Whole-Genome-Based Survey for Polyphyletic Serovars of Salmonella enterica subsp. enterica Provides New Insights into Public Health Surveillance

 , , and
, , and
Abstract

1. Introduction
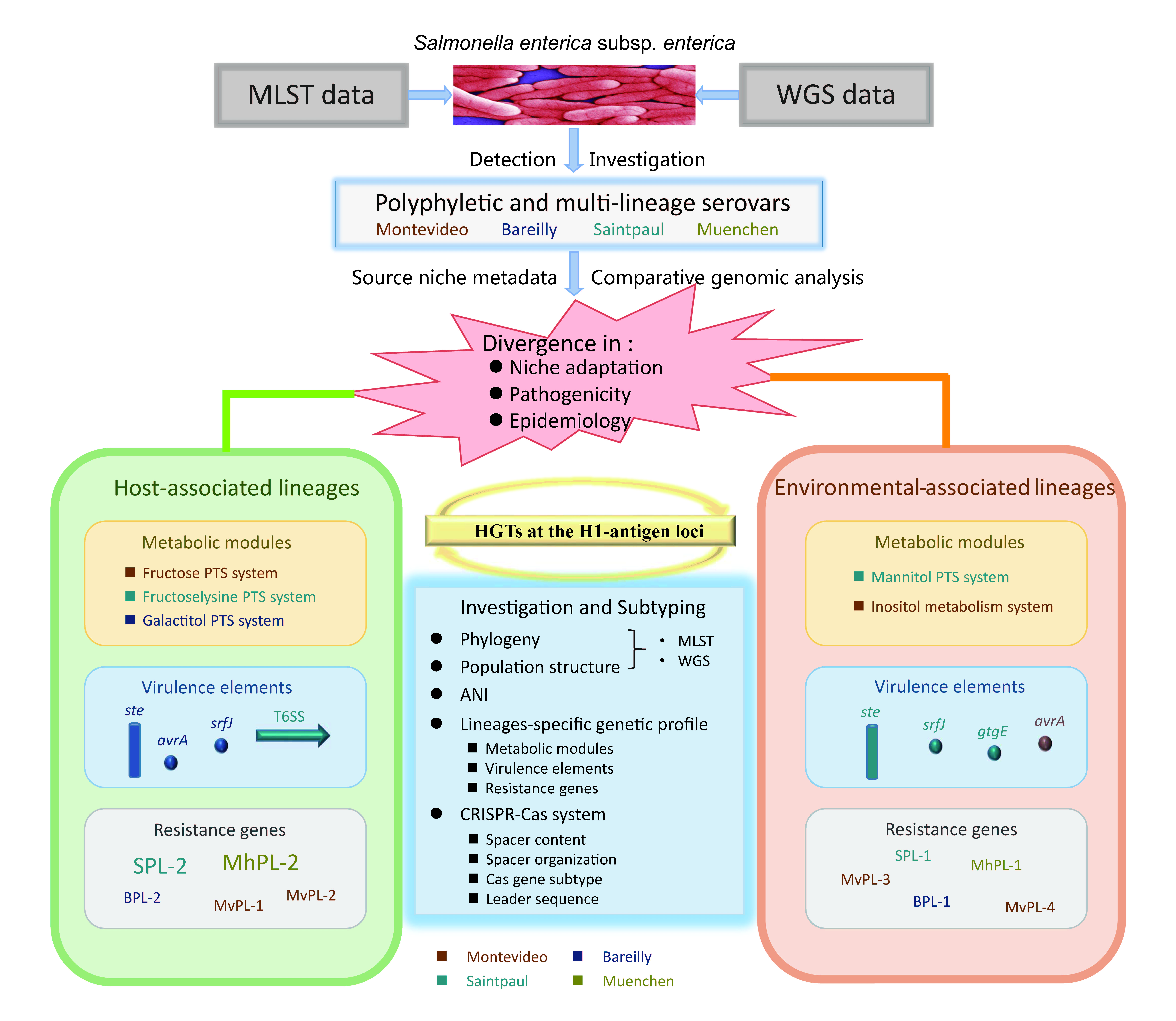
2. Results and Discussion
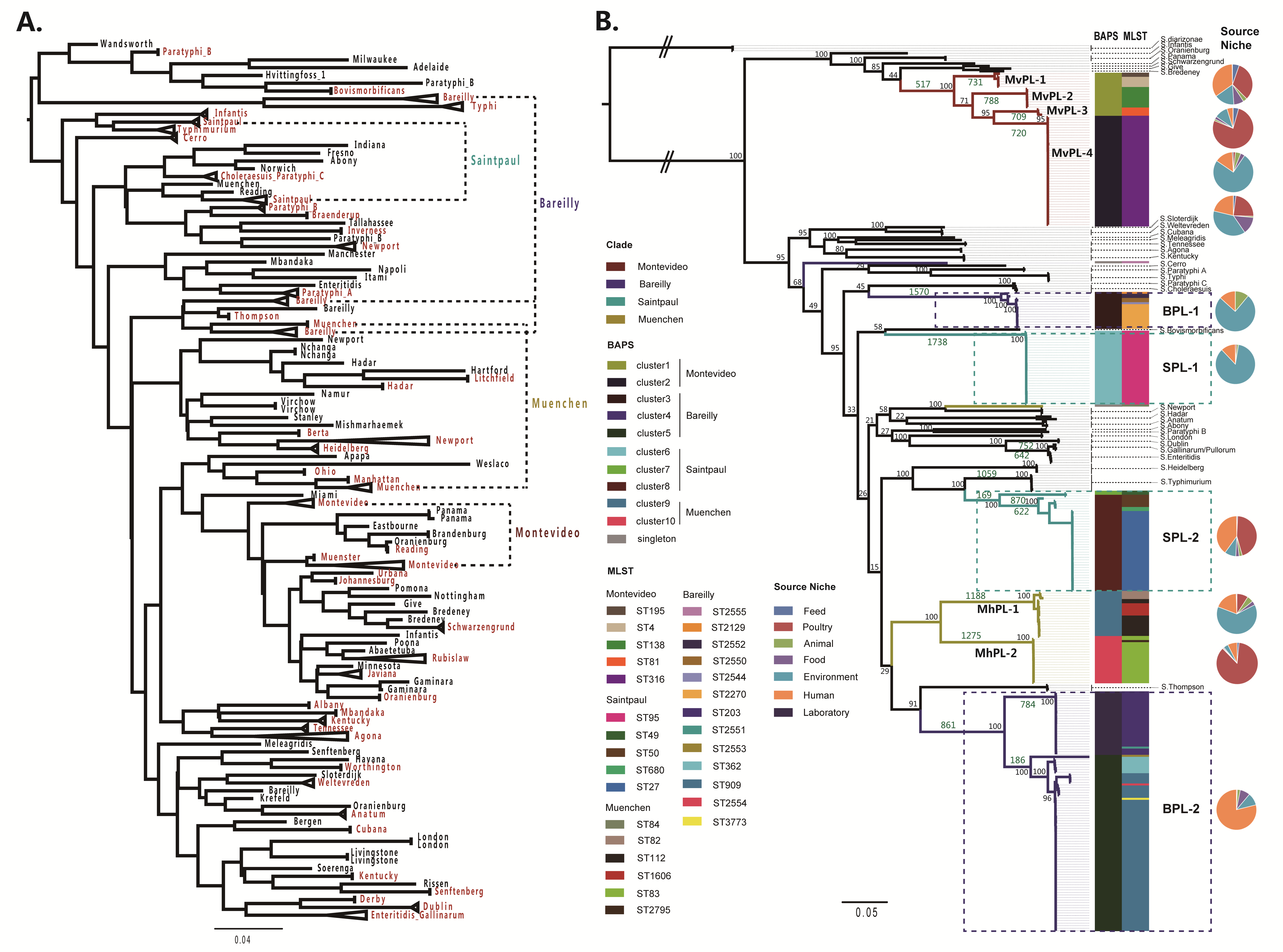
2.1. Identification of Putative Polyphyletic Serovars Based on MLST Phylogeny
2.2. Extensive Genetic Diversity Is Revealed by Whole-Genome-Based Phylogeny, Population Structure, and Average Nucleotide Identity (ANI)
2.3. Characterizing the Core and Pan Genomes Exhibits the Open Pan Genome and the Small Core Genome
2.4. The Source Niche Metadata and Lineage-Specific Genomic Contents Reveal the Potential Differentiation in Niche Adaptation
2.5. The Virulence Profile Indicates the Divergence in Pathogenicity between Distinct Lineages
2.6. Differences in the Antimicrobial Resistance (AMR) Profile at the Inter-Lineage Level within Saintpaul and Muenchen
2.7. Polyphyletic Serovars Are the Result of Recombination Events at the H1-Antigen Loci
2.8. Polyphyletic and Multi-Lineage Serovars Are Clinically Important Salmonella Serovars
2.9. CRISPR-Cas System: A High-Resolution Subtyping Method for Polyphyletic and/or Multi-Lineage Serovar
3. Conclusions
4. Materials and Methods
4.1. Data Collection
4.2. Construction of MLST Tree
4.3. Identification of Gene Orthologous Group
4.4. The Phylogenetic Analysis and Population Genetic Analysis Based on Core Genome Single-Nucleotide Variants (cgSNVs)
4.5. Whole Genome Average Nucleotide Identity
4.6. Core and Pan Genome Analysis
4.7. Lineage-Specific Core Genome Comparison
4.8. Identification of Virulence-Related Elements and Antimicrobial Resistance Genes
4.9. Phylogenetic Analysis of O, H1, and H2-Antigen
4.10. CRISPR-Cas Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| WGS | whole-genome sequencing |
| MLST | multi-locus sequence typing |
| CDC | Centers for Disease Control and Prevention |
| NCBI | National Center for Biotechnology Information |
| SPIs | Salmonella pathogenicity islands |
| ML | maximum-likelihood |
| ANI | average nucleotide identity |
| STs | sequence types |
| T3SS | type three secretion system |
| T6SS | type six secretion system |
| AMR | antimicrobial resistance |
| MDR | multidrug resistant |
| GTR | General Time Reversible |
| cgSNVs | core genome single-nucleotide variants |
References
- Elgea, P.V.; Loeckaertb, A.C.; Arrowc, P.B. Emergence of Salmonella Epidemics: The problems related to Salmonella enterica serotype Enteritidis and multiple antibiotic resistance in other major serotypes. Vet. Res. 2005, 36, 267–288. [Google Scholar] [CrossRef]
