Irradiation-Induced Upregulation of miR-711 Inhibits DNA Repair and Promotes Neurodegeneration Pathways

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Anti-Apoptotic, DNA-Repair and Neuronal Marker Genes Are Downregulated while Pro-Apoptotic Genes, Pro-Senescence Genes and miR-711 Are Upregulated in the Cortex, Hippocampus and Purified Neurons after Brain Irradiation
2.2. IR Induces Activation of DNA-Damage and p53 Pathways in Primary Cortical Neurons
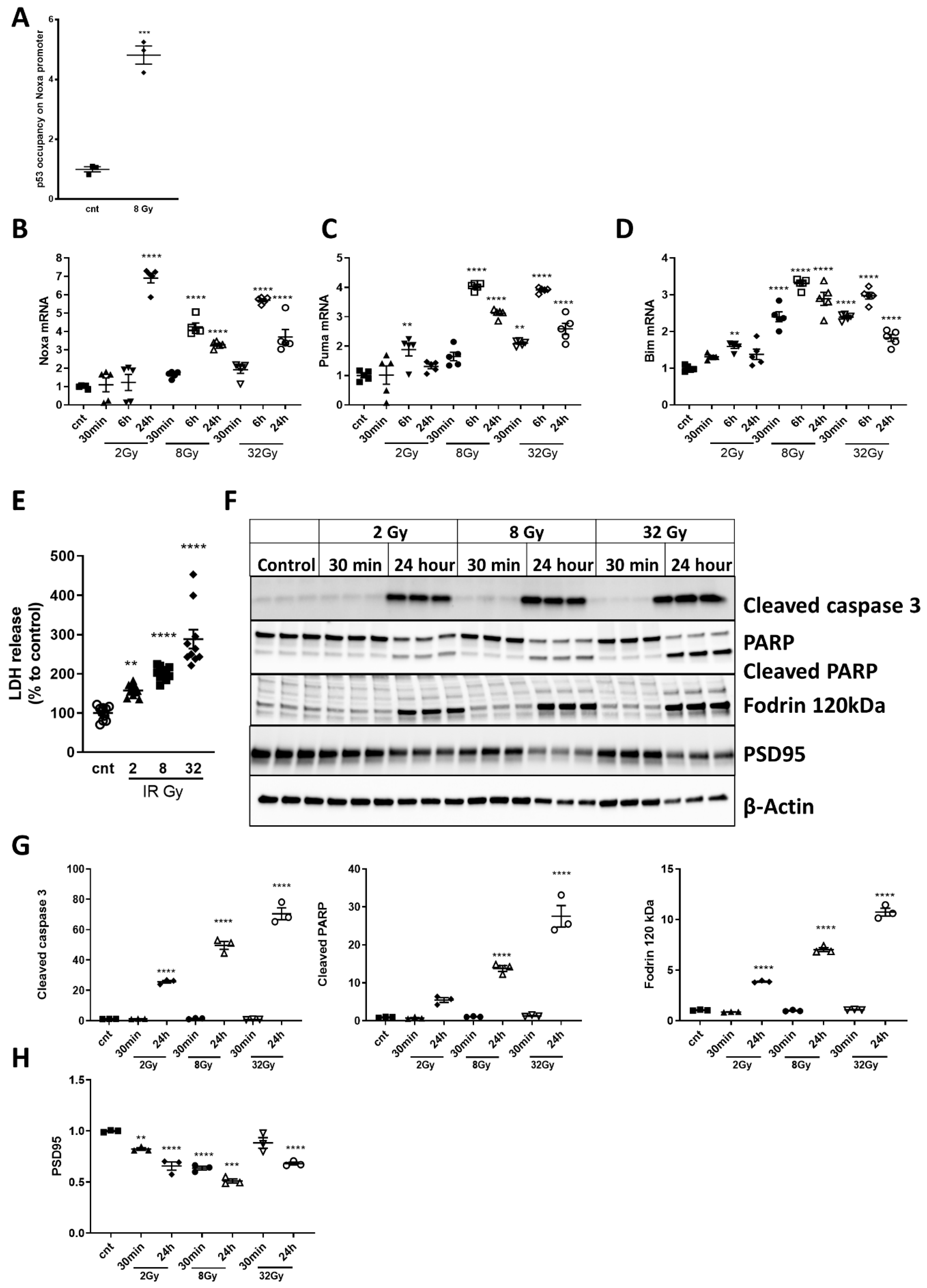
2.3. Noxa, Puma and Bim Are Upregulated after Irradiation
2.4. Irradiation Induces Dose-Dependent Neuronal Cell Death and Time- and Dose-Dependent Activation of Caspase-Dependent Apoptosis
2.5. Irradiation CausesUupregulation of miR-711 and Downregulation of Its Target Genes Expression
2.6. miR-711 Inhibition Attenuates IR-Induced Downregulation of Akt and Ang-1
2.7. miR-711 Inhibition Attenuates Intrinsic Apoptosis After Irradiation
2.8. miR-711 Inhibition Attenuates IR-Induced DNA Damage Markers, p53 Activation, and Neuronal Apoptosis and Senescence Markers
2.9. miR-711 Inhibition Attenuates Irradiation-Induced Downregulation of DNA Repair Molecules Rad50 and Rad54l2
2.10. Effect of miR-711 Inhibition on miR-23a-3p Expression
3. Discussion
4. Methods
4.1. Animals and Radiation Delivery and Quality Control
4.2. In Vitro Cell Culture
4.3. Immunohistochemistry
4.4. RCNs Irradiation
4.5. Cell Death Assays
4.6. RNA-Interacting Protein Immunoprecipitation (RIP) Using AGO2-Specific Antibodies
4.7. RNA Isolation
4.8. qPCR
4.9. miR Reverse Transcription and qPCR
4.10. Isolation of Neurons from the Mouse Brain and qPCR
4.11. ChIP Assay
4.12. Cell Lysates Preparation and Western Blot
4.13. Antibodies
4.14. Subcellular Fractionation
4.15. Statistical Analysis and General Methods
4.16. Experimental Setup
4.16.1. Experiment 1
4.16.2. Experiment 2
4.16.3. Experiment 3
4.16.4. Experiment 4
4.16.5. Experiment 5
4.16.6. Experiment 6
4.16.7. Experiment 7
4.16.8. Experiment 8
4.16.9. Experiment 9
4.16.10. Experiment 10
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alan Mitteer, R.; Wang, Y.; Shah, J.; Gordon, S.; Fager, M.; Butter, P.P.; Jun Kim, H.; Guardiola-Salmeron, C.; Carabe-Fernandez, A.; Fan, Y. Proton beam radiation induces DNA damage and cell apoptosis in glioma stem cells through reactive oxygen species. Sci. Rep. 2015, 5, 13961. [Google Scholar] [CrossRef] [PubMed]
- Gobbel, G.T.; Bellinzona, M.; Vogt, A.R.; Gupta, N.; Fike, J.R.; Chan, P.H. Response of postmitotic neurons to X-irradiation: Implications for the role of DNA damage in neuronal apoptosis. J. Neurosci. 1998, 18, 147–155. [Google Scholar] [CrossRef]
- Gobbel, G.T.; Chan, P.H. Neuronal death is an active, caspase-dependent process after moderate but not severe DNA damage. J. Neurochem. 2001, 76, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Pospisil, P.; Kazda, T.; Bulik, M.; Dobiaskova, M.; Burkon, P.; Hynkova, L.; Slampa, P.; Jancalek, R. Hippocampal proton MR spectroscopy as a novel approach in the assessment of radiation injury and the correlation to neurocognitive function impairment: Initial experiences. Radiat. Oncol. 2015, 10, 211. [Google Scholar] [CrossRef]
