Anti-CXCL4 Antibody Reactivity Is Present in Systemic Sclerosis (SSc) and Correlates with the SSc Type I Interferon Signature

 ,
,  ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
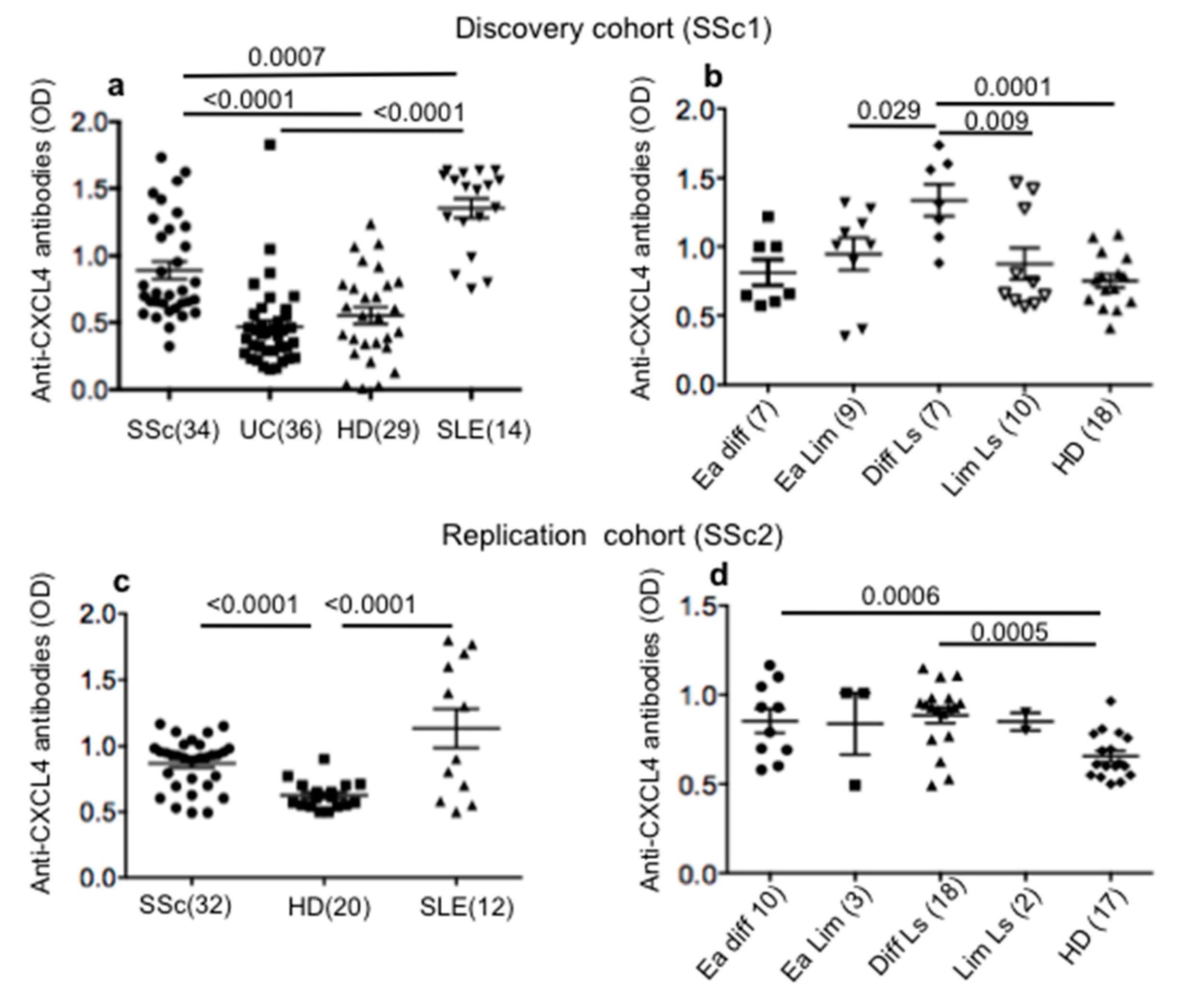
2.1. SSc Patients Show Consistent Antibody Reactivity to CXCL4
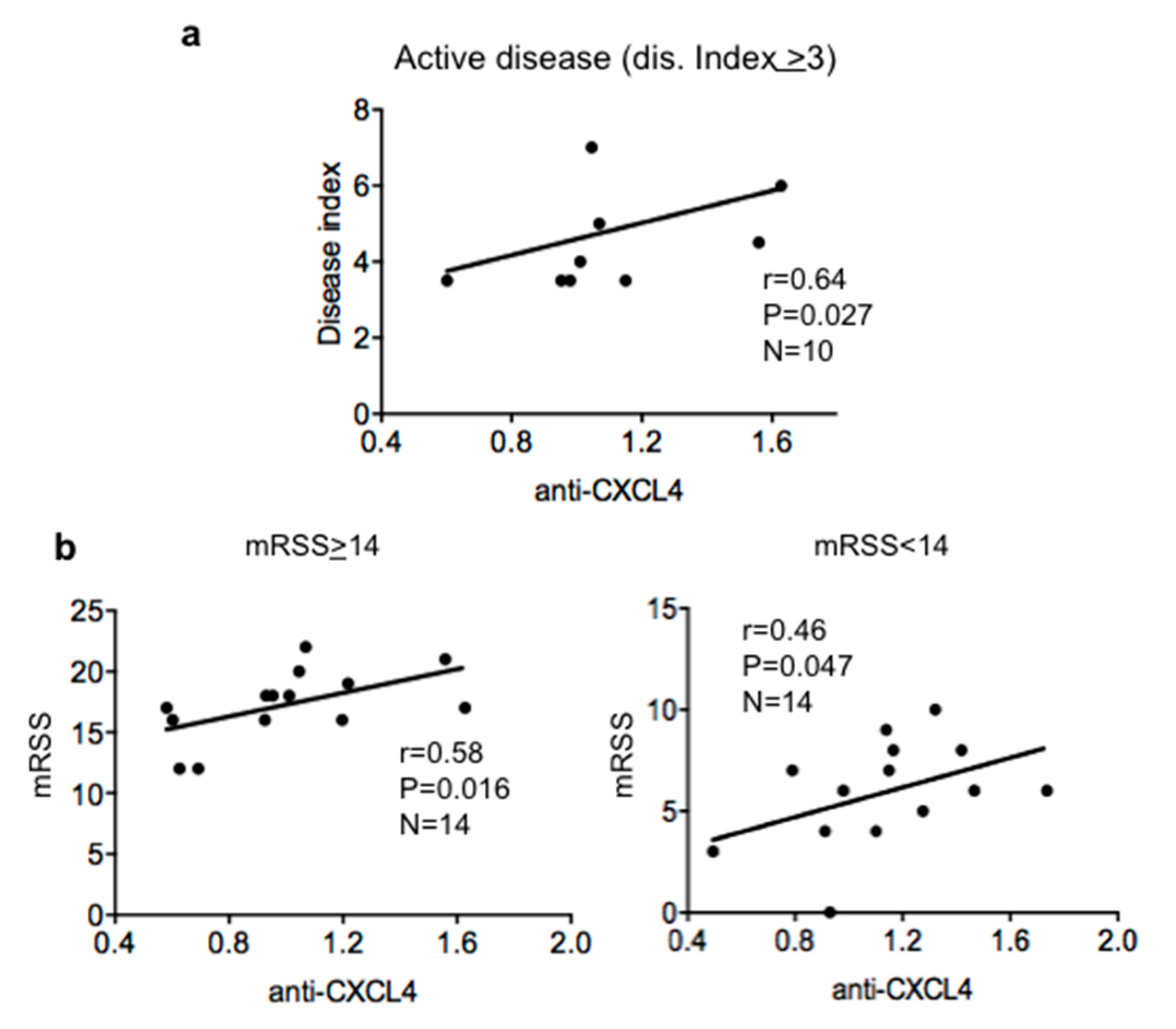
2.2. Antibody Reactivity to CXCL4 Correlates with Disease Score and Modified Rodnan Skin Score (mRSS) in Patients with Active Disease
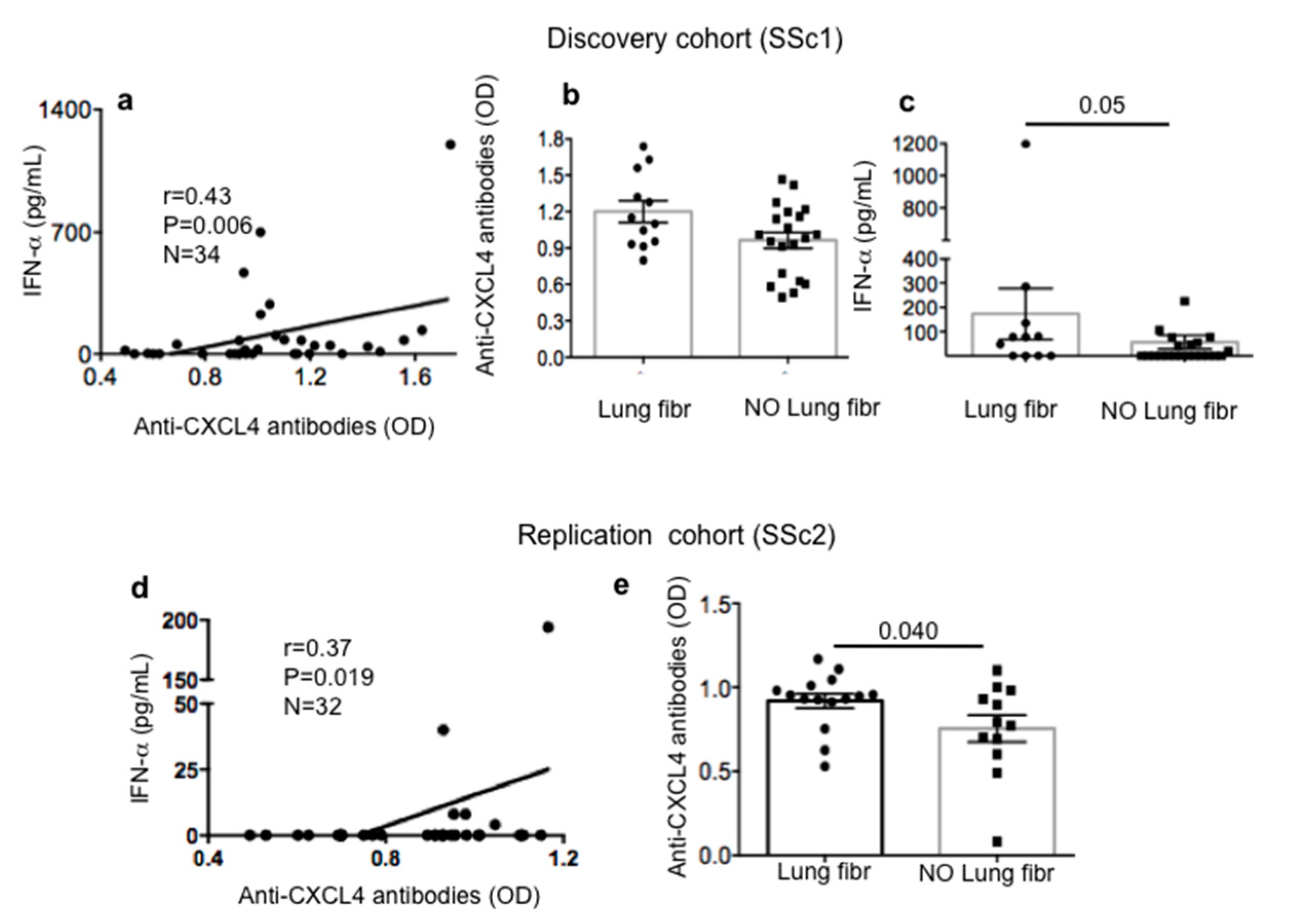
2.3. Anti-CXCL4 Antibody Reactivity in SSc Patients Correlates with Serum/Plasma IFN-α and Is Higher in SSc Patients with Lung Fibrosis
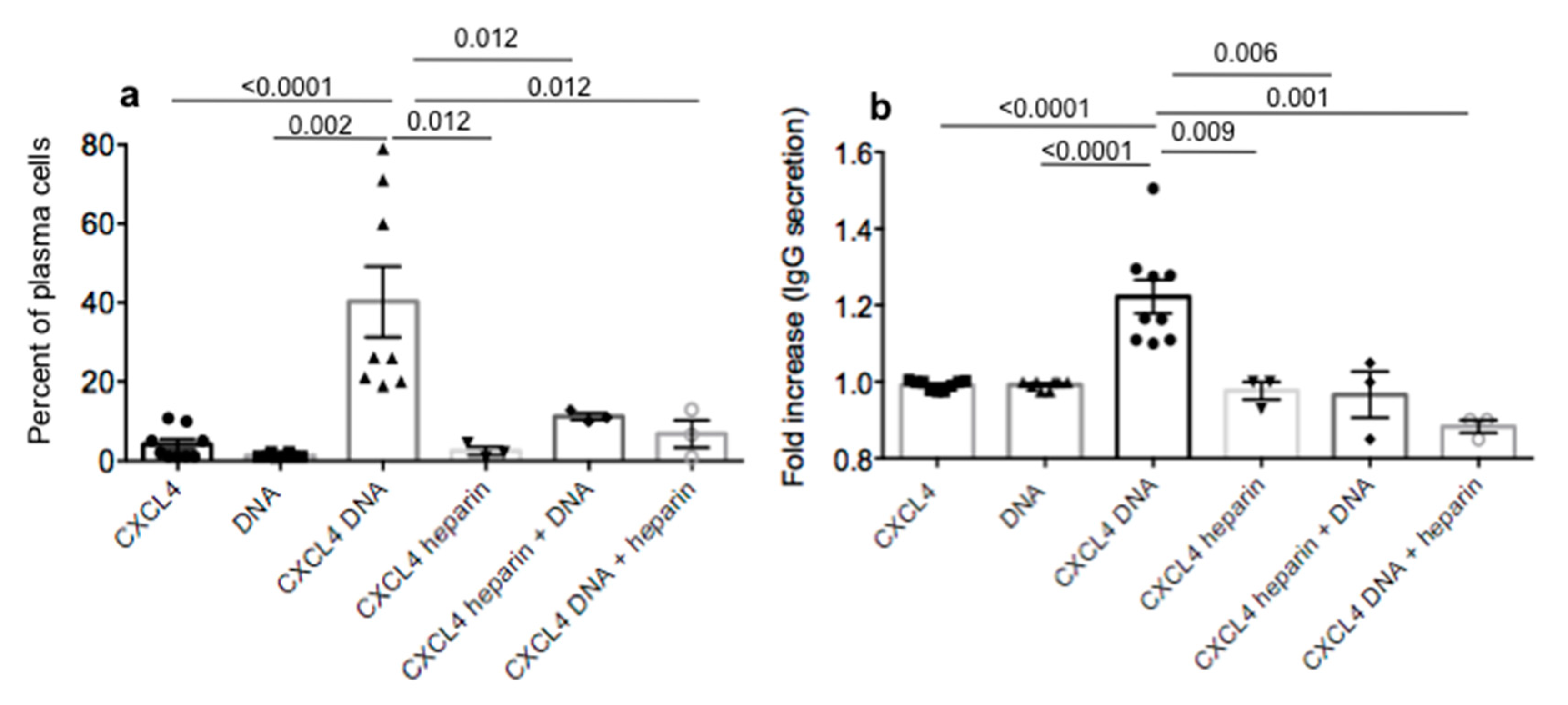
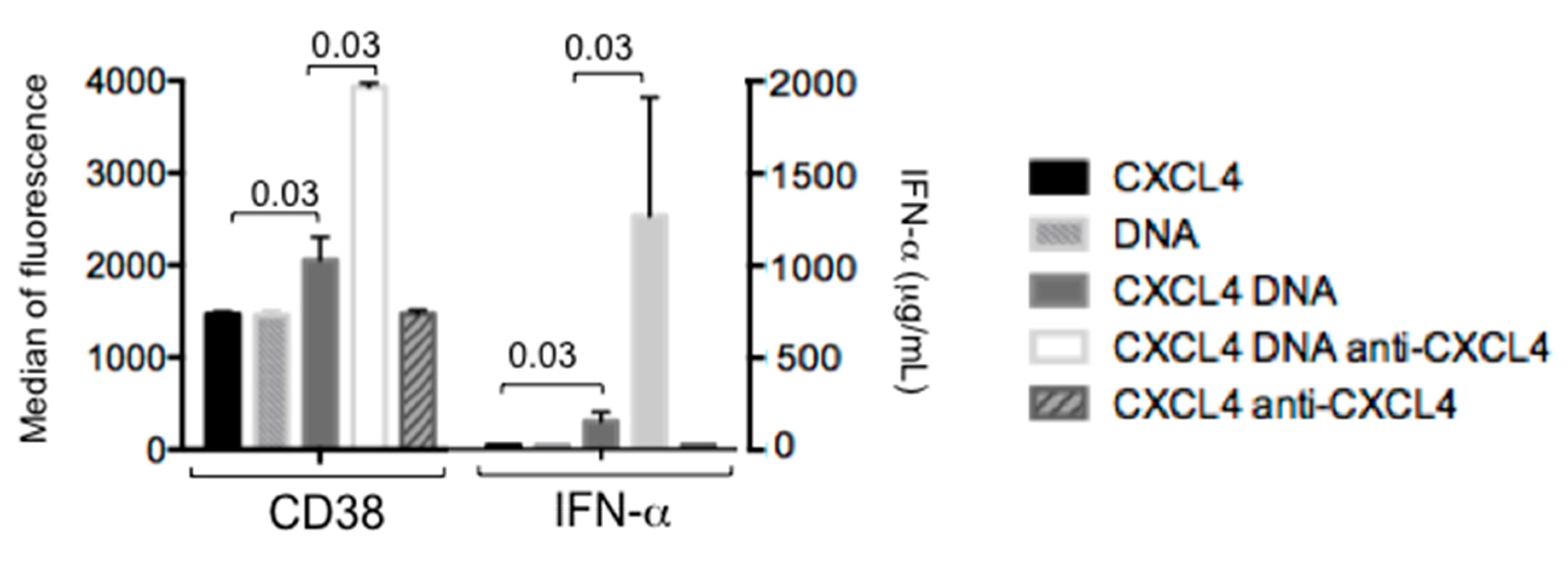
2.4. CXCL4 in Complex with Nucleic Acids Stimulates Memory B-Cells to Become Antibody-Secreting Plasma Cells
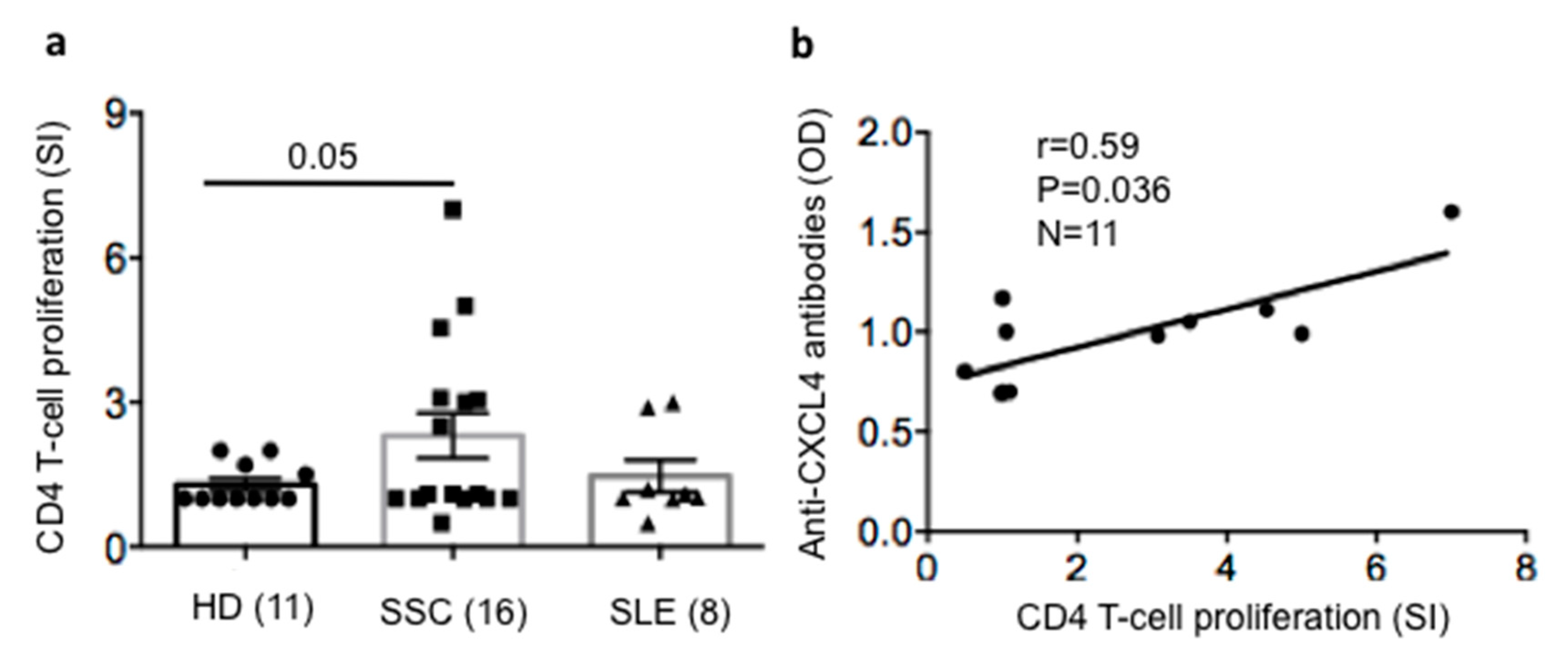
2.5. SSc T-Cells Proliferate to CXCL4 and Their Proliferation Correlates with Anti-CXCL4 Antibody Reactivity
2.6. Autoantibodies to CXCL4 Can Implement IFN-α Production by pDCs
3. Discussion
4. Materials and Methods
4.1. Human Study and Samples
4.2. Antigens, Antibodies for Functional Assays and Flow Cytometry
4.3. T-Cell Proliferation Assay
4.4. Production of Human/Bacterial DNA/RNA
4.5. Isolation and Stimulation of pDCs
4.6. IFN-α Determination in pDC Cell Cultures and Sera/Plasma
4.7. Isolation of B-Cells from Buffy Coats
4.8. Stimulation of Purified B-Cells
4.9. ELISA for Anti-CXCL4 Autoantibodies and Human IgG Detection in Sera/Plasma and Culture Supernatants, Respectively
