Postsynthetic On-Column 2′ Functionalization of RNA by Convenient Versatile Method

 , ,
, ,
Abstract
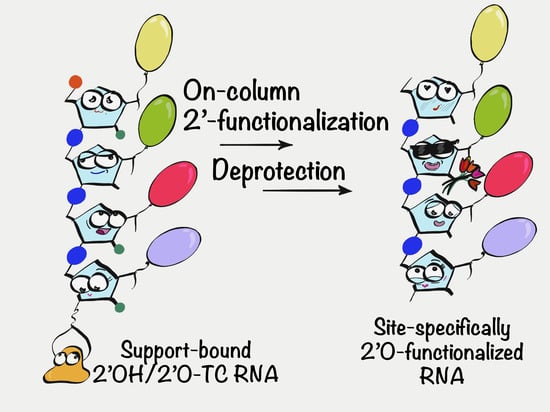
1. Introduction
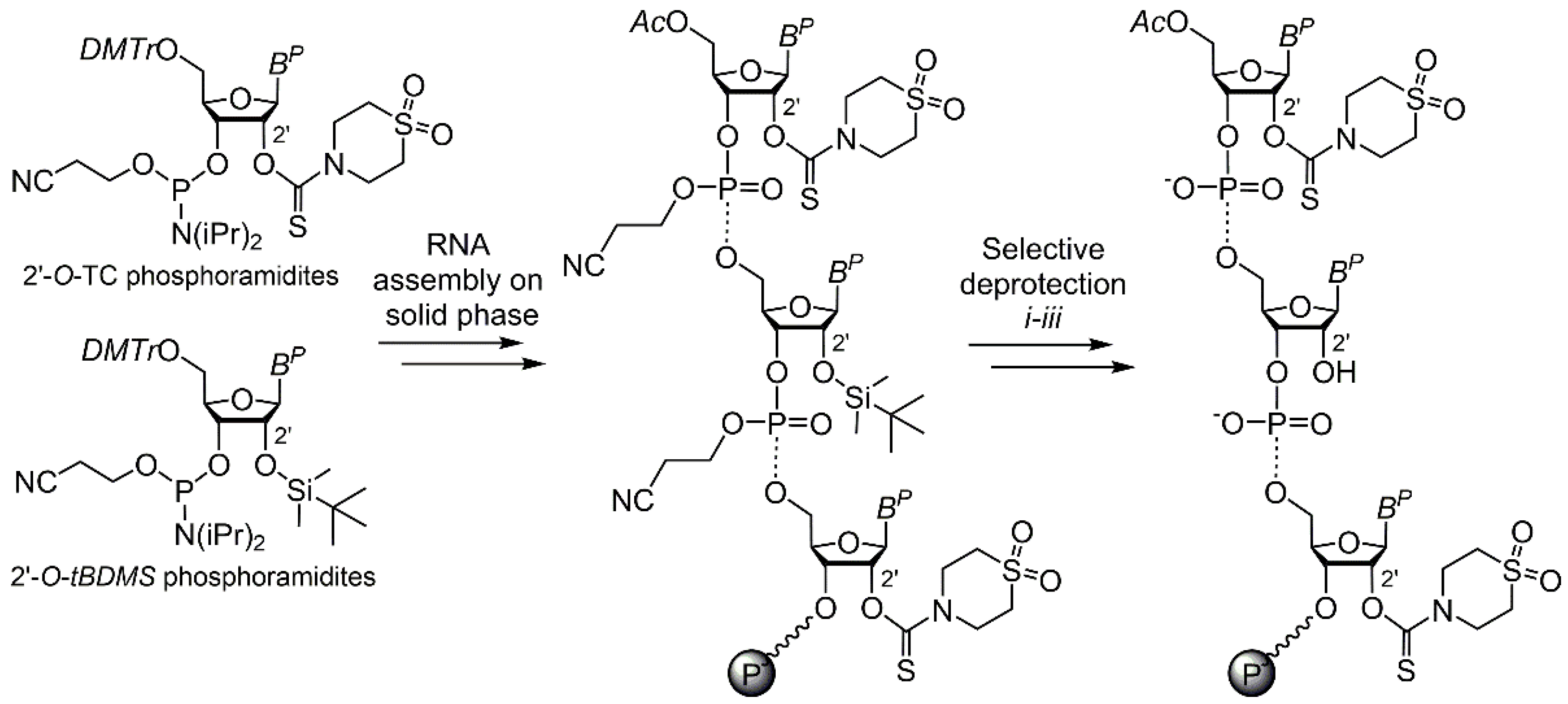
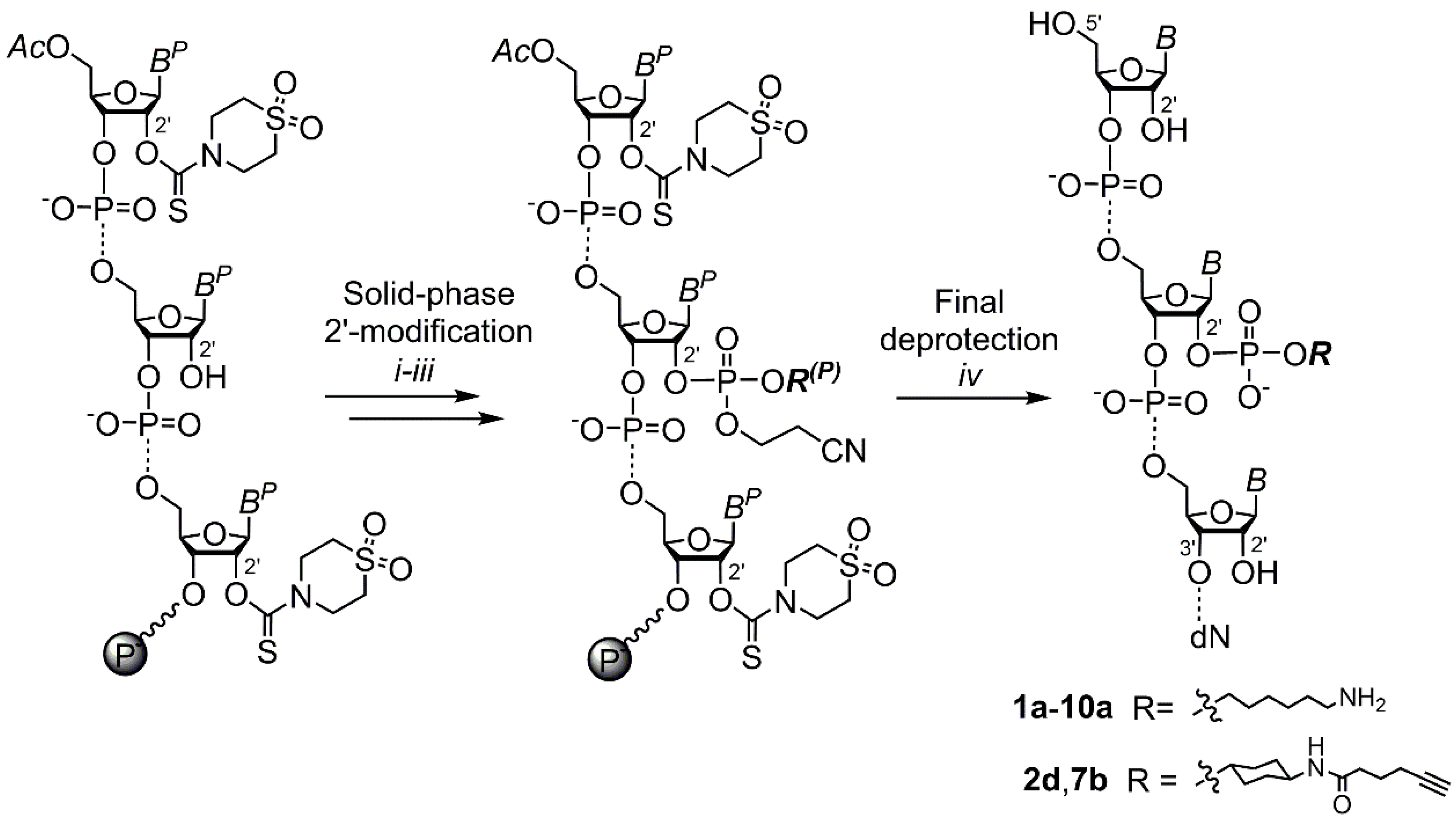
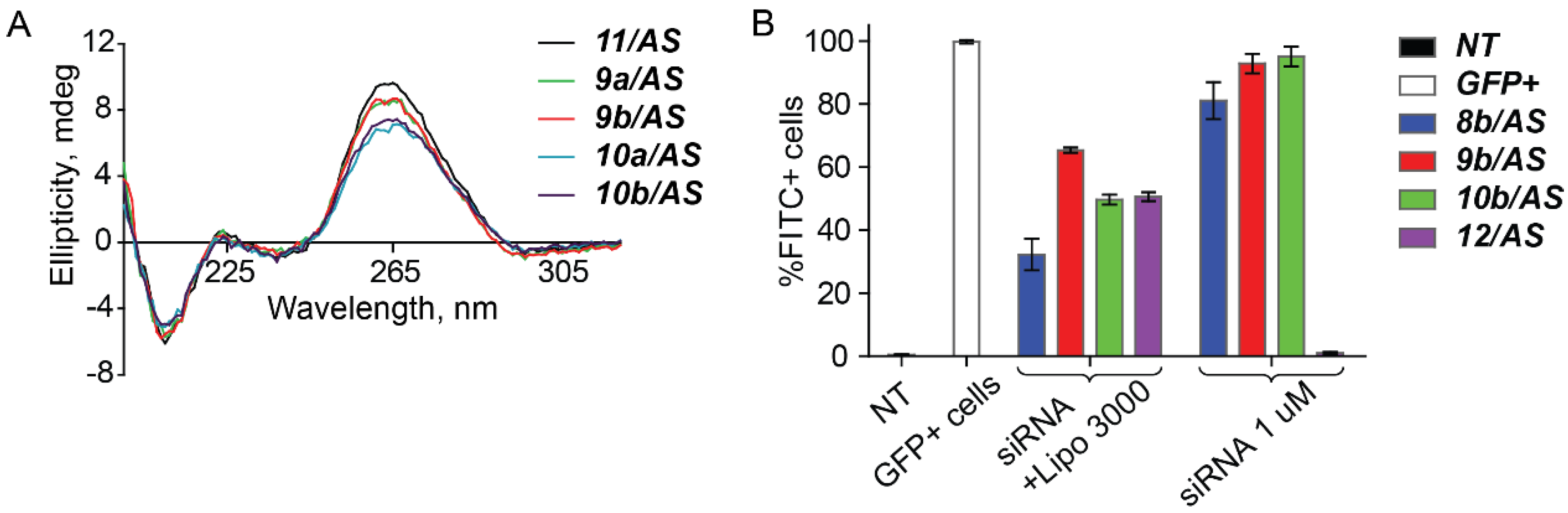
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Oligoribonucleotide Synthesis and Selective Partial Deprotection
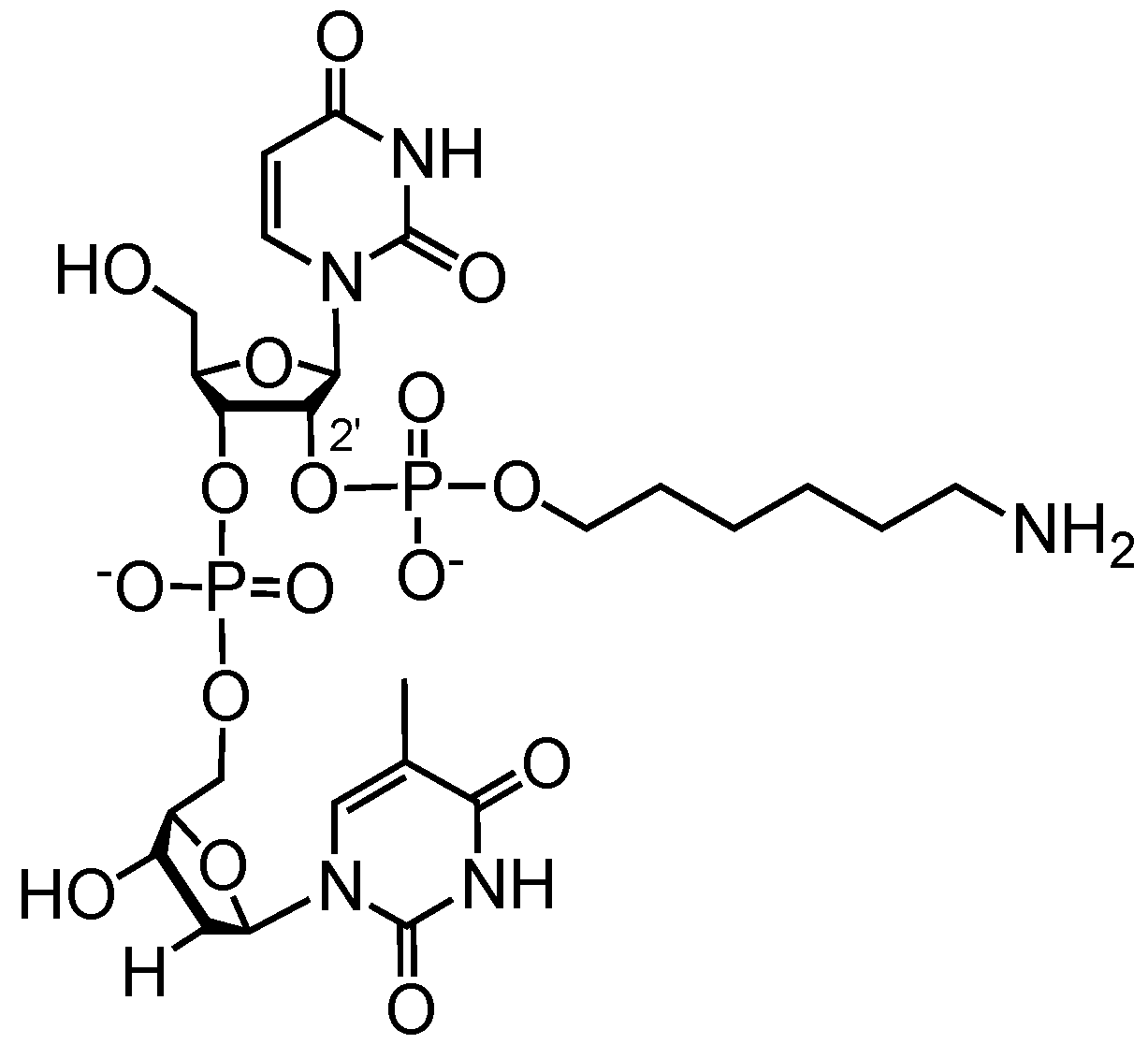
3.3. Solid-Phase 2′ Modification by Modifying Phosphoramidite
3.4. Final Deprotection and Purification of Oligoribonucleotides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RNA | Ribonucleic acid |
| siRNAs | Small interfering RNAs |
| miRNAs | MicroRNA |
| sgRNAs | Single guide RNA |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 |
| MALDI-TOF | Matrix-assisted laser desorption/ionization |
| ESI | Electrospray ionization |
| PAGE | Polyacrylamide gel electrophoresis |
| RP-HPLC | Reversed phase high performance liquid chromatography |
| ON | Oligonucleotide |
References
- Fauster, K.; Hartl, M.; Santner, T.; Aigner, M.; Kreutz, C.; Bister, K.; Ennifar, E.; Micura, R. 2′-Azido RNA, a Versatile Tool for Chemical Biology: Synthesis, X-ray Structure, siRNA Applications, Click Labeling. ACS Chem. Biol. 2012, 7, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Pei, F.; Zhang, J.; Wu, J.; Feng, M.; Wang, Y.; Jin, H.; Zhang, L.; Tang, X. Synthesis of Site-Specifically Phosphate-Caged siRNAs and Evaluation of Their RNAi Activity and Stability. Chem. Eur. J. 2014, 20, 12114–12122. [Google Scholar] [CrossRef]
- Meade, B.R.; Gogoi, K.; Hamil, A.S.; Palm-Apergi, C.; van den Berg, A.; Hagopian, J.C.; Springer, A.D.; Eguchi, A.; Kacsinta, A.D.; Dowdy, C.F.; et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat. Biotechnol. 2014, 32, 1256–1261. [Google Scholar] [CrossRef]
- Selvam, C.; Mutisya, D.; Prakash, S.; Ranganna, K.; Thilagavathi, R. Therapeutic potential of chemically modified siRNA: Recent trends. Chem. Biol. Drug Des. 2017, 90, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Dovydenko, I.; Tarassov, I.; Venyaminova, A.; Entelis, N. Method of Carrier-Free Delivery of Therapeutic RNA Importable Into Human Mitochondria: Lipophilic Conjugates With Cleavable Bonds. Biomaterials 2016, 76, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Granqvist, L.; Virta, P. 2′-O-[(4-CF3-triazol-1-yl)methyl] Uridine—A Sensitive 19F NMR Sensor for the Detection of RNA Secondary Structures. J. Org. Chem. 2015, 80, 7961–7970. [Google Scholar] [CrossRef]
- Neuner, S.; Santner, T.; Kreutz, C.; Micura, R. The “Speedy” Synthesis of Atom-Specific 15N Imino/Amido-Labeled RNA. Chem. Eur. J. 2015, 21, 11634–11643. [Google Scholar] [CrossRef]
