Bioinformatics and Molecular Insights to Anti-Metastasis Activity of Triethylene Glycol Derivatives

 ,
, 

Abstract
1. Introduction
2. Results
2.1. TEG Derivatives Inhibited the Migration and Invasion Potential of Cancer Cells
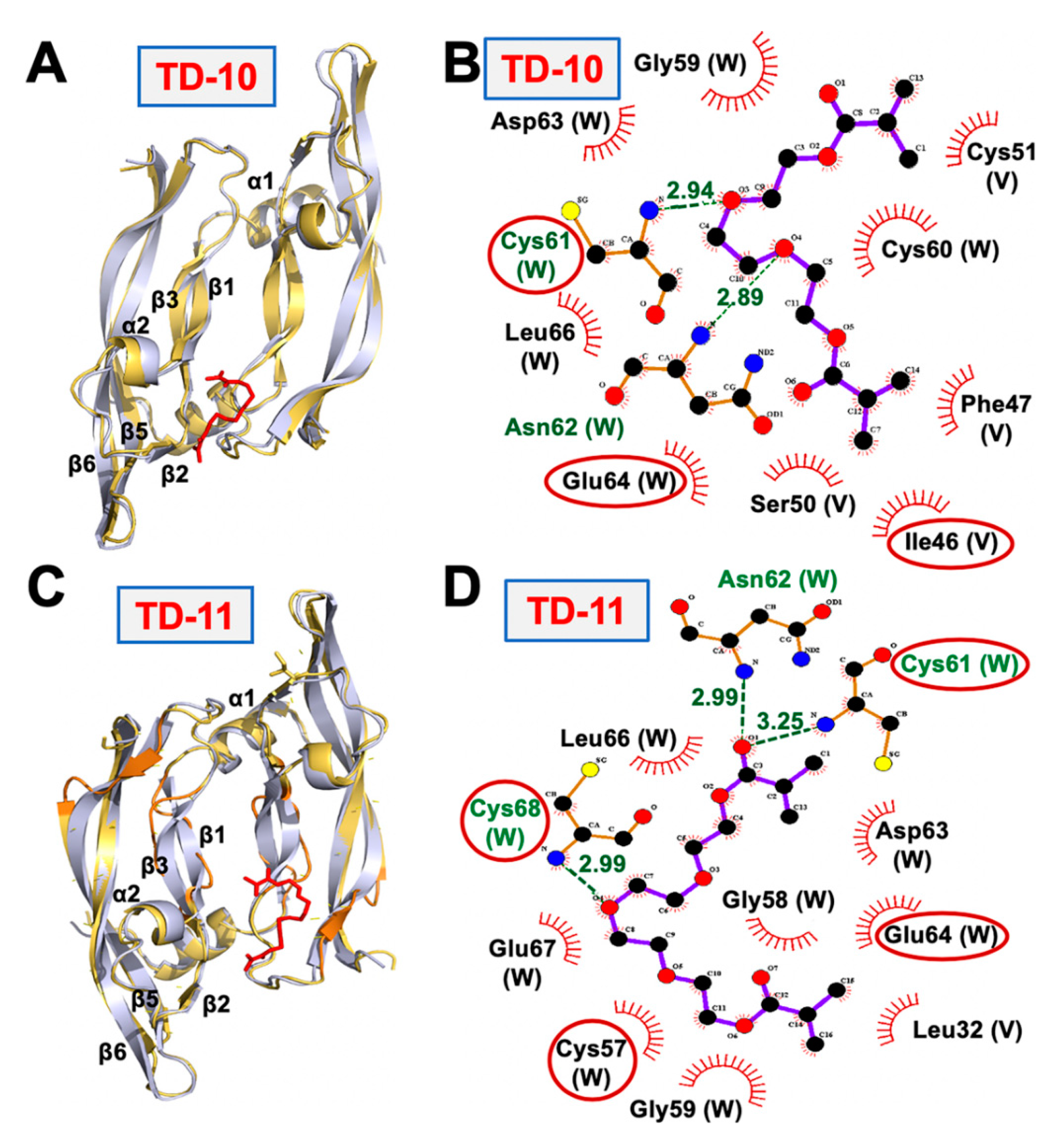
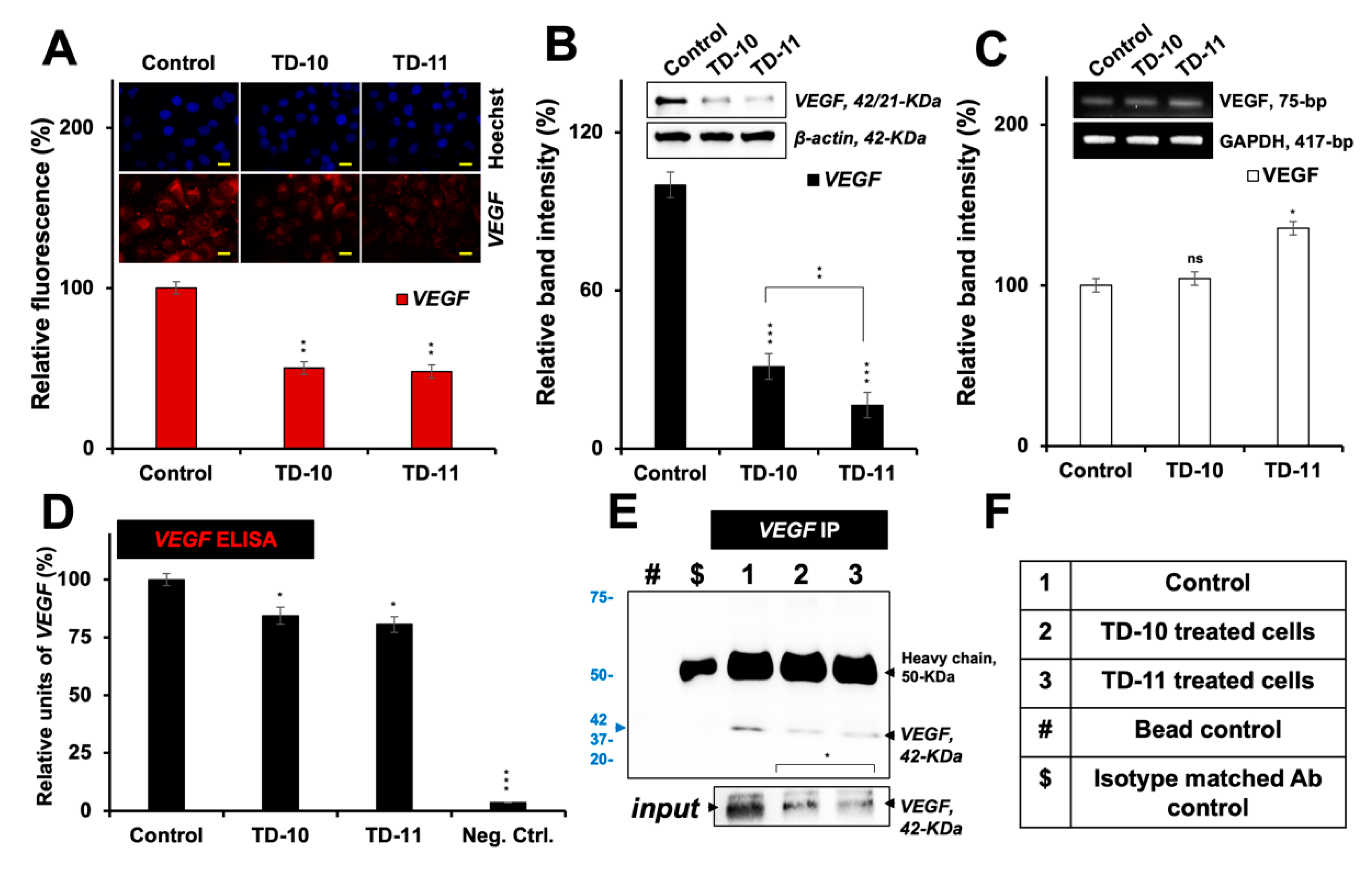
2.2. Molecular Docking and Experimental Analysis of the Effect of TD-10 and TD-11 on VEGFA-VEGFR Complexes
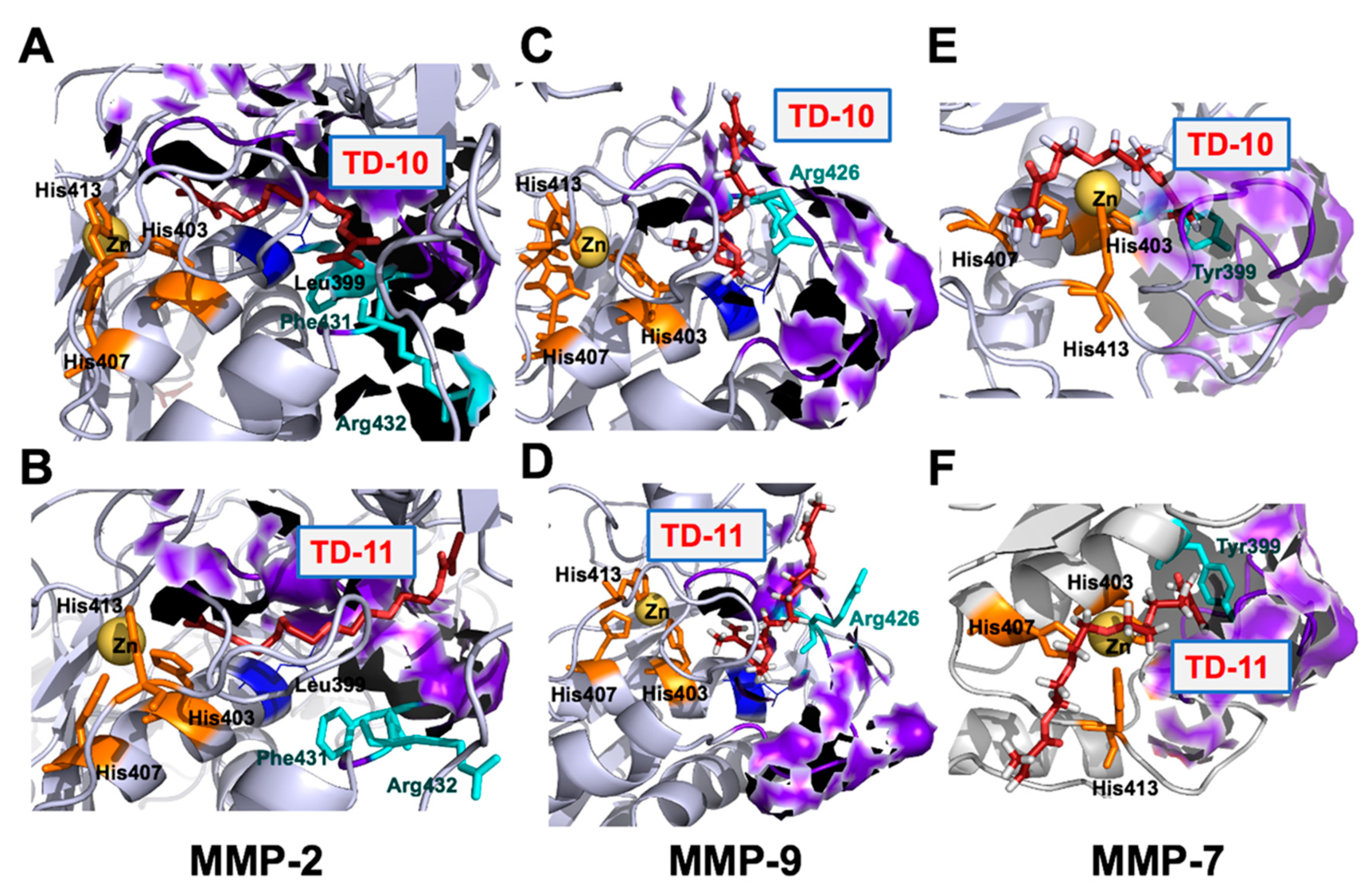
2.3. Molecular Docking and Experimental Analyses of Interactions of TD-10 and TD-11 with MMP-2, MMP-7 and MMP-9
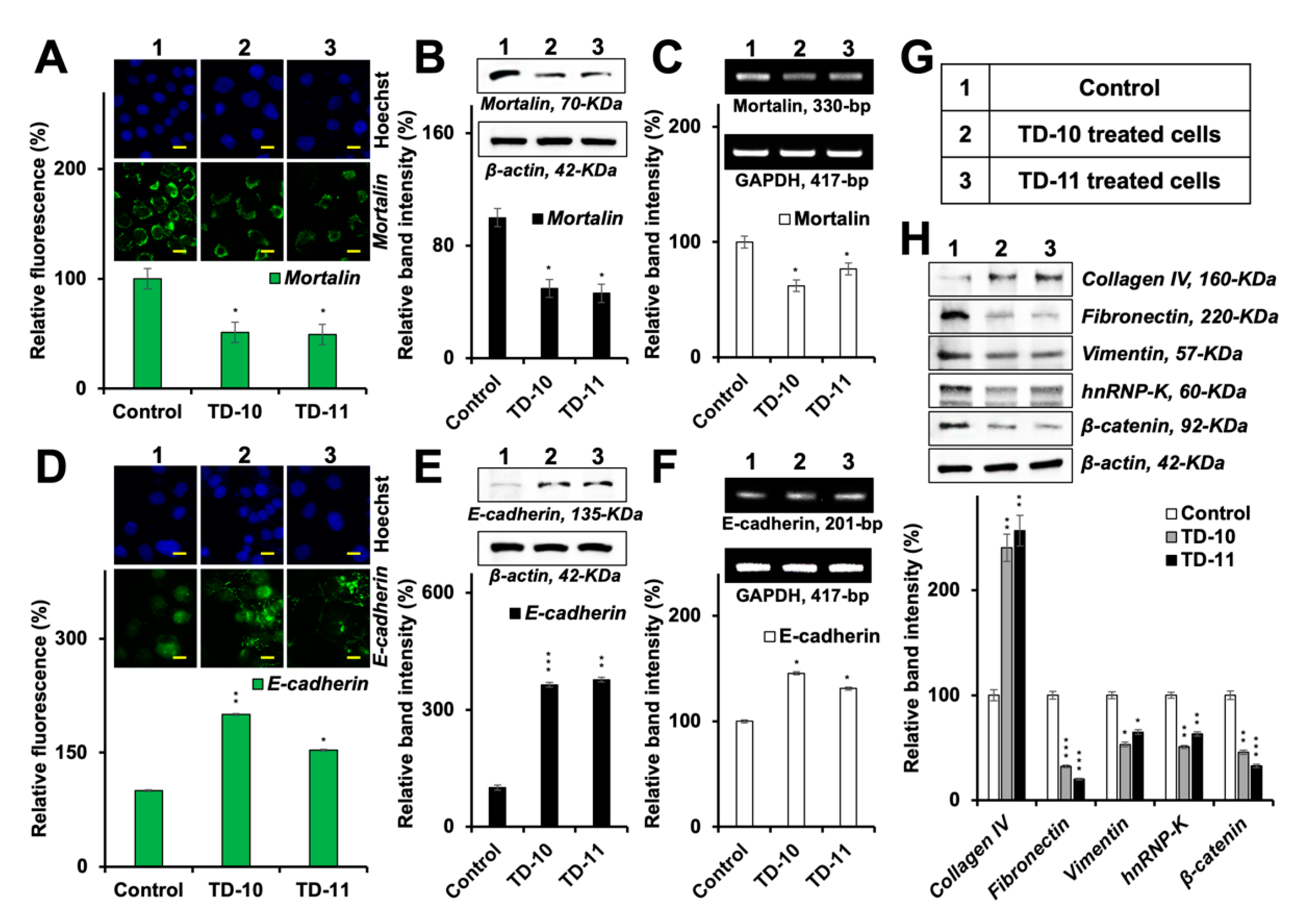
2.4. Molecular Docking and Experimental Analyses of Interactions of TD-10 and TD-11 with Vimentin and Mortalin
3. Discussion
4. Materials and Methods
4.1. Cell Line and Reagents
4.2. Dose Titration
4.3. Cell Migration and Invasion Assays
4.4. Molecular Docking and Simulations to Check the Effect of TD-10 and TD-11 with MMPs (MMP-2, MMP-7 and MMP-9) Family of Protein and VEGFA Protein and VEGFA-VEGFR-1 Complex
4.5. Immunostaining and Cell Congregation Analyses
4.6. Western Blotting
4.7. RT-PCR
4.8. VEGF Analyses
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EMT | Epithelial-to-mesenchymal transformation |
| MMP | Matrix metalloprotease |
| TD-10 | Triethylene glycol dimethacrylate-10 |
| TD-11 | Triethylene glycol dimethacrylate-11 |
| TEG | Triethylene glycol |
| VEGF | Vascular endothelial growth factor |
References
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental Determinants of Pancreatic Cancer. Physiol. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Hasanzadeh, M. Cell-specific frequency as a new hallmark to early detection of cancer and efficient therapy: Recording of cancer voice as a new horizon. Biomed. Pharmacothe. 2020, 122, 109770. [Google Scholar] [CrossRef]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer. Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Hua, W.; Ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFbeta-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell. Mol. Life. Sci. 2019, 77, 2103–2123. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef]
- Jaszai, J.; Schmidt, M.H.H. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells 2019, 8, 1102. [Google Scholar] [CrossRef]