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gabrielli, A.; Avvedimento, E.V.; Krieg, T. Scleroderma. N. Engl. J. Med. 2009, 360, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis—A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Frasca, L.; Lande, R. Toll-like receptors in mediating pathogenesis in systemic sclerosis review. Clin. Exp. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Maehara, T.; Kaneko, N.; Perugino, C.A.; Mattoo, H.; Kers, J.; Allard-Chamard, H.; Mahajan, V.S.; Liu, H.; Murphy, J.H.; Ghebremichael, M.; et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J. Clin. Investig. 2020, 130, 2451–2464. [Google Scholar] [CrossRef]
- Cutolo, M.; Soldano, S.; Smith, V. Pathophysiology of systemic sclerosis: Current understanding and new insights. Expert Rev. Clin. Immunol. 2019, 15, 753–764. [Google Scholar] [CrossRef]
- O’Reilly, S. Toll-like receptors in systemic sclerosis: An emerging target. Immunol. Lett. 2018, 195, 2–8. [Google Scholar] [CrossRef] [PubMed]
- van Bon, L.; Affandi, A.J.; Broen, J.; Christmann, R.B.; Marijnissen, R.J.; Stawski, L.; Farina, G.A.; Stifano, G.; Mathes, A.L.; Cossu, M.; et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N. Engl. J. Med. 2014, 370, 433–443. [Google Scholar] [CrossRef]
- Ah Kioon, M.D.; Tripodo, C.; Fernandez, D.; Kirou, K.A.; Spiera, R.F.; Crow, M.K.; Gordon, J.K.; Barrat, F.J. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Kim, D.; Peck, A.; Santer, D.; Patole, P.; Schwartz, S.M.; Molitor, J.A.; Arnett, F.C.; Elkon, K.B. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I: Association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum. 2008, 58, 2163–2173. [Google Scholar] [CrossRef]