- Torgerson, P.; Devleesschauwer, B.; Gargouri, N.; Fürst, T.; Budke, C.; Carabin, H.; Kirk, M.D.; Angulo, F.J.; Havelaar, A.; De Silva, N.R.; et al. World Health Organization Estimates of the Global and Regional Disease Burden of 11 Foodborne Parasitic Diseases, 2010: A Data Synthesis. PLoS Med. 2015, 12, e1001920. [Google Scholar] [CrossRef] [PubMed]
- Grimont, P.A.D.; Weill, F.-X. Antigenic formulae of the Salmonella serovars. In WHO Collaborating Centre for Reference and Research on Salmonella, 9th ed.; Institut Pasteur: Paris, France, 2007. [Google Scholar]
- Desai, P.T.; Porwollik, S.; Long, F.; Cheng, P.; Wollam, A.; Clifton, S.W.; Weinstock, G.M. Evolutionary Genomics of Salmonella enterica Subspecies. MBio 2013, 4, 00579-12. [Google Scholar] [CrossRef] [PubMed]
- Achtman, M.; Wain, J.; Weill, F.-X.; Nair, S.; Zhou, Z.; Sangal, V.; Krauland, M.; Hale, J.L.; Harbottle, H.; Uesbeck, A.; et al. Multilocus Sequence Typing as a Replacement for Serotyping in Salmonella enterica. PLoS Pathog. 2012, 8, e1002776. [Google Scholar] [CrossRef] [PubMed]
- Timme, R.E.; Pettengill, J.B.; Allard, M.W.; Strain, E.; Barrangou, R.; Wehnes, C.; Van Kessel, J.S.; Karns, J.S.; Musser, S.M.; Brown, E.W. Phylogenetic Diversity of the Enteric Pathogen Salmonella enterica subsp. enterica Inferred from Genome-Wide Reference-Free SNP Characters. Genome Boil. Evol. 2013, 5, 2109–2123. [Google Scholar] [CrossRef]
- E Holt, K.; Parkhill, J.; Mazzoni, C.J.; Roumagnac, P.; Weill, F.-X.; Goodhead, I.; Rance, R.; Baker, S.; Maskell, D.J.; Wain, J.; et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat. Genet. 2008, 40, 987–993. [Google Scholar] [CrossRef]
- Beltran, P.; Musser, J.M.; Helmuth, R.; Farmer, J.J.; Frerichs, W.M.; Wachsmuth, I.K.; Ferris, K.; McWhorter, A.C.; Wells, J.G.; Cravioto, A. Toward a population genetic analysis of Salmonella: Genetic diversity and relationships among strains of serotypes S. choleraesuis, S. derby, S. dublin, S. enteritidis, S. heidelberg, S. infantis, S. newport, and S. typhimurium. Proc. Natl. Acad. Sci. USA 1988, 85, 7753–7757. [Google Scholar] [CrossRef]
- Cao, G.; Meng, J.; Strain, E.; Stones, R.; Pettengill, J.; Zhao, S.; McDermott, P.; Brown, E.; Allard, M. Phylogenetics and Differentiation of Salmonella Newport Lineages by Whole Genome Sequencing. PLoS ONE 2013, 8, e55687. [Google Scholar] [CrossRef]
- Sangal, V.; Harbottle, H.; Mazzoni, C.J.; Helmuth, R.; Guerra, B.; Didelot, X.; Paglietti, B.; Rabsch, W.; Brisse, S.; Weill, F.-X.; et al. Evolution and Population Structure of Salmonella enterica Serovar Newport. J. Bacteriol. 2010, 192, 6465–6476. [Google Scholar] [CrossRef]