- Jeltsch, M.; Leppanen, V.M.; Saharinen, P.; Alitalo, K. Receptor tyrosine kinase-mediated angiogenesis. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [PubMed]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Herrmann, S.M.S.; Herrmann, J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J. Am. Soc. Hypertens. 2018, 12, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Loureiro, R.M.; D’Amore, P.A. Transcriptional regulation of vascular endothelial growth factor in cancer. Cytokine Growth Factor Rev. 2005, 16, 77–89. [Google Scholar] [CrossRef]
- Bao, P.; Kodra, A.; Tomic-Canic, M.; Golinko, M.S.; Ehrlich, H.P.; Brem, H. The role of vascular endothelial growth factor in wound healing. J. Surg. Res. 2009, 153, 347–358. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. S3), 4–10. [Google Scholar] [CrossRef]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef]
- Ansari, A.M.; Shaikh, S.; Muteeb, G.; Mohd, S.; Rizvi, D.; Shakil, S.; Alam, A.; Tripathi, R.; Ghazal, F.; Regman, A.; et al. Role of Matrix Metalloproteinases in Cancer. In Advances in Protein Chemistry; Ashraf, M.G., Sheikh, A.I., Eds.; OMICS: Foster City, CA, USA, 2013; pp. 1–19. [Google Scholar]
- Quintero-Fabian, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argaez, V.; Lara-Riegos, J.; Ramirez-Camacho, M.A.; Alvarez-Sanchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef]
- Huang, S.T.; Chang, C.C.; Pang, J.S.; Huang, H.S.; Chou, S.C.; Kao, M.C.; You, H.L. Drynaria fortunei Promoted Angiogenesis Associated With Modified MMP-2/TIMP-2 Balance and Activation of VEGF Ligand/Receptors Expression. Front. Pharmacol. 2018, 9, 979. [Google Scholar] [CrossRef] [PubMed]
- Manicone, A.M.; McGuire, J.K. Matrix metalloproteinases as modulators of inflammation. Semin. Cell Dev. Biol. 2008, 19, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour Cell Secretome in Chemoresistance and Tumour Recurrence. Trends Cancer 2020, 6, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Mitschke, J.; Burk, U.C.; Reinheckel, T. The role of proteases in epithelial-to-mesenchymal cell transitions in cancer. Cancer Metastasis Rev. 2019, 38, 431–444. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Huang, J.; Liu, Y. Matrix Metalloproteinase 2 Knockdown Suppresses the Proliferation of HepG2 and Huh7 Cells and Enhances the Cisplatin Effect. Open Med. 2019, 14, 384–391. [Google Scholar] [CrossRef]
