Expression Profiling and Functional Characterization of miR-26a and miR-130a in Regulating Zhongwei Goat Hair Development via the TGF-β/SMAD Pathway

 ,
,
Abstract
1. Introduction
2. Results
2.1. Identification of miRNAs in Goat Hair Follicles
2.2. Analysis and Validation of DE miRNAs
2.3. Prediction and Functional Annotation of the Target Genes of Identified miRNAs
2.4. Integrated miRNA and mRNA Analysis Identified Target Genes Involved in Curly Wool Development
2.5. Isolation and Identification of Dermal Papilla Cells In Vitro
2.6. Effects of miR-26a and miR-130a on Cell Proliferation
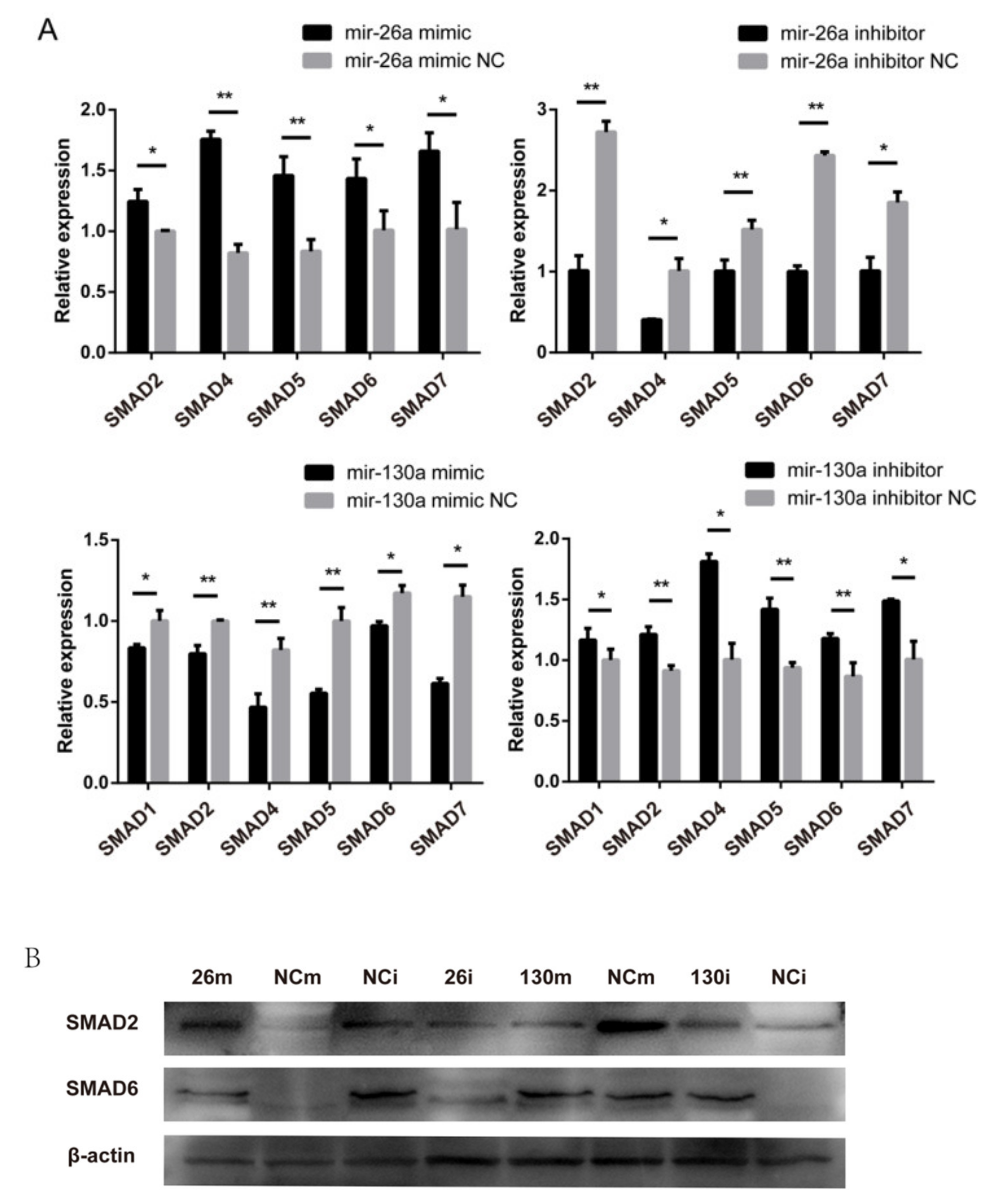
2.7. Effects of miR-26a and miR-130a on the TGF-β/SMAD Pathway
2.8. Dual Luciferase Reporter Gene Detection
3. Discussion
4. Materials and Methods
4.1. Animals and Samples
4.2. Total RNA Extraction and Small RNA Library Preparation
4.3. Small RNA Sequencing and Read Processing
4.4. The Sequencing Results for Small RNA Were Verified by RT-qPCR
4.5. Target Gene Prediction and Functional Notation of Differentially Expressed miRNAs
4.6. Differentially Expressed miRNA-Target Gene Interaction Network Analysis
4.7. Dermal Papilla Cell Separation and Culture
4.8. Dermal Papilla Cell Identification
4.9. Overexpression and Inhibition of miR-26a and miR-130a
4.10. Cell Proliferation Assay
4.11. RNA Extraction, Reverse Transcription and RT-qPCR
4.12. Western Blot Analysis
4.13. Luciferase Reporter Gene Assay
4.14. Statistical Analysis
4.15. Availability of Data and Materials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| miRNA | microRNA |
| DE | different expression |
| Seq | sequence |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PAGE | polyacrylamide gel electrophoresis |
| RT-qPCR | Quantitative real time polymerase chain reaction |
References
- Auber, L. VII.—The anatomy of follicles producing wool-fibres, with special reference to keratinization. Earth Environ. Sci. Trans. R. Soc. Edinb. 1952, 62, 191–254. [Google Scholar] [CrossRef]
- Nay, T.; Jackson, N. Effect of changes in nutritional level on the depth and curvature of wool follicles in Australian Merino sheep. Aust. J. Agric. Res. 1973, 24, 439–447. [Google Scholar] [CrossRef]
- Thibaut, S.; Gaillard, O.; Bouhanna, P.; Cannell, D.W.; Bernard, B.A. Human hair shape is programmed from the bulb. Br. J. Dermatol. 2005, 152, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.C.; Orwin, D.F.; Gillespie, J.M. Structure and biochemistry of mammalian hard keratin. Electron. Microsc. Rev. 1991, 4, 47–83. [Google Scholar] [CrossRef]
- Teh, M.T.; Blaydon, D.; Ghali, L.R.; Briggs, V.; Edmunds, S.; Pantazi, E.; Barnes, M.R.; Leigh, I.M.; Kelsell, D.P.; Philpott, M.P. Role for WNT16B in human epidermal keratinocyte proliferation and differentiation. J. Cell Sci. 2007, 120 Pt 2, 330–339. [Google Scholar] [CrossRef]
- Harland, D.P.; Vernon, J.A.; Woods, J.L.; Nagase, S.; Itou, T.; Koike, K.; Scobie, D.A.; Grosvenor, A.J.; Dyer, J.M.; Clerens, S. Intrinsic curvature in wool fibres is determined by the relative length of orthocortical and paracortical cells. J. Exp. Biol. 2018. [Google Scholar] [CrossRef]