- Krasheninina, O.A.; Novopashina, D.S.; Lomzov, A.A.; Venyaminova, A.G. 2′-Bispyrene-Modified 2′-O -Methyl RNA Probes as Useful Tools for the Detection of RNA: Synthesis, Fluorescent Properties, and Duplex Stability. ChemBioChem 2014, 15, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Zatsepin, T.S.; Romanova, E.A.; Oretskaya, T.S. Nucleosides and oligonucleotides containing 2′-reactive groups: Synthesis and applications. Russ. Chem. Rev. 2004, 73, 701–733. [Google Scholar] [CrossRef]
- Azéma, L.; Bathany, K.; Rayner, B. 2′-O-Appended Polyamines that Increase Triple-Helix-Forming Oligonucleotide Affinity are Selected by Dynamic Combinatorial Chemistry. ChemBioChem 2010, 11, 2513–2516. [Google Scholar] [CrossRef]
- Jeong, J.H.; Mok, H.; Oh, Y.-K.; Park, T.G. siRNA Conjugate Delivery Systems. Bioconjug. Chem. 2009, 20, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Biscans, A.; Bertrand, J.-R.; Dubois, J.; Rüger, J.; Vasseur, J.-J.; Sczakiel, G.; Dupouy, C.; Debart, F. Lipophilic 2′-O-Acetal Ester RNAs: Synthesis, Thermal Duplex Stability, Nuclease Resistance, Cellular Uptake, and siRNA Activity after Spontaneous Naked Delivery. ChemBioChem 2016, 17, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Biscans, A.; Rouanet, S.; Vasseur, J.-J.; Dupouy, C.; Debart, F. A versatile post-synthetic method on a solid support for the synthesis of RNA containing reduction-responsive modifications. Org. Biomol. Chem. 2016, 14, 7010–7017. [Google Scholar] [CrossRef] [PubMed]
- Aigner, M.; Hartl, M.; Fauster, K.; Steger, J.; Bister, K.; Micura, R. Chemical Synthesis of Site-Specifically 2′-Azido-Modified RNA and Potential Applications for Bioconjugation and RNA Interference. ChemBioChem 2011, 12, 47–51. [Google Scholar] [CrossRef]
- Matsuda, S.; Keiser, K.; Nair, J.K.; Charisse, K.; Manoharan, R.M.; Kretschmer, P.; Peng, C.G.; Kel’in, A.V.; Kandasamy, P.; Willoughby, J.L.S.; et al. siRNA Conjugates Carrying Sequentially Assembled Trivalent N-Acetylgalactosamine Linked Through Nucleosides Elicit Robust Gene Silencing In Vivo in Hepatocytes. ACS Chem. Biol. 2015, 10, 1181–1187. [Google Scholar] [CrossRef]
- Steinmeyer, J.; Walter, H.-K.; Bichelberger, M.A.; Schneider, V.; Kubař, T.; Rönicke, F.; Olshausen, B.; Nienhaus, K.; Nienhaus, G.U.; Schepers, U. “siRNA traffic lights”: Arabino-configured 2′-anchors for fluorescent dyes are key for dual color readout in cell imaging. Org. Biomol. Chem. 2018, 16, 3726–3731. [Google Scholar] [CrossRef]
- Pradère, U.; Brunschweiger, A.; Gebert, L.F.R.; Lucic, M.; Roos, M.; Hall, J. Chemical Synthesis of Mono- and Bis-Labeled Pre-MicroRNAs. Angew. Chem. Int. Ed. Engl. 2013, 52, 12028–12032. [Google Scholar] [CrossRef]
- Pradère, U.; Hall, J. Site-Specific Difunctionalization of Structured RNAs Yields Probes for microRNA Maturation. Bioconjug. Chem. 2016, 27, 681–687. [Google Scholar] [CrossRef]