- Yuan, S.; Lin, L.S.; Gan, R.H.; Huang, L.; Wu, X.T.; Zhao, Y.; Su, B.H.; Zheng, D.; Lu, Y.G. Elevated matrix metalloproteinase 7 expression promotes the proliferation, motility and metastasis of tongue squamous cell carcinoma. BMC Cancer 2020, 20, 33. [Google Scholar] [CrossRef]
- Zhu, L.; Zheng, X.; Du, Y.; Xing, Y.; Xu, K.; Cui, L. Matrix metalloproteinase-7 may serve as a novel biomarker for cervical cancer. OncoTargets Ther. 2018, 11, 4207–4220. [Google Scholar] [CrossRef]
- Hawinkels, L.J.; Zuidwijk, K.; Verspaget, H.W.; de Jonge-Muller, E.S.; van Duijn, W.; Ferreira, V.; Fontijn, R.D.; David, G.; Hommes, D.W.; Lamers, C.B.; et al. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur. J. Cancer 2008, 44, 1904–1913. [Google Scholar] [CrossRef]
- Webb, A.H.; Gao, B.T.; Goldsmith, Z.K.; Irvine, A.S.; Saleh, N.; Lee, R.P.; Lendermon, J.B.; Bheemreddy, R.; Zhang, Q.; Brennan, R.C.; et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer 2017, 17, 434. [Google Scholar] [CrossRef]
- Li, H.; Qiu, Z.; Li, F.; Wang, C. The relationship between MMP-2 and MMP-9 expression levels with breast cancer incidence and prognosis. Oncol. Lett. 2017, 14, 5865–5870. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Young, D.A.; Barter, M.J.; Wilkinson, D.J. Recent advances in understanding the regulation of metalloproteinases. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Krzeski, P.; Buckland-Wright, C.; Balint, G.; Cline, G.A.; Stoner, K.; Lyon, R.; Beary, J.; Aronstein, W.S.; Spector, T.D. Development of musculoskeletal toxicity without clear benefit after administration of PG-116800, a matrix metalloproteinase inhibitor, to patients with knee osteoarthritis: A randomized, 12-month, double-blind, placebo-controlled study. Arthritis Res. Ther. 2007, 9, R109. [Google Scholar] [CrossRef]
- Bloomston, M.; Zervos, E.E.; Rosemurgy, A.S., 2nd. Matrix metalloproteinases and their role in pancreatic cancer: A review of preclinical studies and clinical trials. Ann. Surg. Oncol. 2002, 9, 668–674. [Google Scholar] [CrossRef]