- Plowman, J.E.; Harland, D.P.; Scobie, D.R.; O’Connell, D.; Thomas, A.; Brorens, P.H.; Richena, M.; Meenken, E.; Phillips, A.J.; Vernon, J.A.; et al. Differences between ultrastructure and protein composition in straight hair fibres. Zoology (Jena) 2019, 133, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Xiang, H.; Zhang, G.; Ding, J.P.; Zhang, Z.J.; Zhang, Y.H.; Han, J.L.; Ma, Y.H.; Zhang, X.R. Identification of complete linkage disequilibrium in the DSG4 gene and its association with wool length and crimp in Chinese indigenous sheep. Genet Mol Res. 2014, 13, 5617–5625. [Google Scholar] [CrossRef]
- Telerman, S.B.; Rognoni, E.; Sequeira, I.; Pisco, A.O.; Lichtenberger, B.M.; Culley, O.J.; Viswanathan, P.; Driskell, R.R.; Watt, F.M. Dermal blimp1 acts downstream of epidermal TGFbeta and Wnt/beta-catenin to regulate hair follicle formation and growth. J. Invest. Dermatol. 2017, 137, 2270–2281. [Google Scholar] [CrossRef]
- Lu, C.P.; Polak, L.; Keyes, B.E.; Fuchs, E. Spatiotemporal antagonism in mesenchymal-epithelial signaling in sweat versus hair fate decision. Science 2016, 354. [Google Scholar] [CrossRef]
- Tomann, P.; Paus, R.; Millar, S.E.; Scheidereit, C.; Schmidt-Ullrich, R. Lhx2 is a direct NF-kappaB target gene that promotes primary hair follicle placode down-growth. Development 2016, 143, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Rishikaysh, P.; Dev, K.; Diaz, D.; Qureshi, W.M.; Filip, S.; Mokry, J. Signaling involved in hair follicle morphogenesis and development. Int. J. Mol. Sci. 2014, 15, 1647–1670. [Google Scholar] [CrossRef] [PubMed]
- Andl, T.; Murchison, E.P.; Liu, F.; Zhang, Y.; Yunta-Gonzalez, M.; Tobias, J.W.; Andl, C.D.; Seykora, J.T.; Hannon, G.J.; Millar, S.E. The miRNA-processing enzyme dicer is essential for the morphogenesis and maintenance of hair follicles. Curr. Biol. 2006, 16, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; O’Carroll, D.; Pasolli, H.A.; Zhang, Z.; Dietrich, F.S.; Tarakhovsky, A.; Fuchs, E. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat. Genet. 2006, 38, 356–362. [Google Scholar] [CrossRef]
- Tang, X.H.; Liu, G.B.; Du, X.Y.; Cao, J.H.; Li, X.Y.; Luo, Z.; Yu, M.; Zhao, S.H. The study on expression of miR-184 and miR-205 at different developmental stages of hair follicle and other tissues of sheep. Chin. J. Anim. Vet. Sci. 2012, 43, 1897–1903. [Google Scholar]
- Kang, X.; Liu, G.; Liu, Y.; Xu, Q.; Zhang, M.; Fang, M. Transcriptome profile at different physiological stages reveals potential mode for curly fleece in Chinese tan sheep. PLoS ONE 2013, 8, e71763. [Google Scholar] [CrossRef]
- Nissimov, J.N.; Das, C.A. Hair curvature: A natural dialectic and review. Biol. Rev. Camb. Philos. Soc. 2014, 89, 723–766. [Google Scholar] [CrossRef]