- Eloranta, M.-L.; Franck-Larsson, K.; Lövgren, T.; Kalamajski, S.; Rönnblom, A.; Rubin, K.; Alm, G.V.; Rönnblom, L. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann. Rheum Dis. 2010, 69, 1396–1402. [Google Scholar] [CrossRef]
- Brkic, Z.; van Bon, L.; Cossu, M.; van Helden-Meeuwsen, C.G.; Vonk, M.C.; Knaapen, H.; van den Berg, W.; Dalm, V.A. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann. Rheum. Dis. 2016, 75, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Lee, E.Y.; Palazzo, R.; Marinari, B.; Pietraforte, I.; Santos, G.S.; Mattenberger, Y.; Spadaro, F.; Stefanantoni, K.; Iannace, N.; et al. CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-alpha production in systemic sclerosis. Nat. Commun. 2019, 10, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.K.; Verstraete, K.; Heyndrickx, I.; Gevaert, E.; Aegerter, H.; Percier, J.-M.; Deswarte, K.; Verschueren, K.H.G.; Dansercoer, A.; Gras, D.; et al. Protein crystallization promotes type 2 immunity and is reversible by antibody treatment. Science 2019, 364. [Google Scholar] [CrossRef] [PubMed]
- Arepally, G.M.; Cines, D.B. Pathogenesis of heparin-induced thrombocytopenia. Transl. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, Y.-P.; Yu, N.; Jia, Y.-X.; Wan, S.-J.; Wang, F.-Y. Serum platelet factor 4 is a reliable activity parameter in adult patients with inflammatory bowel disease: A pilot study. Medicine 2017, 96. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Tanaka, Y.; Okazaki, Y.; Kaburaki, J.; Ikeda, Y.; Kuwana, M. Heparin-dependent and -independent anti-platelet factor 4 autoantibodies in patients with systemic lupus erythematosus. Rheumatology 2012, 51, 1721–1728. [Google Scholar] [CrossRef]
