Adenosine A2A and A3 Receptors Are Able to Interact with Each Other. A Further Piece in the Puzzle of Adenosine Receptor-Mediated Signaling

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
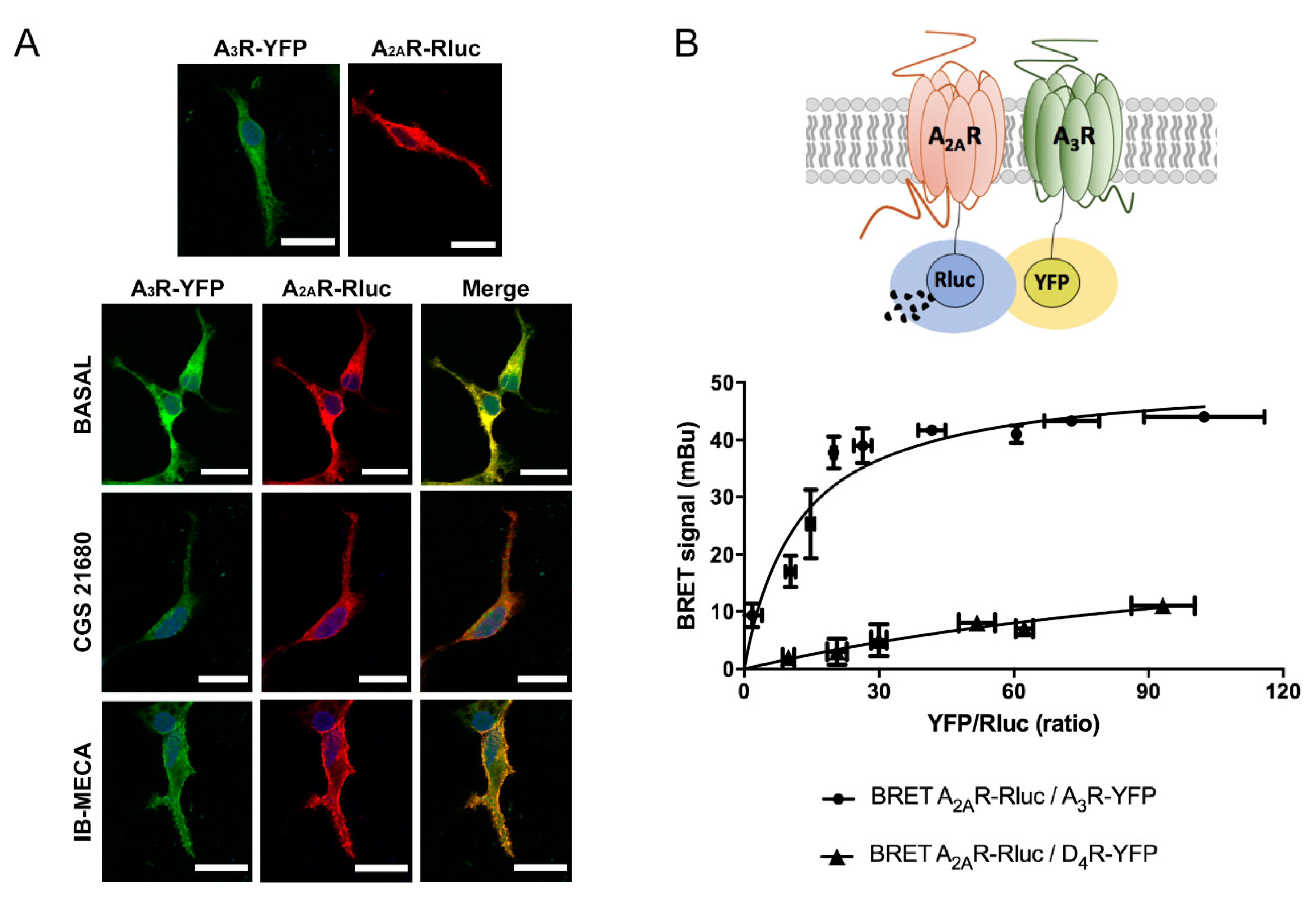