- Bai, L.; Gong, H.; Zhou, H.; Tao, J.; Hickford, J. A nucleotide substitution in the ovine KAP20-2 gene leads to a premature stop codon that affects wool fibre curvature. Anim. Genet. 2018, 49, 357–358. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, G.; Zhang, R.; Guo, J.; Li, C.; Martin, G.; Chen, Y.; Wang, X. Comparative proteomic analyses using iTRAQ-labeling provides insights into fiber diversity in sheep and goats. J. Proteom. 2018, 172, 82–88. [Google Scholar] [CrossRef]
- Rong, E.G.; Yang, H.; Zhang, Z.W.; Wang, Z.P.; Yan, X.H.; Li, H.; Wang, N. Association of methionine synthase gene polymorphisms with wool production and quality traits in Chinese Merino population. J. Anim. Sci. 2015, 93, 4601–4609. [Google Scholar] [CrossRef]
- Xiao, P.; Zhong, T.; Liu, Z.; Ding, Y.; Guan, W.; He, X.; Pu, Y.; Jiang, L.; Ma, Y.; Zhao, Q. Integrated analysis of methylome and transcriptome changes reveals the underlying regulatory signatures driving curly wool transformation in Chinese Zhongwei goats. Front. Genet. 2019, 10, 1263. [Google Scholar] [CrossRef]
- Pedrioli, D.M.; Karpanen, T.; Dabouras, V.; Jurisic, G.; van de Hoek, G.; Shin, J.W.; Marino, D.; Kalin, R.E.; Leidel, S.; Cinelli, P.; et al. miR-31 functions as a negative regulator of lymphatic vascular lineage-specific differentiation in vitro and vascular development in vivo. Mol. Cell. Biol. 2010, 30, 3620–3634. [Google Scholar] [CrossRef] [PubMed]
- Mecklenburg, L.; Tobin, D.J.; Muller-Rover, S.; Handjiski, B.; Wendt, G.; Peters, E.M.; Pohl, S.; Moll, I.; Paus, R. Active hair growth (anagen) is associated with angiogenesis. J. Invest. Dermatol. 2000, 114, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Ahmed, M.I.; Vlahov, N.V.; Fessing, M.Y.; Gill, J.H.; Sharov, A.A.; Botchkareva, N.V. Micro-RNA-31 controls hair cycle-associated changes in gene expression programs of the skin and hair follicle. Faseb. J. 2010, 24, 3869–3881. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, H.; Li, H.; Zhao, Y.; Lai, S.; Yu, X.; Cai, T.; Du, C.; Zhang, W.; Li, J. Identification of conserved and novel microRNAs in cashmere goat skin by deep sequencing. PLoS ONE 2012, 7, e50001. [Google Scholar] [CrossRef]
- Botchkareva, N.V. MicroRNA/mRNA regulatory networks in the control of skin development and regeneration. Cell Cycle 2012, 11, 468–474. [Google Scholar] [CrossRef]
- Westgate, G.E.; Ginger, R.S.; Green, M.R. The biology and genetics of curly hair. Exp. Dermatol. 2017, 26, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Nie, Q.; Su, Y.; Xie, X.; Luo, W.; Jia, X.; Zhang, X. MicroRNA profile analysis on duck feather follicle and skin with high-throughput sequencing technology. Gene 2013, 519, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Sun, W.; Yin, J.; Lv, X.; Bao, J.; Yu, J.; Wang, L.; Jin, C.; Hu, L. Screening candidate microRNAs (miRNAs) in different lambskin hair follicles in Hu sheep. PLoS ONE 2017, 12, e0176532. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, W.; Wu, J.; Li, J.; Yashizawa, M. A subset of skin-expressed microRNAs with possible roles in goat and sheep hair growth based on expression profiling of mammalian microRNAs. Omics 2007, 11, 385–396. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Q.; Wang, H.; Huang, M.; Shi, J.; Han, F.; Cai, W.; Li, Y.; He, T.; Hu, D. MicroRNA-130a has pro-fibroproliferative potential in hypertrophic scar by targeting CYLD. Arch. Biochem. Biophys. 2019, 671, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Icli, B.; Nabzdyk, C.S.; Lujan-Hernandez, J.; Cahill, M.; Auster, M.E.; Wara, A.K.; Sun, X.; Ozdemir, D.; Giatsidis, G.; Orgill, D.P.; et al. Regulation of impaired angiogenesis in diabetic dermal wound healing by microRNA-26a. J. Mol. Cell. Cardiol. 2016, 91, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Lei, M.; Tang, H.; Yan, H.; Wen, X.; Zhang, W.; Tan, R.; Wang, D.; Wu, J. Hoxc13 is a crucial regulator of murine hair cycle. Cell Tissue Res. 2016, 364, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Siegenthaler, J.A.; Dowell, R.D.; Yi, R. Foxc1 reinforces quiescence in self-renewing hair follicle stem cells. Science 2016, 351, 613–617. [Google Scholar] [CrossRef]
- Lay, K.; Kume, T.; Fuchs, E. FOXC1 maintains the hair follicle stem cell niche and governs stem cell quiescence to preserve long-term tissue-regenerating potential. Proc. Natl. Acad. Sci. USA 2016, 113, E1506–E1515. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Z.; O’Loughlin, E.; Wang, L.; Fan, X.; Lai, E.C.; Yi, R. MicroRNA-205 controls neonatal expansion of skin stem cells by modulating the PI(3)K pathway. Nat. Cell Biol. 2013, 15, 1153–1163. [Google Scholar] [CrossRef]
- Harel, S.; Higgins, C.A.; Cerise, J.E.; Dai, Z.; Chen, J.C.; Clynes, R.; Christiano, A.M. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci. Adv. 2015, 1, e1500973. [Google Scholar] [CrossRef]
- Kiso, M.; Hamazaki, T.S.; Itoh, M.; Kikuchi, S.; Nakagawa, H.; Okochi, H. Synergistic effect of PDGF and FGF2 for cell proliferation and hair inductive activity in murine vibrissal dermal papilla in vitro. J. Dermatol. Sci. 2015, 79, 110–118. [Google Scholar] [CrossRef]
- Massague, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Kandyba, E.; Hazen, V.M.; Kobielak, A.; Butler, S.J.; Kobielak, K. SMAD1 and 5 but not SMAD8 establish stem cell quiescence which is critical to transform the premature hair follicle during morphogenesis toward the postnatal state. Stem Cells 2014, 32, 534–547. [Google Scholar] [CrossRef]
- He, W.; Li, A.G.; Wang, D.; Han, S.; Zheng, B.; Goumans, M.J.; Ten, D.P.; Wang, X.J. Overexpression of SMAD7 results in severe pathological alterations in multiple epithelial tissues. EMBO J. 2002, 21, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.B.; Zheng-Yuanquan, X.U. TGF-β and hair follicle development. Chin. J. Derm. 2006, 177–178. [Google Scholar]
- Driskell, R.R.; Clavel, C.; Rendl, M.; Watt, F.M. Hair follicle dermal papilla cells at a glance. J. Cell Sci. 2011, 124 Pt 8, 1179–1182. [Google Scholar] [CrossRef]