- Tikka, T.; Usenius, T.; Tenhunen, M.; Keinanen, R.; Koistinaho, J. Tetracycline derivatives and ceftriaxone, a cephalosporin antibiotic, protect neurons against apoptosis induced by ionizing radiation. J. Neurochem. 2001, 78, 1409–1414. [Google Scholar] [CrossRef]
- Li, L.; Wang, W.; Welford, S.; Zhang, T.; Wang, X.; Zhu, X. Ionizing radiation causes increased tau phosphorylation in primary neurons. J. Neurochem. 2014, 131, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Imperato, J.P.; Paleologos, N.A.; Vick, N.A. Effects of treatment on long-term survivors with malignant astrocytomas. Annals Neurol. 1990, 28, 818–822. [Google Scholar] [CrossRef]
- DeAngelis, L.M.; Delattre, J.Y.; Posner, J.B. Radiation-induced dementia in patients cured of brain metastases. Neurology 1989, 39, 789–796. [Google Scholar] [CrossRef]
- Feng, X.; Liu, S.; Chen, D.; Rosi, S.; Gupta, N. Rescue of cognitive function following fractionated brain irradiation in a novel preclinical glioma model. Elife 2018, 7. [Google Scholar] [CrossRef]
- Sharma, N.K.; Sharma, R.; Mathur, D.; Sharad, S.; Minhas, G.; Bhatia, K.; Anand, A.; Ghosh, S.P. Role of Ionizing Radiation in Neurodegenerative Diseases. Front Aging Neurosci. 2018, 10, 134. [Google Scholar] [CrossRef]
- Guida, P.; Vazquez, M.E.; Otto, S. Cytotoxic effects of low- and high-LET radiation on human neuronal progenitor cells: Induction of apoptosis and TP53 gene expression. Radiat. Res. 2005, 164, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Bhatt, D.; Oltvai, Z.N.; Greenberger, J.S.; Bahar, I. Significance of p53 dynamics in regulating apoptosis in response to ionizing radiation, and polypharmacological strategies. Sci. Rep. 2014, 4, 6245. [Google Scholar] [CrossRef] [PubMed]
- Akhter, R.; Sanphui, P.; Biswas, S.C. The essential role of p53-up-regulated modulator of apoptosis (Puma) and its regulation by FoxO3a transcription factor in beta-amyloid-induced neuron death. J. Biol. Chem. 2014, 289, 10812–10822. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- De Guzman, A.E.; Ahmed, M.; Li, Y.Q.; Wong, C.S.; Nieman, B.J. p53 Loss Mitigates Early Volume Deficits in the Brains of Irradiated Young Mice. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 511–520. [Google Scholar] [CrossRef]
- Thotala, D.; Karvas, R.M.; Engelbach, J.A.; Garbow, J.R.; Hallahan, A.N.; DeWees, T.A.; Laszlo, A.; Hallahan, D.E. Valproic acid enhances the efficacy of radiation therapy by protecting normal hippocampal neurons and sensitizing malignant glioblastoma cells. Oncotarget 2015, 6, 35004–35022. [Google Scholar] [CrossRef] [PubMed]
- Yazlovitskaya, E.M.; Edwards, E.; Thotala, D.; Fu, A.; Osusky, K.L.; Whetsell, W.O., Jr.; Boone, B.; Shinohara, E.T.; Hallahan, D.E. Lithium treatment prevents neurocognitive deficit resulting from cranial irradiation. Cancer Res. 2006, 66, 11179–11186. [Google Scholar] [CrossRef]
- Khasraw, M.; Ashley, D.; Wheeler, G.; Berk, M. Using lithium as a neuroprotective agent in patients with cancer. BMC Med. 2012, 10, 131. [Google Scholar] [CrossRef]