- Menzi, M.; Pradère, U.; Wang, Y.; Fischer, M.; Baumann, F.; Bigatti, M.; Hall, J. Site-Specific Labeling of MicroRNA Precursors: A Structure–Activity Relationship Study. ChemBioChem 2016, 17, 2012–2017. [Google Scholar] [CrossRef]
- Fonvielle, M.; Mellal, D.; Patin, D.; Lecerf, M.; Blanot, D.; Bouhss, A.; Santarem, M.; Mengin-Lecreulx, D.; Sollogoub, M.; Arthur, M.; et al. Efficient Access to Peptidyl-RNA Conjugates for Picomolar Inhibition of Non-ribosomal FemXWv Aminoacyl Transferase. Chem. Eur. J. 2013, 19, 1357–1363. [Google Scholar] [CrossRef]
- Damha, M.J.; Ganeshan, K.; Hudson, R.H.; Zabarylo, S.V. Solid-phase synthesis of branched oligoribonucleotides related to messenger RNA splicing intermediates. Nucleic Acids Res. 1992, 20, 6565–6573. [Google Scholar] [CrossRef] [PubMed]
- Carriero, S.; Damha, M. Template-Mediated Synthesis of Lariat RNA and DNA. J. Org. Chem. 2003, 68, 8328–8338. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Damha, M.J. A Novel Approach to the Synthesis of DNA and RNA Lariats. J. Org. Chem. 2007, 72, 9491–9500. [Google Scholar] [CrossRef] [PubMed]
- Katolik, A.; Johnsson, R.; Montemayor, E.; Lackey, J.G.; Hart, P.J.; Damha, M.J. Regiospecific Solid-Phase Synthesis of Branched Oligoribonucleotides That Mimic Intronic Lariat RNA Intermediates. J. Org. Chem. 2014, 79, 963–975. [Google Scholar] [CrossRef]
- Braich, R.S.; Damha, M.J. Regiospecific Solid-Phase Synthesis of Branched Oligonucleotides. Effect of Vicinal 2‘,5‘- (or 2‘,3‘-) and 3‘,5‘-Phosphodiester Linkages on the Formation of Hairpin DNA. Bioconjug. Chem. 1997, 8, 370–377. [Google Scholar] [CrossRef]
- Plangger, R.; Juen, M.A.; Hoernes, T.P.; Nußbaumer, F.; Kremser, J.; Strebitzer, E.; Klingler, D.; Erharter, K.; Tollinger, M.; Erlacher, M.D.; et al. Branch site bulge conformations in domain 6 determine functional sugar puckers in group II intron splicing. Nucleic Acids Res. 2019, 47, 11430–11440. [Google Scholar] [CrossRef]
- Kadina, A.; Kietrys, A.M.; Kool, E.T. RNA Cloaking by Reversible Acylation. Angew. Chem. Int. Ed. 2018, 57, 3059–3063. [Google Scholar] [CrossRef]
- Velema, W.A.; Kietrys, A.M.; Kool, E.T. RNA Control by Photoreversible Acylation. J. Am. Chem. Soc. 2018, 140, 3491–3495. [Google Scholar] [CrossRef]
- Velema, W.A.; Kool, E.T. The chemistry and applications of RNA 2′-OH acylation. Nat. Rev. Chem. 2020, 4, 22–37. [Google Scholar] [CrossRef]
- Cieślak, J.; Ausín, C.; Grajkowski, A.; Beaucage, S.L. Convenient and Efficient Approach to the Permanent or Reversible Conjugation of RNA and DNA Sequences with Functional Groups. Curr. Protoc. Nucleic Acid Chem. 2012, 50, 4.52.1–4.52.36. [Google Scholar] [CrossRef]
- Cieslak, J.; Grajkowski, A.; Ausín, C.; Gapeev, A.; Beaucage, S.L. Permanent or Reversible Conjugation of 2′-O- Or 5′-O-aminooxymethylated Nucleosides With Functional Groups as a Convenient and Efficient Approach to the Modification of RNA and DNA Sequences. Nucleic Acids Res. 2012, 40, 2312–2329. [Google Scholar] [CrossRef] [PubMed]