- Jahoda, C.A.; Oliver, R.F.; Reynolds, A.J.; Forrester, J.C.; Gillespie, J.W.; Cserhalmi-Friedman, P.B.; Christiano, A.M.; Horne, K.A. Trans-species hair growth induction by human hair follicle dermal papillae. Exp. Dermatol. 2001, 10, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Du, H.; Ju, Y.; Wang, J.; Long, C.Q.; Gao, J.; Liu, H.Y. Isolation and cultivation of Tibet miniature pig dermal papilla cells in vitro. J. Pract. Dermatol. 2013, 6, 1–2. [Google Scholar]
- He, X.; Chao, Y.; Zhou, G.; Chen, Y. Fibroblast growth factor 5-short (FGF5s) inhibits the activity of FGF5 in primary and secondary hair follicle dermal papilla cells of cashmere goats. Gene 2016, 575 Pt 2, 393–398. [Google Scholar] [CrossRef]
- Bai, Y.; Sun, H.T.; Hu, H.M.; Yang, L.P.; Gao, S.X.; Jiang, W.X. Study on isolation, culture, identification and growth characteristics of dermal papilla cells in angora rabbit. J. Domest. Anim. Ecol. 2017, 38, 20–26. [Google Scholar]
- Kwack, M.H.; Yang, J.M.; Won, G.H.; Kim, M.K.; Kim, J.C.; Sung, Y.K. Establishment and characterization of five immortalized human scalp dermal papilla cell lines. Biochem. Biophys. Res. Commun. 2018, 496, 346–351. [Google Scholar] [CrossRef]
- Hunt, D.P.; Morris, P.N.; Sterling, J.; Anderson, J.A.; Joannides, A.; Jahoda, C.; Compston, A.; Chandran, S. A highly enriched niche of precursor cells with neuronal and glial potential within the hair follicle dermal papilla of adult skin. Stem Cells 2008, 26, 163–172. [Google Scholar] [CrossRef]
- Cui, Z.F. Isolation, Culture and Identification of Goat Hair Follicles and Remodeling of Hair Follicles In Vitro; Shandong Agricultural University: Taian, Shandong, China, 2013. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009, 37, D93–D97. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef] [PubMed]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Klimke, W.; Maglott, D.R. NCBI Reference Sequences: Current status, policy and new initiatives. Nucleic Acids Res. 2009, 37, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome. Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microrNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | D108_Count | D45_Count | D108_TPM | D45_TPM | FDR | log2FC |
|---|---|---|---|---|---|---|
| chi-let-7a-5p | 52 | 7 | 9.8407 | 1.4161 | 1.61 × 10−05 | −3.0405 |
| chi-miR-451-5p | 55 | 9 | 10.4084 | 1.8207 | 4.72 × 10−05 | −2.7954 |
| chi-miR-26a-5p | 162 | 11 | 24.4730 | 4.5195 | 8.50 × 10−03 | −2.7095 |
| chi-miR-10a-5p | 40 | 7 | 7.5697 | 1.4161 | 7.96 × 10−04 | −2.6681 |
| chi-miR-1468-5p | 31 | 6 | 5.8666 | 1.2138 | 7.28 × 10−03 | −2.5028 |
| chi-miR-204-5p | 122 | 27 | 23.0877 | 5.4620 | 5.33 × 10−06 | −2.4410 |
| chi-miR-30a-3p | 640 | 146 | 121.1160 | 29.5350 | 1.56 × 10−08 | −2.4293 |
| chi-miR-9-3p | 72 | 16 | 13.6255 | 3.2367 | 1.26 × 10−04 | −2.4089 |
| chi-miR-197-3p | 45 | 12 | 8.5160 | 2.4275 | 8.07 × 10−03 | −2.1288 |
| chi-miR-144-3p | 167 | 50 | 31.6037 | 10.1147 | 1.06 × 10−04 | −2.0246 |