- Jakubowska, K.; Pryczynicz, A.; Januszewska, J.; Sidorkiewicz, I.; Kemona, A.; Niewinski, A.; Lewczuk, L.; Kedra, B.; Guzinska-Ustymowicz, K. Expressions of Matrix Metalloproteinases 2, 7, and 9 in Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Dis. Markers 2016, 2016, 9895721. [Google Scholar] [CrossRef]
- Oh, E.; Garg, S.; Liu, Y.; Afzal, S.; Gao, R.; Yun, C.O.; Kaul, S.C.; Wadhwa, R. Identification and Functional Characterization of Anti-metastasis and Anti-angiogenic Activities of Triethylene Glycol Derivatives. Front. Oncol. 2018, 8, 552. [Google Scholar] [CrossRef]
- Muller, Y.A.; Christinger, H.W.; Keyt, B.A.; de Vos, A.M. The crystal structure of vascular endothelial growth factor (VEGF) refined to 1.93 A resolution: Multiple copy flexibility and receptor binding. Structure 1997, 5, 1325–1338. [Google Scholar] [CrossRef]
- Muller, Y.A.; Li, B.; Christinger, H.W.; Wells, J.A.; Cunningham, B.C.; de Vos, A.M. Vascular endothelial growth factor: Crystal structure and functional mapping of the kinase domain receptor binding site. Proc. Natl. Acad. Sci. USA 1997, 94, 7192–7197. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Fabre, B.; Ramos, A.; de Pascual-Teresa, B. Targeting matrix metalloproteinases: Exploring the dynamics of the s1’ pocket in the design of selective, small molecule inhibitors. J. Med. Chem. 2014, 57, 10205–10219. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Shah, N.; Lee, J.S.; Katiyar, S.P.; Li, L.; Oh, E.; Sundar, D.; Yun, C.O.; Wadhwa, R.; Kaul, S.C. Withanone-rich combination of Ashwagandha withanolides restricts metastasis and angiogenesis through hnRNP-K. Mol. Cancer. Ther. 2014, 13, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Kaus-Drobek, M.; Mucke, N.; Szczepanowski, R.H.; Wedig, T.; Czarnocki-Cieciura, M.; Polakowska, M.; Herrmann, H.; Wyslouch-Cieszynska, A.; Dadlez, M. Vimentin S-glutathionylation at Cys328 inhibits filament elongation and induces severing of mature filaments in vitro. FEBS J. 2020. [Google Scholar] [CrossRef]
- Na, Y.; Kaul, S.C.; Ryu, J.; Lee, J.S.; Ahn, H.M.; Kaul, Z.; Kalra, R.S.; Li, L.; Widodo, N.; Yun, C.O.; et al. Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 2016, 76, 2754–2765. [Google Scholar] [CrossRef]
- Collins, C.; Denisin, A.K.; Pruitt, B.L.; Nelson, W.J. Changes in E-cadherin rigidity sensing regulate cell adhesion. Proc. Natl. Acad. Sci. USA 2017, 114, E5835–E5844. [Google Scholar] [CrossRef]