- Connor, T.R.; Owen, S.V.; Langridge, G.C.; Connell, S.; Nair, S.; Reuter, S.; Dallman, T.J.; Corander, J.; Tabing, K.C.; Le Hello, S.; et al. What’s in a Name? Species-Wide Whole-Genome Sequencing Resolves Invasive and Noninvasive Lineages of Salmonella enterica Serotype Paratyphi B. MBio 2016, 7, e00527-16. [Google Scholar] [CrossRef]
- Zheng, J.; Luo, Y.; Reed, E.; Bell, R.; Brown, E.W.; Hoffmann, M. Whole-Genome Comparative Analysis of Salmonella enterica Serovar Newport Strains Reveals Lineage-Specific Divergence. Genome Boil. Evol. 2017, 9, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Haley, B.J.; Kim, S.W.; Pettengill, J.; Luo, Y.; Karns, J.S.; Van Kessel, J.A.S. Genomic and Evolutionary Analysis of Two Salmonella enterica Serovar Kentucky Sequence Types Isolated from Bovine and Poultry Sources in North America. PLoS ONE 2016, 11, e0161225. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.-F.; Mohamed, K.; Fan, Y.; Achtman, M.; The Agama Study Group; Brown, D.; Chattaway, M.; Dallman, T.; Delahay, R.; et al. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2019, 30, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Octavia, S.; Tanaka, M.M.; Sintchenko, V.; Lan, R. Defining the Core Genome of Salmonella enterica Serovar Typhimurium for Genomic Surveillance and Epidemiological Typing. J. Clin. Microbiol. 2015, 53, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Bottacini, F.; Motherway, M.O.; Kuczynski, J.; O’Connell, K.J.; Serafini, F.; Duranti, S.; Milani, C.; Turroni, F.; Lugli, G.A.; Zomer, A.; et al. Comparative genomics of the Bifidobacterium breve taxon. BMC Genom. 2014, 15, 170. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Nguyen, S.V.; Harhay, D.M.; Bono, J.L.; Smith, T.P.L.; Fields, P.I.; Dinsmore, B.A.; Santovenia, M.; Wang, R.; Bosilevac, J.M.; Harhay, G.P. Comparative genomics of Salmonella enterica serovar Montevideo reveals lineage-specific gene differences that may influence ecological niche association. Microb. Genom. 2018, 4, e000202. [Google Scholar] [CrossRef]
- Geerse, R.H.; Izzo, F.; Postma, P.W. The PEP: Fructose phosphotransferase system in Salmonella typhimurium: FPr combines Enzyme IIIFru and pseudo-HPr activities. Mol. Genet. Genom. 1989, 216, 517–525. [Google Scholar] [CrossRef]
- Nolle, N.; Felsl, A.; Heermann, R.; Fuchs, T.M. Genetic Characterization of the Galactitol Utilization Pathway of Salmonella enterica Serovar Typhimurium. J. Bacteriol. 2016, 199, e00595-16. [Google Scholar] [CrossRef]