- Ambacher, K.K.; Pitzul, K.B.; Karajgikar, M.; Hamilton, A.; Ferguson, S.S.; Cregan, S.P. The JNK- and AKT/GSK3beta- signaling pathways converge to regulate Puma induction and neuronal apoptosis induced by trophic factor deprivation. PLoS ONE 2012, 7, e46885. [Google Scholar] [CrossRef]
- Zareen, N.; Biswas, S.C.; Greene, L.A. A feed-forward loop involving Trib3, Akt and FoxO mediates death of NGF-deprived neurons. Cell Death Differ. 2013, 20, 1719–1730. [Google Scholar] [CrossRef][Green Version]
- Valable, S.; Bellail, A.; Lesne, S.; Liot, G.; Mackenzie, E.T.; Vivien, D.; Bernaudin, M.; Petit, E. Angiopoietin-1-induced PI3-kinase activation prevents neuronal apoptosis. FASEB J. 2003, 17, 443–445. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Koh, G.Y.; Koh, J.Y. Angiopoietin-1 blocks neurotoxic zinc entry into cortical cells via PIP2 hydrolysis-mediated ion channel inhibition. Neurobiol. Dis. 2015, 81, 203–213. [Google Scholar] [CrossRef]
- Sabirzhanov, B.; Faden, A.I.; Aubrecht, T.; Henry, R.; Glaser, E.; Stoica, B.A. MicroRNA-711-Induced Downregulation of Angiopoietin-1 Mediates Neuronal Cell Death. J. Neurotrauma 2018, 35, 2462–2481. [Google Scholar] [CrossRef]
- Cheng, Z.; Zheng, Y.Z.; Li, Y.Q.; Wong, C.S. Cellular Senescence in Mouse Hippocampus After Irradiation and the Role of p53 and p21. J. Neuropathol. Exp. Neurol. 2017, 76, 260–269. [Google Scholar] [CrossRef]
- Casafont, I.; Palanca, A.; Lafarga, V.; Berciano, M.T.; Lafarga, M. Effect of ionizing radiation in sensory ganglion neurons: Organization and dynamics of nuclear compartments of DNA damage/repair and their relationship with transcription and cell cycle. Acta Neuropathol. 2011, 122, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic. Acids. Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Sabirzhanov, B.; Stoica, B.A.; Zhao, Z.; Loane, D.J.; Wu, J.; Dorsey, S.G.; Faden, A.I. miR-711 upregulation induces neuronal cell death after traumatic brain injury. Cell Death Differ. 2016, 23, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, X.; Tang, X.; Wang, P.; Wang, H.; Wang, Y. MiR-21 is continually elevated long-term in the brain after exposure to ionizing radiation. Radiat Res. 2012, 177, 124–128. [Google Scholar] [CrossRef]
- Koturbash, I.; Zemp, F.; Kolb, B.; Kovalchuk, O. Sex-specific radiation-induced microRNAome responses in the hippocampus, cerebellum and frontal cortex in a mouse model. Mutat. Res. 2011, 722, 114–118. [Google Scholar] [CrossRef]
- Kempf, S.J.; Buratovic, S.; von Toerne, C.; Moertl, S.; Stenerlow, B.; Hauck, S.M.; Atkinson, M.J.; Eriksson, P.; Tapio, S. Ionising radiation immediately impairs synaptic plasticity-associated cytoskeletal signalling pathways in HT22 cells and in mouse brain: An in vitro/in vivo comparison study. PLoS ONE 2014, 9, e110464. [Google Scholar] [CrossRef]