- Melsens, K.F.; De Keyser, S.; Decuman, Y.; Piette, E.; Vandecasteele, E.; Smith, V. Disease activity indices in systemic sclerosis: A systematic. Clin. Exp. Rheumatol. 2016, 34. [Google Scholar]
- Kiefer, K.; Oropallo, M.A.; Cancro, M.P.; Marshak-Rothstein, A. Role of type I interferons in the activation of autoreactive B cells. Immunol. Cell Biol. 2012, 90, 498–504. [Google Scholar] [CrossRef]
- Tellier, J.; Nutt, S.L. The programming of an antibody-secreting machine. J. Immunol. 2019, 49, 30–33. [Google Scholar]
- Gestermann, N.; Di Domizio, J.; Lande, R.; Demaria, O.; Frasca, L.; Feldmeyer, L.; Di Lucca, J.; Gilliet, M. Netting neutrophils activate autoreactive B cells in Lupus. J. Immunol. 2018, 200, 3364–3371. [Google Scholar] [CrossRef]
- Capolunghi, F.; Rosado, M.M.; Cascioli, S.; Girolami, E.; Bordasco, S.; Vivarelli, M.; Ruggiero, B.; Cortis, E.; Insalaco, A.; Fantò, N.; et al. Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology 2010, 49, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-H.; Lee, H.; Yamamoto, M.; Jones, L.A.; Dayalan, J.; Hopkins, R.; Zhou, X.J.; Yarovinsky, F.; Connolly, J.E.; de Lafaille, M.A.C.; et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J. Immunol. 2012, 189, 5786–5796. [Google Scholar] [CrossRef]