- Chaudhuri, R.R.; Morgan, E.; Peters, S.E.; Pleasance, S.J.; Hudson, D.L.; Davies, H.M.; Wang, J.; Van Diemen, P.M.; Buckley, A.; Bowen, A.J.; et al. Comprehensive Assignment of Roles for Salmonella Typhimurium Genes in Intestinal Colonization of Food-Producing Animals. PLoS Genet. 2013, 9, e1003456. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.A.; Phillips, R.S.; Kilgore, P.B.; Smith, G.L.; Hoover, T.R. A Mannose Family Phosphotransferase System Permease and Associated Enzymes Are Required for Utilization of Fructoselysine and Glucoselysine in Salmonella enterica Serovar Typhimurium. J. Bacteriol. 2015, 197, 2831–2839. [Google Scholar] [CrossRef]
- Ali, M.M.; Newsom, D.L.; González, J.F.; Sabag-Daigle, A.; Stahl, C.; Steidley, B.; Dubena, J.; Dyszel, J.L.; Smith, J.N.; Dieye, Y.; et al. Fructose-Asparagine Is a Primary Nutrient during Growth of Salmonella in the Inflamed Intestine. PLoS Pathog. 2014, 10, e1004209. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Shiraiwa, Y. Salt-Regulated Mannitol Metabolism in Algae. Mar. Biotechnol. 2005, 7, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Groisillier, A.; Labourel, A.; Michel, G.; Tonon, T. The Mannitol Utilization System of the Marine Bacterium Zobellia galactanivorans. Appl. Environ. Microbiol. 2015, 81, 1799–1812. [Google Scholar] [CrossRef]
- Byer, T.; Wang, J.; Zhang, M.; Vather, N.; Blachman, A.; Visser, B.; Liu, J.M. MtlR negatively regulates mannitol utilization by Vibrio cholerae. Microbiology 2017, 163, 1902–1911. [Google Scholar] [CrossRef]
- Sand, M.; Rodrigues, M.; González, J.M.; De Crécy-Lagard, V.; Santos, H.; Müller, V.; Averhoff, B.; Crécy-Lagard, V. Mannitol-1-phosphate dehydrogenases/phosphatases: A family of novel bifunctional enzymes for bacterial adaptation to osmotic stress. Environ. Microbiol. 2014, 17, 711–719. [Google Scholar] [CrossRef]
- Kröger, C.; Stolz, J.; Fuchs, T.M. myo-Inositol transport by Salmonella enterica serovar Typhimurium. Microbiology 2009, 156, 128–138. [Google Scholar] [CrossRef]
- Kröger, C.; Fuchs, T.M. Characterization of the myo-Inositol Utilization Island of Salmonella enterica serovar Typhimurium. J. Bacteriol. 2008, 191, 545–554. [Google Scholar] [CrossRef]
- Rothhardt, J.E.; Kröger, C.; Broadley, S.P.; Fuchs, T.M. The orphan regulator ReiD of S almonella enterica is essential for myo -inositol utilization. Mol. Microbiol. 2014, 94, 700–712. [Google Scholar] [CrossRef]
- Pezoa, D.; Yang, H.-J.; Blondel, C.J.; Santiviago, C.A.; Andrews-Polymenis, H.; Contreras, I. The Type VI Secretion System Encoded in SPI-6 Plays a Role in Gastrointestinal Colonization and Systemic Spread of Salmonella enterica serovar Typhimurium in the Chicken. PLoS ONE 2013, 8, e63917. [Google Scholar] [CrossRef] [PubMed]