- Biscans, A.; Bos, M.; Martin, A.R.; Ader, N.; Sczakiel, G.; Vasseur, J.-J.; Dupouy, C.; Debart, F. Direct Synthesis of Partially Modified 2′-O-pivaloyloxymethyl RNAs by a Base-Labile Protecting Group Strategy and Their Potential for Prodrug-Based Gene-Silencing Applications. ChemBioChem 2014, 15, 2674–2679. [Google Scholar] [CrossRef]
- Lavergne, T.; Baraguey, C.; Dupouy, C.; Parey, N.; Wuensche, W.; Sczakiel, G.; Vasseur, J.-J.; Debart, F. Synthesis and Preliminary Evaluation of pro-RNA 2′-O-Masked with Biolabile Pivaloyloxymethyl Groups in an RNA Interference Assay. J. Org. Chem. 2011, 76, 5719–5731. [Google Scholar] [CrossRef]
- Biscans, A.; Rouanet, S.; Bertrand, J.-R.; Vasseur, J.-J.; Dupouy, C.; Debart, F. Synthesis, binding, nuclease resistance and cellular uptake properties of 2′-O-acetalester-modified oligonucleotides containing cationic groups. Bioorg. Med. Chem. 2015, 23, 5360–5368. [Google Scholar] [CrossRef]
- Gauthier, F.; Claveau, S.; Bertrand, J.-R.; Vasseur, J.-J.; Dupouy, C.; Debart, F. Gymnotic Delivery and Gene Silencing Activity of Reduction-Responsive siRNAs Bearing Lipophilic Disulfide-Containing Modifications at 2′-position. Bioorg. Med. Chem. 2018, 26, 4635–4643. [Google Scholar] [CrossRef]
- Gauthier, F.; Bertrand, J.-R.; Vasseur, J.-J.; Dupouy, C.; Debart, F. Conjugation of Doxorubicin to siRNA Through Disulfide-based Self-immolative Linkers. Molecules 2020, 25, 2714. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, D.J.; Timár, Z.; Myerson, J.; Sierzchala, A.B.; Turner, J.; Ferreira, F.; Kupihár, Z.; Dellinger, G.; Hill, K.W.; Powell, J.A.; et al. Streamlined Process for the Chemical Synthesis of RNA Using 2′-O-Thionocarbamate-Protected Nucleoside Phosphoramidites in the Solid Phase. J. Am. Chem. Soc. 2011, 133, 11540–11556. [Google Scholar] [CrossRef] [PubMed]
- Usman, N.; Ogilvie, K.K.; Jiang, M.Y.; Cedergren, R.J. The automated chemical synthesis of long oligoribuncleotides using 2′-O-silylated ribonucleoside 3′-O-phosphoramidites on a controlled-pore glass support: Synthesis of a 43-nucleotide sequence similar to the 3′-half molecule of an Escherichia coli formylmethionine tRNA. J. Am. Chem. Soc. 1987, 109, 7845–7854. [Google Scholar] [CrossRef]
- Wu, X.; Pitsch, S. Synthesis and Pairing Properties of Oligoribonucleotide Analogues Containing a Metal-Binding Site Attached to beta-D-allofuranosyl Cytosine. Nucleic Acids Res. 1998, 26, 4315–4323. [Google Scholar] [CrossRef]
- Pitsch, S.; Weiss, P.A.; Jenny, L.; Stutz, A.; Wu, X. Reliable Chemical Synthesis of Oligoribonucleotides (RNA) with 2′-O-[(Triisopropylsilyl)oxy]methyl(2′-O-tom)-Protected Phosphoramidites. Helv. Chim. Acta 2001, 84, 3773–3795. [Google Scholar] [CrossRef]