- Qin, L.; Waseem, T.C.; Sahoo, A.; Bieerkehazhi, S.; Zhou, H.; Galkina, E.V.; Nurieva, R. Insights Into the Molecular Mechanisms of T Follicular Helper-Mediated Immunity and Pathology. Front. Immunol. 2018, 9, 1884. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 5, 5621–5635. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Palazzo, R.; Gestermann, N.; Jandus, G.; Falchi, M.; Spadaro, F.; Riccieri, V.; James, E.A.; Butera, A.; Boirivant, M.; et al. Native/citrullinated LL37-specific T-cells help autoantibody production in Systemic Lupus Erythematosus. Sci. Rep. 2020, 10, 5851–5864. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.; Meenagh, A.; Single, R.; McNally, M.; Kelly, P.; Nelson, M.P.; Meyer, D.; Lancaster, A.; Thomson, G.; Middleton, D. High Resolution HLA-DRB1 Identification of a Caucasian Population. Hum. Immunol. 2004, 65, 66–77. [Google Scholar] [CrossRef]
- Niu, Z.; Zhang, P.; Tong, T. Value of HLA-DR genotype in systemic lupus erythematosus and lupus nephritis: A meta-analysis. Int. J. Rheum. Dis. 2015, 18, 17–28. [Google Scholar] [CrossRef]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A Long Journey From Activation Markers to Multifunctional Molecules. Clin. Cytom. 2013, 84, 207–217. [Google Scholar] [CrossRef]
- Bauvois, B.; Durant, L.; Laboureau, J.; Barthelemy, E.; Rouillard, D.; Boulla, G.P.; Deterre, P. Upregulation of CD38 Gene Expression in Leukemic B Cells by Interferon Types I and II. J. Interferon Cytokine Res. 1999, 19, 1059–1066. [Google Scholar] [CrossRef]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of the CXC chemokines platelet factor-4 (CXCL4/PF-4) and its variant (CXCL4L1/PF-4var) in inflammation, angiogenesis and cancer. Cytokine Growth Factor Rev. 2011, 22, 1–18. [Google Scholar] [CrossRef]