- Gieraltowski, L.; Julián, E.; Pringle, J.; Macdonald, K.; Quilliam, D.; Marsden-Haug, N.; Saathoff-Huber, L.; Von Stein, D.; Kissler, B.; Parish, M.; et al. Nationwide outbreak of Salmonella Montevideo infections associated with contaminated imported black and red pepper: Warehouse membership cards provide critical clues to identify the source. Epidemiol. Infect. 2012, 141, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.; Luo, Y.; Strain, E.; Li, C.; Keys, C.E.; Son, I.; Stones, R.; Musser, S.M.; Brown, E.W. High resolution clustering of Salmonella enterica serovar Montevideo strains using a next-generation sequencing approach. BMC Genom. 2012, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Lienau, E.K.; Strain, E.; Wang, C.; Zheng, J.; Ottesen, A.; Keys, C.E.; Hammack, T.S.; Musser, S.M.; Brown, E.W.; Allard, M.; et al. Identification of a Salmonellosis Outbreak by Means of Molecular Sequencing. N. Engl. J. Med. 2011, 364, 981–982. [Google Scholar] [CrossRef]
- Hoffmann, M.; Luo, Y.; Monday, S.R.; Gonzalez-Escalona, N.; Ottesen, A.; Muruvanda, T.; Wang, C.; Kastanis, G.; Keys, C.; Janies, D.; et al. Tracing Origins of theSalmonellaBareilly Strain Causing a Food-borne Outbreak in the United States. J. Infect. Dis. 2015, 213, 502–508. [Google Scholar] [CrossRef]
- Edwards, P.R.; Bruner, D.W. The Occurrence of Multiple Types of Paratyphoid Bacilli in Infections of Fowls, with Special Reference to Two New Salmonella Species. J. Infect. Dis. 1940, 66, 218–221. [Google Scholar] [CrossRef]
- O’Mahony, M.; Cowden, J.; Smyth, B.; Lynch, D.; Hall, M.; Rowe, B.; Teare, E.L.; Tettmar, R.E.; Rampling, A.M.; Coles, M.; et al. An outbreak of Salmonella saint-paul infection associated with beansprouts. Epidemiol. Infect. 1990, 104, 229–235. [Google Scholar] [CrossRef]
- Baggesen, D.L.; Wegener, H.C.; Christensen, J.P. Typing of Salmonella enterica serovar Saintpaul: An outbreak investigation. APMIS 1996, 104, 411–418. [Google Scholar] [CrossRef]
- Munnoch, S.A.; Ward, K.; Sheridan, S.; Fitzsimmons, G.J.; Shadbolt, C.T.; Piispanen, J.P.; Wang, Q.; Ward, T.J.; Worgan, T.L.M.; Oxenford, C.; et al. A multi-state outbreak of Salmonella Saintpaul in Australia associated with cantaloupe consumption. Epidemiol. Infect. 2008, 137, 367–374. [Google Scholar] [CrossRef]
- Lehmacher, A.; Bockemühl, J.; Aleksić, S. Nationwide outbreak of human salmonellosis in Germany due to contaminated paprika and paprika-powdered potato chips. Epidemiol. Infect. 1995, 115, 501–511. [Google Scholar] [CrossRef]