- Gloushankova, N.A.; Rubtsova, S.N.; Zhitnyak, I.Y. Cadherin-mediated cell-cell interactions in normal and cancer cells. Tissue Barriers 2017, 5, e1356900. [Google Scholar] [CrossRef]
- Haq, M.; Shaeii, A.E.; Zervos, E.E.; Rosemurgy, A.S. In vitro and in vivo matrix metalloproteinase production by pancreatic cancer cells and by distant organs. Int. J. Surg. Investig. 2000, 1, 459–465. [Google Scholar]
- Itakura, J.; Ishiwata, T.; Friess, H.; Fujii, H.; Matsumoto, Y.; Buchler, M.W.; Korc, M. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clin. Cancer Res. 1997, 3, 1309–1316. [Google Scholar]
- Wu, P.K.; Hong, S.K.; Park, J.I. Steady-State Levels of Phosphorylated Mitogen-Activated Protein Kinase Kinase 1/2 Determined by Mortalin/HSPA9 and Protein Phosphatase 1 Alpha in KRAS and BRAF Tumor Cells. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Muramatsu, T.; Tanabe, M.; Inazawa, J. Identification and characterization of transforming growth factor beta-induced in circulating tumor cell subline from pancreatic cancer cell line. Cancer Sci. 2018, 109, 3623–3633. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, Y.; Tang, T.; Zhu, Q.; Shi, W.; Yin, X.; Xing, Y.; Shen, Y.; Pan, Y.; Jin, L. Transcriptome Profiling of Panc-1 Spheroid Cells with Pancreatic Cancer Stem Cells Properties Cultured by a Novel 3D Semi-Solid System. Cell. Physiol. Biochem. 2018, 47, 2109–2125. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Cai, J.B.; Dong, R.Z.; Liu, L.X.; Zhang, C.; Zhang, P.F.; Zou, H.; Xie, N.; Zhang, L.; Zhang, X.Y.; et al. Mortalin promotes cell proliferation and epithelial mesenchymal transition of intrahepatic cholangiocarcinoma cells in vitro. J. Clin. Pathol. 2017, 70, 677–683. [Google Scholar] [CrossRef]
- Kenny, H.A.; Kaur, S.; Coussens, L.M.; Lengyel, E. The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J. Clin. Investig. 2008, 118, 1367–1379. [Google Scholar] [CrossRef]
- Zheng, G.; Lyons, J.G.; Tan, T.K.; Wang, Y.; Hsu, T.T.; Min, D.; Succar, L.; Rangan, G.K.; Hu, M.; Henderson, B.R.; et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am. J. Pathol. 2009, 175, 580–591. [Google Scholar] [CrossRef]