- Kempf, S.J.; Casciati, A.; Buratovic, S.; Janik, D.; von Toerne, C.; Ueffing, M.; Neff, F.; Moertl, S.; Stenerlow, B.; Saran, A.; et al. The cognitive defects of neonatally irradiated mice are accompanied by changed synaptic plasticity, adult neurogenesis and neuroinflammation. Molecular Neurodegen. 2014, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Kempf, S.J.; Moertl, S.; Sepe, S.; von Toerne, C.; Hauck, S.M.; Atkinson, M.J.; Mastroberardino, P.G.; Tapio, S. Low-dose ionizing radiation rapidly affects mitochondrial and synaptic signaling pathways in murine hippocampus and cortex. J. Proteome Res. 2015, 14, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Balentova, S.; Adamkov, M. Molecular, Cellular and Functional Effects of Radiation-Induced Brain Injury: A Review. Int. J. Mol. Sci. 2015, 16, 27796–27815. [Google Scholar] [CrossRef]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 2008, 45, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Sabirzhanov, B.; Makarevich, O.; Barrett, J.; Jackson, I.L.; Faden, A.I.; Stoica, B.A. Down-Regulation of miR-23a-3p Mediates Irradiation-Induced Neuronal Apoptosis. Int. J. Mol. Sci. 2020, 21, 3695. [Google Scholar] [CrossRef]
- Vauzour, D.; Vafeiadou, K.; Rice-Evans, C.; Cadenas, E.; Spencer, J.P. Inhibition of cellular proliferation by the genistein metabolite 5,7,3’,4’-tetrahydroxyisoflavone is mediated by DNA damage and activation of the ATR signalling pathway. Arch. Biochem. Biophys. 2007, 468, 159–166. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Nam, C.; Doi, K.; Nakayama, H. Etoposide induces G2/M arrest and apoptosis in neural progenitor cells via DNA damage and an ATM/p53-related pathway. Histol. Histopathol. 2010, 25, 485–493. [Google Scholar] [CrossRef]
- Ho, D.H.; Kim, H.; Kim, J.; Sim, H.; Ahn, H.; Kim, J.; Seo, H.; Chung, K.C.; Park, B.J.; Son, I.; et al. Leucine-Rich Repeat Kinase 2 (LRRK2) phosphorylates p53 and induces p21(WAF1/CIP1) expression. Mol. Brain 2015, 8, 54. [Google Scholar] [CrossRef]
- Yakovlev, A.G.; Di Giovanni, S.; Wang, G.; Liu, W.; Stoica, B.; Faden, A.I. BOK and NOXA are essential mediators of p53-dependent apoptosis. J. Biol. Chem. 2004, 279, 28367–28374. [Google Scholar] [CrossRef]
- Sabirzhanov, B.; Zhao, Z.; Stoica, B.A.; Loane, D.J.; Wu, J.; Borroto, C.; Dorsey, S.G.; Faden, A.I. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J. Neurosci. 2014, 34, 10055–10071. [Google Scholar] [CrossRef] [PubMed]
- Siman, R.; McIntosh, T.K.; Soltesz, K.M.; Chen, Z.; Neumar, R.W.; Roberts, V.L. Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiol. Dis. 2004, 16, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Putkonen, N.; Kukkonen, J.P.; Mudo, G.; Putula, J.; Belluardo, N.; Lindholm, D.; Korhonen, L. Involvement of cyclin-dependent kinase-5 in the kainic acid-mediated degeneration of glutamatergic synapses in the rat hippocampus. European J. Neurosci. 2011, 34, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Bellone, C.; Viviani, B.; Marinovich, M.; Meli, E.; Pellegrini-Giampietro, D.E.; Cattabeni, F.; Di Luca, M. Lack of PSD-95 drives hippocampal neuronal cell death through activation of an alpha CaMKII transduction pathway. European J. Neurosci. 