| chi-miR-187-5p | 28 | 108 | 5.7319 | 38.6300 | 1.18 × 10−06 | 2.4468 |
| chi-miR-424-5p | 232 | 576 | 47.4927 | 206.0269 | 1.24 × 10−05 | 1.8399 |
| chi-miR-100-5p | 16 | 46 | 3.2754 | 16.4535 | 5.98 × 10−03 | 2.0036 |
| chi-miR-542-5p | 17 | 50 | 3.4801 | 17.8843 | 3.25 × 10−03 | 2.0390 |
| chi-miR-301a-3p | 22 | 71 | 4.5036 | 25.3957 | 1.85 × 10−04 | 2.1827 |
| unconservative_NW_017189517.1_39211 | 1514 | 5051 | 309.9306 | 1806.6695 | 1.77 × 10−09 | 2.2690 |
| chi-miR-2332 | 2 | 16 | 0.3021 | 6.5738 | 3.35 × 10−03 | 3.5404 |
| chi-miR-215-5p | 124 | 508 | 23.4662 | 102.7657 | 1.18 × 10−03 | 1.7239 |
| chi-miR-10b-5p | 13 | 55 | 1.9639 | 22.5973 | 8.80 × 10−05 | 3.0961 |
| chi-miR-16a-5p | 7 | 32 | 1.4330 | 11.4459 | 1.40 × 10−03 | 2.5969 |
| chi-miR-17-3p | 7 | 33 | 1.4330 | 11.8036 | 9.25 × 10−04 | 2.6405 |
| chi-miR-3432-5p | 7 | 68 | 1.3247 | 13.7560 | 1.03 × 10−04 | 2.8494 |
| chi-miR-128-3p | 8 | 81 | 1.5140 | 16.3859 | 2.06 × 10−05 | 2.9235 |
| unconservative_Chr1_79 | 25 | 100 | 5.1177 | 35.7686 | 1.19 × 10−06 | 2.4952 |
| chi-miR-19a | 6 | 75 | 1.1355 | 15.1721 | 7.83 × 10−06 | 3.1885 |
| chi-miR-130-5p | 340 | 1195 | 69.6013 | 427.4342 | 7.22 × 10−10 | 2.3422 |
| chi-miR-18a-3p | 6 | 86 | 1.2283 | 30.7610 | 1.07 × 10−12 | 4.2068 |
| chi-miR-2284b | 4 | 44 | 0.6043 | 18.0778 | 5.27 × 10−07 | 4.2491 |
| MicroRNA Name | Sense (5′–3′) |
|---|---|
| miR-26a | UUCAAGUAAUCCAGGAUAGGCU |
| miR-130a | GCUCUUUUCACAUUGUGCUACU |
| microRNA mimics NC | UUGUACUACACAAAAGUACUG |
| mircoRNA inhibitor NC | CAGUACUUUUGUGUAGUACAA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Y.; Xue, X.; Liu, Z.; Ye, Y.; Xiao, P.; Pu, Y.; Guan, W.; Mwacharo, J.M.; Ma, Y.; Zhao, Q. Expression Profiling and Functional Characterization of miR-26a and miR-130a in Regulating Zhongwei Goat Hair Development via the TGF-β/SMAD Pathway. Int. J. Mol. Sci. 2020, 21, 5076. https://doi.org/10.3390/ijms21145076
Ding Y, Xue X, Liu Z, Ye Y, Xiao P, Pu Y, Guan W, Mwacharo JM, Ma Y, Zhao Q. Expression Profiling and Functional Characterization of miR-26a and miR-130a in Regulating Zhongwei Goat Hair Development via the TGF-β/SMAD Pathway. International Journal of Molecular Sciences. 2020; 21(14):5076. https://doi.org/10.3390/ijms21145076
Chicago/Turabian StyleDing, Yangyang, Xianglan Xue, Zhanfa Liu, Yong Ye, Ping Xiao, Yabin Pu, Weijun Guan, Joram Mwashigadi Mwacharo, Yuehui Ma, and Qianjun Zhao. 2020. "Expression Profiling and Functional Characterization of miR-26a and miR-130a in Regulating Zhongwei Goat Hair Development via the TGF-β/SMAD Pathway" International Journal of Molecular Sciences 21, no. 14: 5076. https://doi.org/10.3390/ijms21145076
APA StyleDing, Y., Xue, X., Liu, Z., Ye, Y., Xiao, P., Pu, Y., Guan, W., Mwacharo, J. M., Ma, Y., & Zhao, Q. (2020). Expression Profiling and Functional Characterization of miR-26a and miR-130a in Regulating Zhongwei Goat Hair Development via the TGF-β/SMAD Pathway. International Journal of Molecular Sciences, 21(14), 5076. https://doi.org/10.3390/ijms21145076