- Taylor, D.N.; Wachsmuth, I.K.; Shangkuan, Y.-H.; Schmidt, E.V.; Barrett, T.J.; Schrader, J.S.; Scherach, C.S.; McGee, H.B.; Feldman, R.A.; Brenner, D.J. Salmonellosis Associated with Marijuana: A multistate outbreak traced by plasmid fingerprinting. N. Engl. J. Med. 1982, 306, 1249–1253. [Google Scholar] [CrossRef]
- Proctor, M.E.; Hamacher, M.; Tortorello, M.L.; Archer, J.R.; Davis, J.P. Multistate Outbreak of Salmonella Serovar Muenchen Infections Associated with Alfalfa Sprouts Grown from Seeds Pretreated with Calcium Hypochlorite. J. Clin. Microbiol. 2001, 39, 3461–3465. [Google Scholar] [CrossRef] [PubMed]
- Gebreyes, W.A.; Thakur, S. Multidrug-Resistant Salmonella enterica Serovar Muenchen from Pigs and Humans and Potential Interserovar Transfer of Antimicrobial Resistance. Antimicrob. Agents Chemother. 2005, 49, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Worley, J.; Meng, M.; Allard, M.W.; Brown, E.W.; Timme, R.E. Salmonella enterica Phylogeny Based on Whole-Genome Sequencing Reveals Two New Clades and Novel Patterns of Horizontally Acquired Genetic Elements. MBio 2018, 9, e02303-18. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Kariyawasam, S.; Jayarao, B.M.; Barrangou, R.; Gerner-Smidt, P.; Ribot, E.M.; Knabel, S.J.; Dudley, E.G. Subtyping Salmonella enterica Serovar Enteritidis Isolates from Different Sources by Using Sequence Typing Based on Virulence Genes and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) ▿ †. Appl. Environ. Microbiol. 2011, 77, 4520–4526. [Google Scholar] [CrossRef]
- Liu, F.; Barrangou, R.; Gerner-Smidt, P.; Ribot, E.M.; Knabel, S.J.; Dudley, E.G. Novel virulence gene and clustered regularly interspaced short palindromic repeat (CRISPR) multilocus sequence typing scheme for subtyping of the major serovars of Salmonella enterica subsp. enterica. Appl. Environ. Microbiol. 2011, 77, 1946–1956. [Google Scholar] [CrossRef]
- Fabre, L.; Zhang, J.; Guigon, G.; Le Hello, S.; Guibert, V.; Accou-Demartin, M.; de Romans, S.; Lim, C.; Roux, C.; Passet, V.; et al. CRISPR typing and subtyping for improved Laboratory surveillance of Salmonella infections. PLoS ONE 2012, 7, e36995. [Google Scholar] [CrossRef]
- Zhang, S.; Den-Bakker, H.C.; Li, S.; Chen, J.; Dinsmore, B.A.; Lane, C.; Lauer, A.C.; Fields, P.I.; Deng, X. SeqSero2: Rapid and Improved Salmonella Serotype Determination Using Whole-Genome Sequencing Data. Appl. Environ. Microbiol. 2019, 85, 1–13. [Google Scholar] [CrossRef]