2002, 16, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Cursons, J.; Pillman, K.A.; Scheer, K.G.; Gregory, P.A.; Foroutan, M.; Hediyeh-Zadeh, S.; Toubia, J.; Crampin, E.J.; Goodall, G.J.; Bracken, C.P.; et al. Combinatorial Targeting by MicroRNAs Co-ordinates Post-transcriptional Control of EMT. Cell Syst. 2018, 7, 77–91.e77. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.M.; Morquette, B.; Abdouh, M.; Unsain, N.; Barker, P.A.; Feinstein, E.; Bernier, G.; Di Polo, A. ASPP1/2 regulate p53-dependent death of retinal ganglion cells through PUMA and Fas/CD95 activation in vivo. J. Neurosci. 2013, 33, 2205–2216. [Google Scholar] [CrossRef]
- Wang, L.; Xing, H.; Tian, Z.; Peng, L.; Li, Y.; Tang, K.; Rao, Q.; Wang, M.; Wang, J. iASPPsv antagonizes apoptosis induced by chemotherapeutic agents in MCF-7 cells and mouse thymocytes. Biochem. Biophys. Res. Commun. 2012, 424, 414–420. [Google Scholar] [CrossRef]
- Suhaili, S.H.; Karimian, H.; Stellato, M.; Lee, T.H.; Aguilar, M.I. Mitochondrial outer membrane permeabilization: A focus on the role of mitochondrial membrane structural organization. Biophys. Rev. 2017, 9, 443–457. [Google Scholar] [CrossRef]
- Metheetrairut, C.; Slack, F.J. MicroRNAs in the ionizing radiation response and in radiotherapy. Curr. Opin. Genet. Dev. 2013, 23, 12–19. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Baltanas, F.C.; Casafont, I.; Lafarga, V.; Weruaga, E.; Alonso, J.R.; Berciano, M.T.; Lafarga, M. Purkinje cell degeneration in pcd mice reveals large scale chromatin reorganization and gene silencing linked to defective DNA repair. J. Biol. Chem. 2011, 286, 28287–28302. [Google Scholar] [CrossRef]
- Rothkamm, K.; Barnard, S.; Moquet, J.; Ellender, M.; Rana, Z.; Burdak-Rothkamm, S. DNA damage foci: Meaning and significance. Environ. Mol. Mutagen. 2015, 56, 491–504. [Google Scholar] [CrossRef]
- Mata-Garrido, J.; Casafont, I.; Tapia, O.; Berciano, M.T.; Lafarga, M. Neuronal accumulation of unrepaired DNA in a novel specific chromatin domain: Structural, molecular and transcriptional characterization. Acta Neuropathol. Commun. 2016, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Kashammer, L.; Saathoff, J.H.; Lammens, K.; Gut, F.; Bartho, J.; Alt, A.; Kessler, B.; Hopfner, K.P. Mechanism of DNA End Sensing and Processing by the Mre11-Rad50 Complex. Mol. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.B.; Hohl, M.; Padjasek, M.; Jeong, E.; Jin, K.S.; Krezel, A.; Petrini, J.H.; Cho, Y. Eukaryotic Rad50 functions as a rod-shaped dimer. Nat. Struct. Mol. Biol. 2017, 24, 248–257. [Google Scholar] [CrossRef]
- De Jager, M.; van Noort, J.; van Gent, D.C.; Dekker, C.; Kanaar, R.; Wyman, C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell 2001, 8, 1129–1135. [Google Scholar] [CrossRef]
- Trujillo, K.M.; Yuan, S.S.; Lee, E.Y.; Sung, P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J. Biol. Chem. 1998, 273, 21447–21450. [Google Scholar] [CrossRef]
- Domanskyi, A.; Virtanen, K.T.; Palvimo, J.J.; Janne, O.A. Biochemical characterization of androgen receptor-interacting protein 4. Biochem. J. 2006, 393, 789–795. [Google Scholar] [CrossRef][Green Version]