- Cowden Dahl, K.D.; Symowicz, J.; Ning, Y.; Gutierrez, E.; Fishman, D.A.; Adley, B.P.; Stack, M.S.; Hudson, L.G. Matrix metalloproteinase 9 is a mediator of epidermal growth factor-dependent e-cadherin loss in ovarian carcinoma cells. Cancer Res. 2008, 68, 4606–4613. [Google Scholar] [CrossRef]
- Han, S.; Han, L.; Sun, H.; Zan, X.; Zhou, Z.; Xu, K.; Yao, Y.; Liu, Q. Kruppellike factor expression and correlation with FAK, MMP9 and Ecadherin expression in hepatocellular carcinoma. Mol. Med. Rep. 2013, 8, 81–88. [Google Scholar] [CrossRef][Green Version]
- Maity, G.; Fahreen, S.; Banerji, A.; Roy Choudhury, P.; Sen, T.; Dutta, A.; Chatterjee, A. Fibronectin-integrin mediated signaling in human cervical cancer cells (SiHa). Mol. Cell. Biochem. 2010, 336, 65–74. [Google Scholar] [CrossRef]
- Pal, S.; Ganguly, K.K.; Chatterjee, A. Extracellular matrix protein fibronectin induces matrix metalloproteinases in human prostate adenocarcinoma cells PC-3. Cell Commun. Adhes. 2013, 20, 105–114. [Google Scholar] [CrossRef]
- Sen, T.; Dutta, A.; Maity, G.; Chatterjee, A. Fibronectin induces matrix metalloproteinase-9 (MMP-9) in human laryngeal carcinoma cells by involving multiple signaling pathways. Biochimie 2010, 92, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, F.; Peterson, M.K.; Caldeira Araujo, H.; Lautenschlager, F.; Gad, A.K.B. Vimentin Diversity in Health and Disease. Cells 2018, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Vinnakota, K.; Zhang, Y.; Selvanesan, B.C.; Topi, G.; Salim, T.; Sand-Dejmek, J.; Jonsson, G.; Sjolander, A. M2-like macrophages induce colon cancer cell invasion via matrix metalloproteinases. J. Cell. Physiol. 2017, 232, 3468–3480. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Yano, K.; Sugimoto, S.; Ishii, G.; Hasebe, T.; Endoh, Y.; Kodama, K.; Goya, M.; Chiba, T.; Ochiai, A. Matrix metalloproteinase-7 facilitates insulin-like growth factor bioavailability through its proteinase activity on insulin-like growth factor binding protein 3. Cancer Res. 2004, 64, 665–671. [Google Scholar] [CrossRef]
- Mok, K.W.; Mruk, D.D.; Cheng, C.Y. rpS6 regulates blood-testis barrier dynamics through Akt-mediated effects on MMP-9. J. Cell Sci. 2014, 127, 4870–4882. [Google Scholar] [CrossRef]