- Ohgi, T.; Masutomi, Y.; Ishiyama, K.; Kitagawa, H.; Shiba, Y.; Yano, J. A New RNA Synthetic Method with a 2‘-O-(2-Cyanoethoxymethyl) Protecting Group. Org. Lett. 2005, 7, 3477–3480. [Google Scholar] [CrossRef]
- Shiba, Y.; Masuda, H.; Watanabe, N.; Ego, T.; Takagaki, K.; Ishiyama, K.; Ohgi, T.; Yano, J. Chemical synthesis of a very long oligoribonucleotide with 2-cyanoethoxymethyl (CEM) as the 2′-O-protecting group: Structural identification and biological activity of a synthetic 110mer precursor-microRNA candidate. Nucleic Acids Res. 2007, 35, 3287–3296. [Google Scholar] [CrossRef] [PubMed]
- Yamakage, S.; Sakatsume, O.; Furuyama, E.; Takaku, H. 1-(2-Chloroethoxy)ethyl group for the protection of 2′-hydroxyl group in the synthesis of oligoribonucleotides. Tetrahedron Lett. 1989, 30, 6361–6364. [Google Scholar] [CrossRef]
- Lavergne, T.; Bertrand, J.-R.; Vasseur, J.-J.; Debart, F. A Base-Labile Group for 2′-OH Protection of Ribonucleosides: A Major Challenge for RNA Synthesis. Chem. Eur. J. 2008, 14, 9135–9138. [Google Scholar] [CrossRef]
- Lavergne, T.; Janin, M.; Dupouy, C.; Vasseur, J.-J.; Debart, F. Chemical Synthesis of RNA With Base-Labile 2′-O-(pivaloyloxymethyl)-protected Ribonucleoside Phosphoramidites. Curr. Protoc. Nucleic Acid Chem. 2010, 43, 3.19.1–3.19.27. [Google Scholar] [CrossRef]
- Semenyuk, A.; Földesi, A.; Johansson, T.; Estmer-Nilsson, C.; Blomgren, P.; Brännvall, M.; Kirsebom, L.A.; Kwiatkowski, M. Synthesis of RNA Using 2‘-O-DTM Protection. J. Am. Chem. Soc. 2006, 128, 12356–12357. [Google Scholar] [CrossRef]
- Kierzek, R. The stability of trisubstituted internucleotide bond in the presence of vicinal 2′-hydroxyl. Chemical synthesis of the uridyl-(2′-phosphate)-3′-5′-uridine. Nucleosides Nucleotides Nucleic Acids 1994, 13, 1757–1768. [Google Scholar] [CrossRef]
- Sekine, M.; Tsuruoka, H.; Iimura, S.; Kusuoku, H.; Wada, T.; Furusawa, K. Studies on Steric and Electronic Control of 2‘−3‘ Phosphoryl Migration in 2‘-Phosphorylated Uridine Derivatives and Its Application to the Synthesis of 2‘-Phosphorylated Oligouridylates. J. Org. Chem. 1996, 61, 4087–4100. [Google Scholar] [CrossRef] [PubMed]
- Wincott, F.; DiRenzo, A.; Shaffer, C.; Grimm, S.; Tracz, D.; Workman, C.; Sweedler, D.; Gonzalez, C.; Scaringe, S.; Usman, N. Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res. 1995, 23, 2677–2684. [Google Scholar] [CrossRef] [PubMed]
- Prhavc, M.; Lesnik, E.A.; Mohan, V.; Manoharan, M. 2′-O-Carbamate-containing oligonucleotides: Synthesis and properties. Tetrahedron Lett. 2001, 42, 8777–8780. [Google Scholar] [CrossRef]
- Korshun, V.A.; Stetsenko, D.A.; Gait, M.J. Novel uridin-2′-yl carbamates: Synthesis, incorporation into oligodeoxyribonucleotides, and remarkable fluorescence properties of 2′-pyren-1-ylmethylcarbamate. J. Chem. Soc. Perkin Trans. 1 2002, 1092–1104. [Google Scholar] [CrossRef]