- Alikhan, N.-F.; Zhou, Z.; Sergeant, M.; Achtman, M. A genomic overview of the population structure of Salmonella. PLoS Genet. 2018, 14, e1007261. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability Article Fast Track. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Boil. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Connor, T.R.; Sirén, J.; Aanensen, D.M.; Corander, J. Hierarchical and Spatially Explicit Clustering of DNA Sequences with BAPS Software. Mol. Boil. Evol. 2013, 30, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Bottacini, F.; Medini, D.; Pavesi, A.; Turroni, F.; Foroni, E.; Riley, D.; Giubellini, V.; Tettelin, H.; Van Sinderen, D.; Ventura, M. Comparative genomics of the genus Bifidobacterium. Microbiology 2010, 156, 3243–3254. [Google Scholar] [CrossRef]
- Seif, Y.; Kavvas, E.S.; Lachance, J.-C.; Yurkovich, J.T.; Nuccio, S.-P.; Fang, X.; Catoiu, E.; Raffatellu, M.; Palsson, B.; Monk, J.M. Genome-scale metabolic reconstructions of multiple Salmonella strains reveal serovar-specific metabolic traits. Nat. Commun. 2018, 9, 3771. [Google Scholar] [CrossRef]
- Sahl, J.W.; Caporaso, J.G.; Rasko, D.A.; Keim, P. The large-scale blast score ratio (LS-BSR) pipeline: A method to rapidly compare genetic content between bacterial genomes. PeerJ 2014, 2, e332. [Google Scholar] [CrossRef]
- Neuert, S.; Nair, S.; Day, M.R.; Doumith, M.; Ashton, P.M.; Mellor, K.C.; Jenkins, C.; Hopkins, K.L.; Woodford, N.; De Pinna, E.; et al. Prediction of Phenotypic Antimicrobial Resistance Profiles From Whole Genome Sequences of Non-typhoidal Salmonella enterica. Front Microbiol. 2018, 9, 592. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Liu, B.; Pop, M. ARDB--Antibiotic Resistance Genes Database. Nucleic Acids Res. 2009, 37, D443–D447. [Google Scholar] [CrossRef]
- Liu, B.; Knirel, Y.A.; Feng, L.; Perepelov, A.V.; Senchenkova, S.N.; Reeves, P.; Wang, L. Structural diversity inSalmonellaO antigens and its genetic basis. FEMS Microbiol. Rev. 2014, 38, 56–89. [Google Scholar] [CrossRef]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.; Hugenholtz, P. CRISPR Recognition Tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank. | Serovar | Serogroup | Number Reported (Total = 32,271) | per 100,000 | MLST Phylogeny | Core Genome Phylogeny | MSTree of MLST Database | Recombination Events |
|---|---|---|---|---|---|---|---|---|
| 1 | Enteritidis | D | 7830 | 16.8 | Monophyly * | - | Monophyly | - |
| 2 | Newport | C2 | 4728 | 10.1 | Polyphyly | Polyphyly [9] | Polyphyly | Recombination of H1 (This study) |
| 3 | Typhimurium | B | 4581 | 9.8 | Monophyly | - | Monophyly | - |
| 4 | Javiana | D | 2719 | 5.8 | Monophyly | - | - | |