- Wu, M.; Assassi, S. The role of type 1 interferon in systemic sclerosis. Front. Immunol. 2013, 4, 266. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, S.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 19–38. [Google Scholar] [CrossRef] [PubMed]
- Bellando-Randone, S.; Matucci-Cerinic, M. From Raynaud’s Phenomenon to Very Early Diagnosis of Systemic Sclerosis—The VEDOSS approach. Curr. Rheumatol. Rev. 2013, 9, 245–248. [Google Scholar] [CrossRef]
- Homey, B.; Alenius, H.; Muller, A.; Soto, H.; Bowman, E.P.; Yuan, W.; McEvoy, L.; Lauerma, A.I.; Assmann, T.; Bunemann, E.; et al. CCL27-CCR10 interactions regulate T cell-mediated skin inflammation. Nat. Med. 2002, 8, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Frasca, L. Personal Observations. UNIGE, Geneva, Switzerland, July 2015. [Google Scholar]
- Scherlinger, M.; Guillotin, V.; Truchetet, M.E.; Contin-Bordes, C.; Sisirak, V.; Duffau, P.; Lazaro, E.; Richez, C.; Blanco, P. Systemic lupus erythematosus and systemic sclerosis: All roads lead to platelets. Autoimmun. Rev. 2018, 17, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Krauel, K.; Jaax, M.; Renne, T.; Helm, C.A.; Hammerschmidt, S.; Delcea, M.; Greinacher, A. Polyphosphates form antigenic complexes with platelet factor 4 (PF4) and enhance PF4- binding to bacteria. Thromb. Haemost. 2015, 114, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Yarovoi, S.V.; Zaitsev, S.V.; Lebedeva, T.; Rauova, L.; Poncz, M.; Arepally, G.M.; Khandelwal, S.; Stepanova, V.; Rux, A.H.; et al. Polyphosphate/platelet factor 4 complexes can mediate heparin-independent platelet activation in heparin-induced thrombocytopenia. Blood Adv. 2016, 1, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef]