- Korshun, V.A.; Stetsenko, D.A.; Gait, M.J. Uridine 2′-carbamates: Facile Tools for Oligonucleotide 2′-functionalization. Curr. Protoc. Nucleic Acid Chem. 2004, 15, 4.21.1–4.21.26. [Google Scholar] [CrossRef] [PubMed]
- Seio, K.; Tawarada, R.; Sasami, T.; Serizawa, M.; Ise, M.; Ohkubo, A.; Sekine, M. Synthesis and hybridization of 2′-O-methyl-RNAs incorporating 2′-O-carbamoyluridine and unique participation of the carbamoyl group in U–G base pair. Bioorg. Med. Chem. 2009, 17, 7275–7280. [Google Scholar] [CrossRef] [PubMed]
- Seio, K.; Tokugawa, M.; Kanamori, T.; Tsunoda, H.; Ohkubo, A.; Sekine, M. Synthesis and properties of cationic 2′-O-[N-(4-aminobutyl)carbamoyl] modified oligonucleotides. Bioorg. Med. Chem. Lett. 2012, 22, 2470–2473. [Google Scholar] [CrossRef] [PubMed]
- El-Sagheer, A.H.; Brown, T. New Strategy for the Synthesis of Chemically Modified RNA Constructs Exemplified by Hairpin and Hammerhead Ribozymes. Proc. Natl. Acad. Sci. USA 2010, 107, 15329–15334. [Google Scholar] [CrossRef]
- Warminski, M.; Kowalska, J.; Jemielity, J. Synthesis of RNA 5′-Azides from 2′-O-Pivaloyloxymethyl-Protected RNAs and Their Reactivity in Azide–Alkyne Cycloaddition Reactions. Org. Lett. 2017, 19, 3624–3627. [Google Scholar] [CrossRef] [PubMed]
- Santner, T.; Hartl, M.; Bister, K.; Micura, R. Efficient Access to 3′-Terminal Azide-Modified RNA for Inverse Click-Labeling Patterns. Bioconjug. Chem. 2014, 25, 188–195. [Google Scholar] [CrossRef]
- Agustin, E.; Asare Okai, P.N.; Khan, I.; Miller, M.R.; Wang, R.; Sheng, J.; Royzen, M. A fast click–slow release strategy towards the HPLC-free synthesis of RNA. Chem. Commun. 2016, 52, 1405–1408. [Google Scholar] [CrossRef]
- Khan, I.; Seebald, L.M.; Robertson, N.M.; Yigit, M.V.; Royzen, M. Controlled in-cell activation of RNA therapeutics using bond-cleaving bio-orthogonal chemistry. Chem. Sci. 2017, 8, 5705–5712. [Google Scholar] [CrossRef]
- Mikkola, S.; Lönnberg, T.; Lönnberg, H. Phosphodiester models for cleavage of nucleic acids. Beilstein J. Org. Chem. 2018, 14, 803–837. [Google Scholar] [CrossRef]
- Tsuruoka, H.; Shohda Ki, K.; Wada, T.; Sekine, M. Kinetics and Mechanism of Facile and Selective Dephosphorylation of 2‘-Phosphorylated and 2‘-Thiophosphorylated Dinucleotides: Neighboring 3‘−5‘ Phosphodiester Promotes 2‘-Dephosphorylation. J. Org. Chem. 1997, 62, 2813–2822. [Google Scholar] [CrossRef] [PubMed]
- Pyshnyi, D.V.; Lomzov, A.A.; Pyshnaya, I.A.; Ivanova, E.M. Hybridization of the Bridged Oligonucleotides with DNA: Thermodynamic and Kinetic Studies. J. Biomol. Struct. Dyn. 2006, 23, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Petersheim, M.; Turner, D.H. Base-stacking and base-pairing contributions to helix stability: Thermodynamics of double-helix formation with CCGG, CCGGp, CCGGAp, ACCGGp, CCGGUp, and ACCGGUp. Biochemistry 1983, 22, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Pattanayek, R.; Sethaphong, L.; Pan, C.; Prhavc, M.; Prakash, T.P.; Manoharan, M.; Egli, M. Structural Rationalization of a Large Difference in RNA Affinity Despite a Small Difference in Chemistry between Two 2‘-O-Modified Nucleic Acid Analogues. J. Am. Chem. Soc. 2004, 126, 15006–15007. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Peng, C.G.; Matsuda, S.; Addepalli, H.; Jayaprakash, K.N.; Alam, M.R.; Mills, K.; Maier, M.A.; Charisse, K.; Sekine, M.; et al. Versatile Site-Specific Conjugation of Small Molecules to siRNA Using Click Chemistry. J. Org. Chem. 2011, 76, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Chernikov, I.V.; Gladkikh, D.V.; Meschaninova, M.I.; Ven’yaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Cholesterol-Containing Nuclease-Resistant siRNA Accumulates in Tumors in a Carrier-free Mode and Silences MDR1 Gene. Mol. Ther. Nucleic Acids 2017, 6, 209–220. [Google Scholar] [CrossRef]
- Nakajima, M.; Kasuya, T.; Yokota, S.; Onishi, R.; Ikehara, T.; Kugimiya, A.; Watanabe, A. Gene Silencing Activity and Hepatic Accumulation of Antisense Oligonucleotides Bearing Cholesterol-Conjugated Thiono Triester at the Gap Region. Nucleic Acid Ther. 2017, 27, 232–237. [Google Scholar] [CrossRef]
- Osborn, M.F.; Khvorova, A. Improving siRNA Delivery In Vivo Through Lipid Conjugation. Nucleic Acid Ther. 2018, 28, 128–136. [Google Scholar] [CrossRef]
- Shmushkovich, T.; Monopoli, K.R.; Homsy, D.; Leyfer, D.; Betancur-Boissel, M.; Khvorova, A.; Wolfson, A.D. Functional features defining the efficacy of cholesterol-conjugated, self-deliverable, chemically modified siRNAs. Nucleic Acids Res. 2018, 46, 10905–10916. [Google Scholar] [CrossRef]
- Ryzhakov, G.; Randow, F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. Embo J. 2007, 26, 3180–3190. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rON | Sequence 1 | R | Isolated Yield/Conversion 2, % | RT 3, min | Molecular Weight | |
|---|---|---|---|---|---|---|
| Found 4 m/z | Calcd m/z | |||||
| 2 | 5′-r(ACGUACGU)dT | - | 12.5/85.1 | 6.3 | 2812.5 | 2813.8 |
| 2a | 5′-r(ACGU*ACGU)dT | a | 7.4/78.5 | 6.4 | 2991.9 | 2992.0 |
| 2b | 5′-r(ACGU*ACGU)dT | b | 3.1/~80 * | n/d | 3405.6 | 3404.6 |
| 2c | 5′-r(ACGU*ACGU)dT | c | 5.6/84.5 | 9.9 | 3523.6 | 3523.7 |
| 2d | 5′-r(ACGU*ACGU)dT | d | 11.2/85.2 | 6.9 | 3084.9 † | 3084.1 |
| 2e | 5′-r(ACGU*ACGU)dT | e | 10.2/81.4 | 13.4 | 3529.5 †,5 | 3486.5 |
| 2f | 5′-r(ACGU*ACGU)dT | f | 10.5/80.3 | 6.4 | 2926.2 | 2925.9 |
| 2g | 5′-r(ACGU*ACGU)dT | g | 5.4/90.9 | 12.8 | 3365.4 | 3367.6 |
| 3a | 5′-r(GUGAA*AUG)dC | a | 12.5/89.0 | 6.4 | 3041.7 | 3041.1 |
| 4a | 5′-r(GUGAU*AUG)dC | a | 18.4/84.6 | 6.4 | 3018.3 | 3018.1 |
| 5a | 5′-r(GUGAC*AUG)dC | a | 20.1/86.7 | 6.4 | 3017.9 | 3017.9 |
| 6a | 5′-r(GUGAG*AUG)dC | a | 19.5/87.4 | 6.5 | 3057.2 | 3057.9 |
| 7a | 5′-r(GCCACAACGUCUAUAUCAU*)dTdT | a | 5.6/78.9 | 7.2 | 6756.2 † | 6753.1 |