- Lynch, C.C.; Vargo-Gogola, T.; Matrisian, L.M.; Fingleton, B. Cleavage of E-Cadherin by Matrix Metalloproteinase-7 Promotes Cellular Proliferation in Nontransformed Cell Lines via Activation of RhoA. J. Oncol. 2010, 2010, 530745. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Pan, S.; Shang, S.; Li, C. Interaction between the Wnt/beta-catenin signaling pathway and the EMMPRIN/MMP-2, 9 route in periodontitis. J. Periodontal. Res. 2018, 53, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Belotti, D.; Paganoni, P.; Manenti, L.; Garofalo, A.; Marchini, S.; Taraboletti, G.; Giavazzi, R. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: Implications for ascites formation. Cancer Res. 2003, 63, 5224–5229. [Google Scholar] [PubMed]
- Ghosh, S.; Basu, M.; Roy, S.S. ETS-1 protein regulates vascular endothelial growth factor-induced matrix metalloproteinase-9 and matrix metalloproteinase-13 expression in human ovarian carcinoma cell line SKOV-3. J. Biol. Chem. 2012, 287, 15001–15015. [Google Scholar] [CrossRef]
- Wadhwa, R.; Kaul, S.C.; Ikawa, Y.; Sugimoto, Y. Identification of a novel member of mouse hsp70 family. Its association with cellular mortal phenotype. J. Biol. Chem. 1993, 268, 6615–6621. [Google Scholar]
- Afzal, S.; Garg, S.; Adiga, D.; Ishida, Y.; Terao, K.; Kaul, S.C.; Wadhwa, R. Anti-stress, Glial- and Neuro-differentiation Potential of Resveratrol: Characterization by Cellular, Biochemical and Imaging Assays. Nutrients 2020, 12, 671. [Google Scholar] [CrossRef] [PubMed]
- Edman, K.; Furber, M.; Hemsley, P.; Johansson, C.; Pairaudeau, G.; Petersen, J.; Stocks, M.; Tervo, A.; Ward, A.; Wells, E.; et al. The discovery of MMP7 inhibitors exploiting a novel selectivity trigger. ChemMedChem 2011, 6, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Morgunova, E.; Tuuttila, A.; Bergmann, U.; Isupov, M.; Lindqvist, Y.; Schneider, G.; Tryggvason, K. Structure of human pro-matrix metalloproteinase-2: Activation mechanism revealed. Science 1999, 284, 1667–1670. [Google Scholar] [CrossRef]
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.; Zhang, Y.M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974. [Google Scholar] [CrossRef] [PubMed]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the Full-length VEGFR-1 Extracellular Domain in Complex with VEGF-A. Structure 2017, 25, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Schrödinger, Release 2018-4: Glide, Desmond; Schrödinger Inc.: New York, NY, USA, 2020.
