells dagger. Cancers 2020, 12, 581. [Google Scholar] [CrossRef]
- Foucher, E.D.; Ghigo, C.; Chouaib, S.; Galon, J.; Iovanna, J.; Olive, D. Pancreatic Ductal Adenocarcinoma: A Strong Imbalance of Good and Bad Immunological Cops in the Tumor Microenvironment. Front. Immunol. 2018, 9, 1044. [Google Scholar] [CrossRef]
- Das, S.; Berlin, J.; Cardin, D. Harnessing the Immune System in Pancreatic Cancer. Curr. Treat. Options Oncol. 2018, 19, 48. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Bleul, C.C.; Fuhlbrigge, R.C.; Casasnovas, J.M.; Aiuti, A.; Springer, T.A. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 1996, 184, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Mazur, A.; Holthoff, E.; Vadali, S.; Kelly, T.; Post, S.R. Cleavage of Type I Collagen by Fibroblast Activation Protein-α Enhances Class A Scavenger Receptor Mediated Macrophage Adhesion. PLoS ONE 2016, 11, e0150287. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Phillips, M.; Alice, A.; Bambina, S.; Zebertavage, L.; Friedman, D.; Cottam, B.; Newell, P.; et al. Blockade of fibroblast activation protein in combination with radiation treatment in murine models of pancreatic adenocarcinoma. PLoS ONE 2019, 14, e0211117. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Yue, W.; Lattime, E.C.; Stein, M.N.; Xu, Q.; Tan, X.L. Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget 2016, 7, 50735–50754. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Andersson, P.; Yang, Y.; Hosaka, K.; Zhang, Y.; Fischer, C.; Braun, H.; Liu, S.; Yu, G.; Liu, S.; Beyaert, R.; et al. Molecular mechanisms of IL-33-mediated stromal interactions in cancer metastasis. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Barnden, M.J.; Allison, J.; Heath, W.R.; Carbone, F.R. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 1998, 76, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Goehrig, D.; Nigri, J.; Samain, R.; Wu, Z.; Cappello, P.; Gabiane, G.; Zhang, X.; Zhao, Y.; Kim, I.S.; Chanal, M.; et al. Stromal protein βig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut 2019, 68, 693–707. [Google Scholar] [CrossRef] [PubMed]
- McClanahan, F.; Riches, J.C.; Miller, S.; Day, W.P.; Kotsiou, E.; Neuberg, D.; Croce, C.M.; Capasso, M.; Gribben, J.G. Mechanisms of PD-L1/PD-1-mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Eµ-TCL1 CLL mouse model. Blood 2015, 126, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Callahan, M.K.; Wolchok, J.D. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 41–53. [Google Scholar] [CrossRef]
- Mucciolo, G.; Roux, C.; Scagliotti, A.; Brugiapaglia, S.; Novelli, F.; Cappello, P. The dark side of immunotherapy: Pancreatic cancer. Cancer Drug Resist. 2020, 3. [Google Scholar] [CrossRef]
- Hilmi, M.; Bartholin, L.; Neuzillet, C. Immune therapies in pancreatic ductal adenocarcinoma: Where are we now? World J. Gastroenterol. 2018, 24, 2137–2151. [Google Scholar] [CrossRef]
- Inman, K.S.; Francis, A.A.; Murray, N.R. Complex role for the immune system in initiation and progression of pancreatic cancer. World J. Gastroenterol. 2014, 20, 11160–11181. [Google Scholar] [CrossRef]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Turtoi, A.; Musmeci, D.; Wang, Y.; Dumont, B.; Somja, J.; Bevilacqua, G.; De Pauw, E.; Delvenne, P.; Castronovo, V. Identification of novel accessible proteins bearing diagnostic and therapeutic potential in human pancreatic ductal adenocarcinoma. J. Proteome Res. 2011, 10, 4302–4313. [Google Scholar] [CrossRef] [PubMed]
- Hogquist, K.A.; Jameson, S.C.; Heath, W.R.; Howard, J.L.; Bevan, M.J.; Carbone, F.R. T cell receptor antagonist peptides induce positive selection. Cell 1994, 76, 17–27. [Google Scholar] [CrossRef]
- Nyakeriga, A.M.; Garg, H.; Joshi, A. TCR-induced T cell activation leads to simultaneous phosphorylation at Y505 and Y394 of p56(lck) residues. Cytom. A 2012, 81, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.B.; Pothula, S.P.; Xu, Z.; Lee, A.K.; Goldstein, D.; Pirola, R.C.; Apte, M.V.; Wilson, J.S. The role of the hepatocyte growth factor/c-MET pathway in pancreatic stellate cell-endothelial cell interactions: Antiangiogenic implications in pancreatic cancer. Carcinogenesis 2014, 35, 1891–1900. [Google Scholar] [CrossRef]