- Zhang, F.P.; Domanskyi, A.; Palvimo, J.J.; Sariola, H.; Partanen, J.; Janne, O.A. An adenosine triphosphatase of the sucrose nonfermenting 2 family, androgen receptor-interacting protein 4, is essential for mouse embryonic development and cell proliferation. Mol. Endocrinol. 2007, 21, 1430–1442. [Google Scholar] [CrossRef][Green Version]
- Guo, Z.; Tian, Y.; Guo, Y.; Li, B.; Liu, X.; Xie, K.; Song, Y.; Wang, D. RAD6B Plays a Critical Role in Neuronal DNA Damage Response to Resist Neurodegeneration. Front. Cell Neurosci. 2019, 13, 392. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef]
- Vujaskovic, Z.; Thrasher, B.A.; Jackson, I.L.; Brizel, M.B.; Brizel, D.M. Radioprotective effects of amifostine on acute and chronic esophageal injury in rodents. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Aubrecht, T.G.; Faden, A.I.; Sabirzhanov, B.; Glaser, E.P.; Roelofs, B.A.; Polster, B.M.; Makarevich, O.; Stoica, B.A. Comparing effects of CDK inhibition and E2F1/2 ablation on neuronal cell death pathways in vitro and after traumatic brain injury. Cell Death Dis. 2018, 9, 1121. [Google Scholar] [CrossRef]
- Ma, C.M.; Coffey, C.W.; DeWerd, L.A.; Liu, C.; Nath, R.; Seltzer, S.M.; Seuntjens, J.P.; American Association of Physicists in, M. AAPM protocol for 40-300 kV x-ray beam dosimetry in radiotherapy and radiobiology. Med. Phys. 2001, 28, 868–893. [Google Scholar] [CrossRef]
- Dmitrieva, N.I.; Burg, M.B. Osmotic stress and DNA damage. Methods Enzymol. 2007, 428, 241–252. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic. Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Sabirzhanov, B.; Stoica, B.A.; Hanscom, M.; Piao, C.S.; Faden, A.I. Over-expression of HSP70 attenuates caspase-dependent and caspase-independent pathways and inhibits neuronal apoptosis. J. Neurochem. 2012, 123, 542–554. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabirzhanov, B.; Makarevich, O.; Barrett, J.P.; Jackson, I.L.; Glaser, E.P.; Faden, A.I.; Stoica, B.A. Irradiation-Induced Upregulation of miR-711 Inhibits DNA Repair and Promotes Neurodegeneration Pathways. Int. J. Mol. Sci. 2020, 21, 5239. https://doi.org/10.3390/ijms21155239
Sabirzhanov B, Makarevich O, Barrett JP, Jackson IL, Glaser EP, Faden AI, Stoica BA. Irradiation-Induced Upregulation of miR-711 Inhibits DNA Repair and Promotes Neurodegeneration Pathways. International Journal of Molecular Sciences. 2020; 21(15):5239. https://doi.org/10.3390/ijms21155239
Chicago/Turabian StyleSabirzhanov, Boris, Oleg Makarevich, James P. Barrett, Isabel L. Jackson, Ethan P. Glaser, Alan I. Faden, and Bogdan A. Stoica. 2020. "Irradiation-Induced Upregulation of miR-711 Inhibits DNA Repair and Promotes Neurodegeneration Pathways" International Journal of Molecular Sciences 21, no. 15: 5239. https://doi.org/10.3390/ijms21155239
APA StyleSabirzhanov, B., Makarevich, O., Barrett, J. P., Jackson, I. L., Glaser, E. P., Faden, A. I., & Stoica, B. A. (2020). Irradiation-Induced Upregulation of miR-711 Inhibits DNA Repair and Promotes Neurodegeneration Pathways. International Journal of Molecular Sciences, 21(15), 5239. https://doi.org/10.3390/ijms21155239