| 5 | I 4, [5],12:i:- | B | 2179 | 4.7 | Monophyly | - | Monophyly | - |
| 6 | Infantis | C1 | 1281 | 2.7 | Monophyly * | - | Monophyly | - |
| 7 | Muenchen | C2 | 1216 | 2.6 | Polyphyly | Multi-lineage (This study) Polyphyly [6] | Monophyly | - |
| 8 | Montevideo | C1 | 1018 | 2.2 | Polyphyly | Multi-lineage (This study) Polyphyly [45] | Polyphyly | - |
| 9 | Braenderup | C1 | 1001 | 2.1 | Monophyly | - | Monophyly | - |
| 10 | Thompson | C1 | 792 | 1.7 | Monophyly | - | - | - |
| 11 | Saintpaul | B | 778 | 1.7 | Polyphyly | Polyphyly (This study) | Polyphyly | Recombination of H1 (This study) |
| 12 | Heidelberg | B | 754 | 1.6 | Monophyly | - | Monophyly | - |
| 13 | Oranienburg | C1 | 692 | 1.5 | Monophyly * | - | Polyphyly | - |
| 14 | Mississippi | G | 536 | 1.1 | - | - | - | |
| 15 | Typhi | D | 423 | 0.9 | Monophyly | - | Monophyly | - |
| 16 | Bareilly | C1 | 412 | 0.9 | Polyphyly | Polyphyly (This study) Polyphyly [45] | - | Recombination of H1 (This study) |
| 17 | Berta | D | 369 | 0.8 | Monophyly | - | - | - |
| 18 | Agona | B | 362 | 0.8 | Monophyly | - | Monophyly | - |
| 19 | Paratyphi B var. L(+) tartrate+ | B | 343 | 0.7 | Polyphyly | Polyphyly [11] | Polyphyly | Recombination of H [11] |
| 20 | Anatum | E1 | 257 | 0.6 | Monophyly | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Z.; Liu, J.; Du, B.; Ruan, H.-H.; Huo, Y.-X.; Du, Y.; Qiao, J. Whole-Genome-Based Survey for Polyphyletic Serovars of Salmonella enterica subsp. enterica Provides New Insights into Public Health Surveillance. Int. J. Mol. Sci. 2020, 21, 5226. https://doi.org/10.3390/ijms21155226
Yin Z, Liu J, Du B, Ruan H-H, Huo Y-X, Du Y, Qiao J. Whole-Genome-Based Survey for Polyphyletic Serovars of Salmonella enterica subsp. enterica Provides New Insights into Public Health Surveillance. International Journal of Molecular Sciences. 2020; 21(15):5226. https://doi.org/10.3390/ijms21155226
Chicago/Turabian StyleYin, Zhiqiu, Jiaheng Liu, Binghai Du, Hai-Hua Ruan, Yi-Xin Huo, Yuhui Du, and Jianjun Qiao. 2020. "Whole-Genome-Based Survey for Polyphyletic Serovars of Salmonella enterica subsp. enterica Provides New Insights into Public Health Surveillance" International Journal of Molecular Sciences 21, no. 15: 5226. https://doi.org/10.3390/ijms21155226
APA StyleYin, Z., Liu, J., Du, B., Ruan, H.-H., Huo, Y.-X., Du, Y., & Qiao, J. (2020). Whole-Genome-Based Survey for Polyphyletic Serovars of Salmonella enterica subsp. enterica Provides New Insights into Public Health Surveillance. International Journal of Molecular Sciences, 21(15), 5226. https://doi.org/10.3390/ijms21155226