- Gollomp, K.; Kim, M.; Johnston, I.; Hayes, V.; Welsh, J.; Arepally, G.M.; Kahn, M.; Lambert, M.P.; Cuker, A.; Cines, D.B.; et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight. 2018, 3, 42. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Basciano, P.A.; Knopman, J.; Bernstein, R.A. Spontaneous heparin-induced thrombocytopenia syndrome: 2 new cases and a proposal for defining this disorder. Blood 2014, 123, 3651–3654. [Google Scholar] [CrossRef]
- Bacsi, S.; De Palma, R.; Visentin, G.P.; Gorski, J.; Aster, R.H. Complexes of heparin and platelet factor 4 specifically stimulate T cells from patients with heparin-induced thrombocytopenia/thrombosis. Blood 1999, 94, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, S.; Rauova, L.; McCracken, E.K.; Goss, C.M.; Sachais, B.S.; McKenzie, S.E.; Reilly, M.P.; Gunn, M.D.; Cines, D.B.; Poncz, M.; et al. PF4/heparin complexes are T cell-dependent antigens. Blood 2005, 106, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Ibañez, D.; Urowitz, M.B. Systemic lupus erythematosus disease activity index 2000. J. Rheumatol. 2002, 29, 288–291. [Google Scholar]
- Rutgeerts, P.; Sandborn, W.J.; Feagan, B.G.; Reinisch, W.; Olson, A.; Johanns, J.; Travers, S.; Rachmilewitz, D.; Hanauer, S.B.; Lichtenstein, G.R.; et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2005, 353, 2462–2476. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lande, R.; Mennella, A.; Palazzo, R.; Pietraforte, I.; Stefanantoni, K.; Iannace, N.; Butera, A.; Boirivant, M.; Pica, R.; Conrad, C.; et al. Anti-CXCL4 Antibody Reactivity Is Present in Systemic Sclerosis (SSc) and Correlates with the SSc Type I Interferon Signature. Int. J. Mol. Sci. 2020, 21, 5102. https://doi.org/10.3390/ijms21145102
Lande R, Mennella A, Palazzo R, Pietraforte I, Stefanantoni K, Iannace N, Butera A, Boirivant M, Pica R, Conrad C, et al. Anti-CXCL4 Antibody Reactivity Is Present in Systemic Sclerosis (SSc) and Correlates with the SSc Type I Interferon Signature. International Journal of Molecular Sciences. 2020; 21(14):5102. https://doi.org/10.3390/ijms21145102
Chicago/Turabian StyleLande, Roberto, Anna Mennella, Raffaella Palazzo, Immacolata Pietraforte, Katia Stefanantoni, Nicoletta Iannace, Alessia Butera, Monica Boirivant, Roberta Pica, Curdin Conrad, and et al. 2020. "Anti-CXCL4 Antibody Reactivity Is Present in Systemic Sclerosis (SSc) and Correlates with the SSc Type I Interferon Signature" International Journal of Molecular Sciences 21, no. 14: 5102. https://doi.org/10.3390/ijms21145102
APA StyleLande, R., Mennella, A., Palazzo, R., Pietraforte, I., Stefanantoni, K., Iannace, N., Butera, A., Boirivant, M., Pica, R., Conrad, C., Chizzolini, C., Riccieri, V., & Frasca, L. (2020). Anti-CXCL4 Antibody Reactivity Is Present in Systemic Sclerosis (SSc) and Correlates with the SSc Type I Interferon Signature. International Journal of Molecular Sciences, 21(14), 5102. https://doi.org/10.3390/ijms21145102