| 7b | 5′-r(GCCACAACGUCUAUAUCAU*)dTdT | d | 6.7/74.2 | 6.9 | 6847.8 † | 6844.1 |
| 8a | 5′-Alk-r(GCCACAACGUCUAUAUCAU*)dTdT | a | 6.0/69.3 | 7.3 | 7024.0 † | 7022.4 |
| 8b | 5′-Fluo-r(GCCACAACGUCUAUAUCAU*)dTdT | b | 3.6/~50 * | n/d | 7894.3 † | 7894.5 |
| 9a | 5′-Alk-r(GCCACAACGUCUAUA*UCAU)dTdT | a | 5.7/76.7 | 7.3 | 7024.0 † | 7022.4 |
| 9b | 5′-Fluo-r(GCCACAACGUCUAUA*UCAU)dTdT | b | 3.5/~45 * | n/d | 7895.0 † | 7894.5 |
| 10a | 5′-Alk-r(GCCACAACGUCU*AUAUCAU)dTdT | a | 5.7/75.6 | 7.3 | 7024.0 † | 7022.4 |
| 10b | 5′-Fluo-r(GCCACAACGUCU*AUAUCAU)dTdT | b | 3.9/~45 * | n/d | 7895.5 † | 7894.5 |
| ON | Sequence 1, 5′–3′ | Tm (Δ Tm 2), °C | |||
|---|---|---|---|---|---|
| Target | 5′-r(GCAUBUCAC) | B = U | A | G | C |
| 3 | 5′-r(GUGAAAUG)dC | 39.0 | 23 (−18.5) | 25.5 (−20.5) | 27.0 (−18.5) |
| 4 | 5′-r(GUGAUAUG)dC | 26.0 (−13.0) | 41.5 | 37.0 (−9.0) | 26.0 (−19.0) |
| 5 | 5′-r(GUGACAUG)dC | 22.5 (−16.5) | 25.5 (−16.0) | 46.0 | 24.0 (−21.0) |
| 6 | 5′-r(GUGAGAUG)dC | 35.5 (−3.5) | 25.0 (−16.5) | 25.5 (−20.5) | 47.5 |
| 3a | 5′-r(GUGAA*AUG)dC | 32.0 | 21.0 (−14.0) | 18.0 (−22.0) | 23.5 (−17.0) |
| 4a | 5′-r(GUGAU*AUG)dC | 17.7 (−14.3) | 35.0 | 28.0 (−12.0) | 21.5 (−19.0) |
| 5a | 5′-r(GUGAC*AUG)dC | 20.0 (−12.0) | 20.0 (−15.0) | 40.0 | 18.0 (−22.5) |
| 6a | 5′-r(GUGAG*AUG)dC | 29.0 (−3.0) | 21.0 (−14.0) | 27.0 (−13.0) | 40.5 |
| S/AS | Sequences | Representation |
|---|---|---|
| 8b/AS | 5′-Fluo-r(GCCACAACGUCUAUAUCAU*)dTdT3′-dTdT-r(CGGUGUUGCAGAUAUAGUA) |  |
| 9b/AS | 5′-Fluo-r(GCCACAACGUCUAUA*UCAU)dTdT 3′-dTdT-r(CGGUGUUGCAGAUAUAGUA) |  |
| 10b/AS | 5′-Fluo-r(GCCACAACGUCU*AUAUCAU)dTdT 3′-dTdT-r(CGGUGUUGCAGAUAUAGUA) |  |
| 12/AS | 5′-Fluo-r(GCCACAACGUCUAUAUCAU)dTdT 3′-dTdT-r(CGGUGUUGCAGAUAUAGUA) |  |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasheninina, O.A.; Fishman, V.S.; Lomzov, A.A.; Ustinov, A.V.; Venyaminova, A.G. Postsynthetic On-Column 2′ Functionalization of RNA by Convenient Versatile Method. Int. J. Mol. Sci. 2020, 21, 5127. https://doi.org/10.3390/ijms21145127
Krasheninina OA, Fishman VS, Lomzov AA, Ustinov AV, Venyaminova AG. Postsynthetic On-Column 2′ Functionalization of RNA by Convenient Versatile Method. International Journal of Molecular Sciences. 2020; 21(14):5127. https://doi.org/10.3390/ijms21145127
Chicago/Turabian StyleKrasheninina, Olga A., Veniamin S. Fishman, Alexander A. Lomzov, Alexey V. Ustinov, and Alya G. Venyaminova. 2020. "Postsynthetic On-Column 2′ Functionalization of RNA by Convenient Versatile Method" International Journal of Molecular Sciences 21, no. 14: 5127. https://doi.org/10.3390/ijms21145127
APA StyleKrasheninina, O. A., Fishman, V. S., Lomzov, A. A., Ustinov, A. V., & Venyaminova, A. G. (2020). Postsynthetic On-Column 2′ Functionalization of RNA by Convenient Versatile Method. International Journal of Molecular Sciences, 21(14), 5127. https://doi.org/10.3390/ijms21145127