- Pausch, T.M.; Aue, E.; Wirsik, N.M.; Freire Valls, A.; Shen, Y.; Radhakrishnan, P.; Hackert, T.; Schneider, M.; Schmidt, T. Metastasis-associated fibroblasts promote angiogenesis in metastasized pancreatic cancer via the CXCL8 and the CCL2 axes. Sci. Rep. 2020, 10, 5420. [Google Scholar] [CrossRef]
- Watanabe, K.; Hasegawa, Y.; Yamashita, H.; Shimizu, K.; Ding, Y.; Abe, M.; Ohta, H.; Imagawa, K.; Hojo, K.; Maki, H.; et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J. Clin. Investig. 2004, 114, 898–907. [Google Scholar] [CrossRef]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef]
- Bakst, R.L.; Wong, R.J. Mechanisms of Perineural Invasion. J. Neurol. Surg. B Skull Base 2016, 77, 96–106. [Google Scholar] [CrossRef]
- Nan, L.; Qin, T.; Xiao, Y.; Qian, W.; Li, J.; Wang, Z.; Ma, J.; Ma, Q.; Wu, Z. Pancreatic Stellate Cells Facilitate Perineural Invasion of Pancreatic Cancer via HGF/c-Met Pathway. Cell Transplant. 2019, 28, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Friess, H.; DiMola, F.F.; Zimmermann, A.; Graber, H.U.; Korc, M.; Büchler, M.W. Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. J. Clin. Oncol. 1999, 17, 2419–2428. [Google Scholar] [CrossRef] [PubMed]
- Bapat, A.A.; Munoz, R.M.; Von Hoff, D.D.; Han, H. Blocking Nerve Growth Factor Signaling Reduces the Neural Invasion Potential of Pancreatic Cancer Cells. PLoS ONE 2016, 11, e0165586. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2015, 28, 831–833. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Pasca di Magliano, M.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Van Mackelenbergh, M.G.; Stroes, C.I.; Spijker, R.; Van Eijck, C.H.J.; Wilmink, J.W.; Bijlsma, M.F.; Van Laarhoven, H.W.M. Clinical Trials Targeting the Stroma in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Cancers (Basel) 2019, 11, 588. [Google Scholar] [CrossRef]
- Torphy, R.J.; Wang, Z.; True-Yasaki, A.; Volmar, K.E.; Rashid, N.; Yeh, B.; Anderson, J.M.; Johansen, J.S.; Hollingsworth, M.A.; Yeh, J.J.; et al. Stromal Content Is Correlated with Tissue Site, Contrast Retention, and Survival in Pancreatic Adenocarcinoma. JCO Precis. Oncol. 2018. [Google Scholar] [CrossRef]
- Wang, J.; Chan, D.K.W.; Sen, A.; Ma, W.W.; Straubinger, R.M. Tumor Priming by SMO Inhibition Enhances Antibody Delivery and Efficacy in a Pancreatic Ductal Adenocarcinoma Model. Mol. Cancer Ther. 2019, 18, 2074–2084. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Semaan, A.; Huang, J.; San Lucas, F.A.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019, 25, 2194–2205. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Muller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Pasca Di Magliano, M.; Welling, T.H.; et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Sakaguchi, M.; Maruyama, S.; Iioka, H.; Putranto, E.W.; Sumardika, I.W.; Tomonobu, N.; Kawasaki, T.; Homma, K.; Kondo, E. Stromal mesenchymal stem cells facilitate pancreatic cancer progression by regulating specific secretory molecules through mutual cellular interaction. J. Cancer 2018, 9, 2916–2929. [Google Scholar] [CrossRef]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; Di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sperb, N.; Tsesmelis, M.; Wirth, T. Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 5486. https://doi.org/10.3390/ijms21155486
Sperb N, Tsesmelis M, Wirth T. Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2020; 21(15):5486. https://doi.org/10.3390/ijms21155486
Chicago/Turabian StyleSperb, Nadine, Miltiadis Tsesmelis, and Thomas Wirth. 2020. "Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 21, no. 15: 5486. https://doi.org/10.3390/ijms21155486
APA StyleSperb, N., Tsesmelis, M., & Wirth, T. (2020). Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences, 21(15), 5486. https://doi.org/10.3390/ijms21155486