- Backe, P.H.; Messias, A.C.; Ravelli, R.B.; Sattler, M.; Cusack, S. X-ray crystallographic and NMR studies of the third KH domain of hnRNP K in complex with single-stranded nucleic acids. Structure 2005, 13, 1055–1067. [Google Scholar] [CrossRef]
- Qin, Z.; Kreplak, L.; Buehler, M.J. Hierarchical structure controls nanomechanical properties of vimentin intermediate filaments. PLoS ONE 2009, 4, e7294. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Omichinski, J.G.; Sakaguchi, K.; Zambrano, N.; Sakamoto, H.; Appella, E.; Gronenborn, A.M. High-resolution structure of the oligomerization domain of p53 by multidimensional NMR. Science 1994, 265, 386–391. [Google Scholar] [CrossRef]
- Amick, J.; Schlanger, S.E.; Wachnowsky, C.; Moseng, M.A.; Emerson, C.C.; Dare, M.; Luo, W.I.; Ithychanda, S.S.; Nix, J.C.; Cowan, J.A.; et al. Crystal structure of the nucleotide-binding domain of mortalin, the mitochondrial Hsp70 chaperone. Protein Sci. Publ. Protein Soc. 2014, 23, 833–842. [Google Scholar] [CrossRef]
- De Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC.: New York, NY, USA, 2017.
- Lu, W.J.; Lee, N.P.; Kaul, S.C.; Lan, F.; Poon, R.T.; Wadhwa, R.; Luk, J.M. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011, 18, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | MMP-2 | MMP-9 | MMP-7 | |||
|---|---|---|---|---|---|---|
| TD-10 | TD-11 | TD-10 | TD-11 | TD-10 | TD-11 | |
| Docking Score (kCal/mol) | −5.80 | −7.78 | −5.70 | −5.81 | −4.98 | −6.15 |
| MM-GBSA Binding energy (kCal/mol) | −63.55 | −74.81 | −58.44 | −45.78 | −15.86 | −28.97 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, V.; Garg, S.; Afzal, S.; Dhanjal, J.K.; Yun, C.-O.; Kaul, S.C.; Sundar, D.; Wadhwa, R. Bioinformatics and Molecular Insights to Anti-Metastasis Activity of Triethylene Glycol Derivatives. Int. J. Mol. Sci. 2020, 21, 5463. https://doi.org/10.3390/ijms21155463
Malik V, Garg S, Afzal S, Dhanjal JK, Yun C-O, Kaul SC, Sundar D, Wadhwa R. Bioinformatics and Molecular Insights to Anti-Metastasis Activity of Triethylene Glycol Derivatives. International Journal of Molecular Sciences. 2020; 21(15):5463. https://doi.org/10.3390/ijms21155463
Chicago/Turabian StyleMalik, Vidhi, Sukant Garg, Sajal Afzal, Jaspreet Kaur Dhanjal, Chae-Ok Yun, Sunil C. Kaul, Durai Sundar, and Renu Wadhwa. 2020. "Bioinformatics and Molecular Insights to Anti-Metastasis Activity of Triethylene Glycol Derivatives" International Journal of Molecular Sciences 21, no. 15: 5463. https://doi.org/10.3390/ijms21155463
APA StyleMalik, V., Garg, S., Afzal, S., Dhanjal, J. K., Yun, C.-O., Kaul, S. C., Sundar, D., & Wadhwa, R. (2020). Bioinformatics and Molecular Insights to Anti-Metastasis Activity of Triethylene Glycol Derivatives. International Journal of Molecular Sciences, 21(15), 5463. https://doi.org/10.3390/ijms21155463