2.1. Molecular Interaction between A2AR and A3R
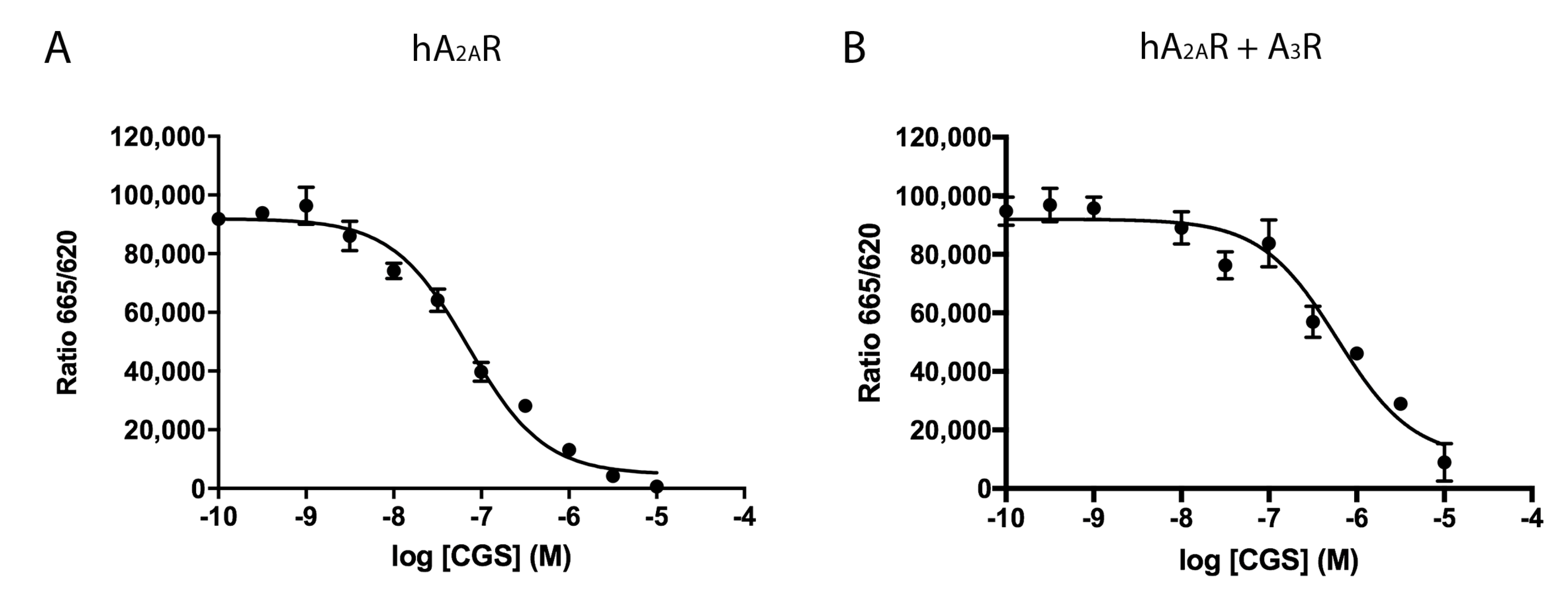
2.2. Effect of the A3R Expression on Homogeneous Ligand Binding to the A2AR
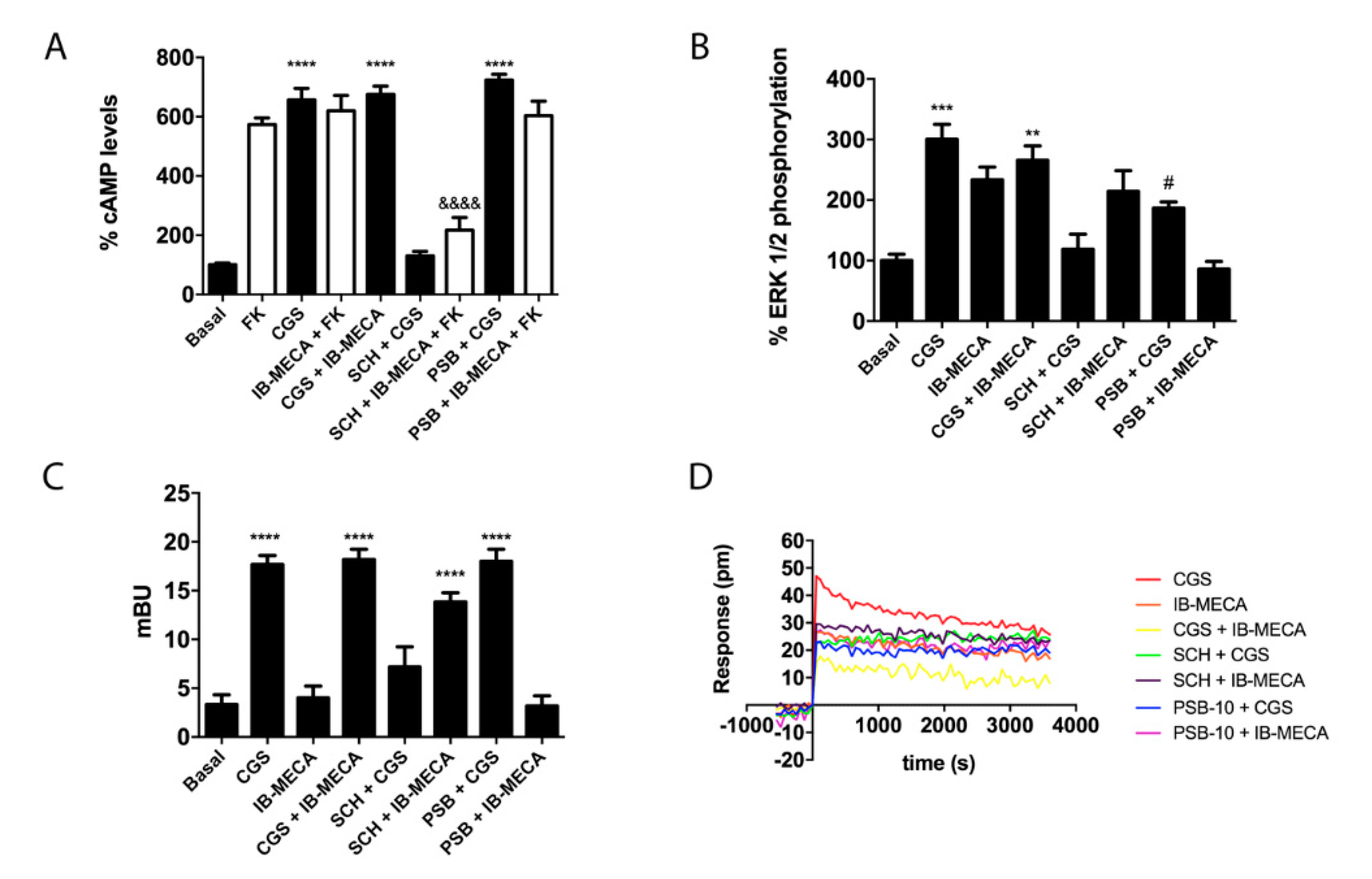
2.3. Functional Characterization of A2AR/A3R-Hets in HEK-293T Cells
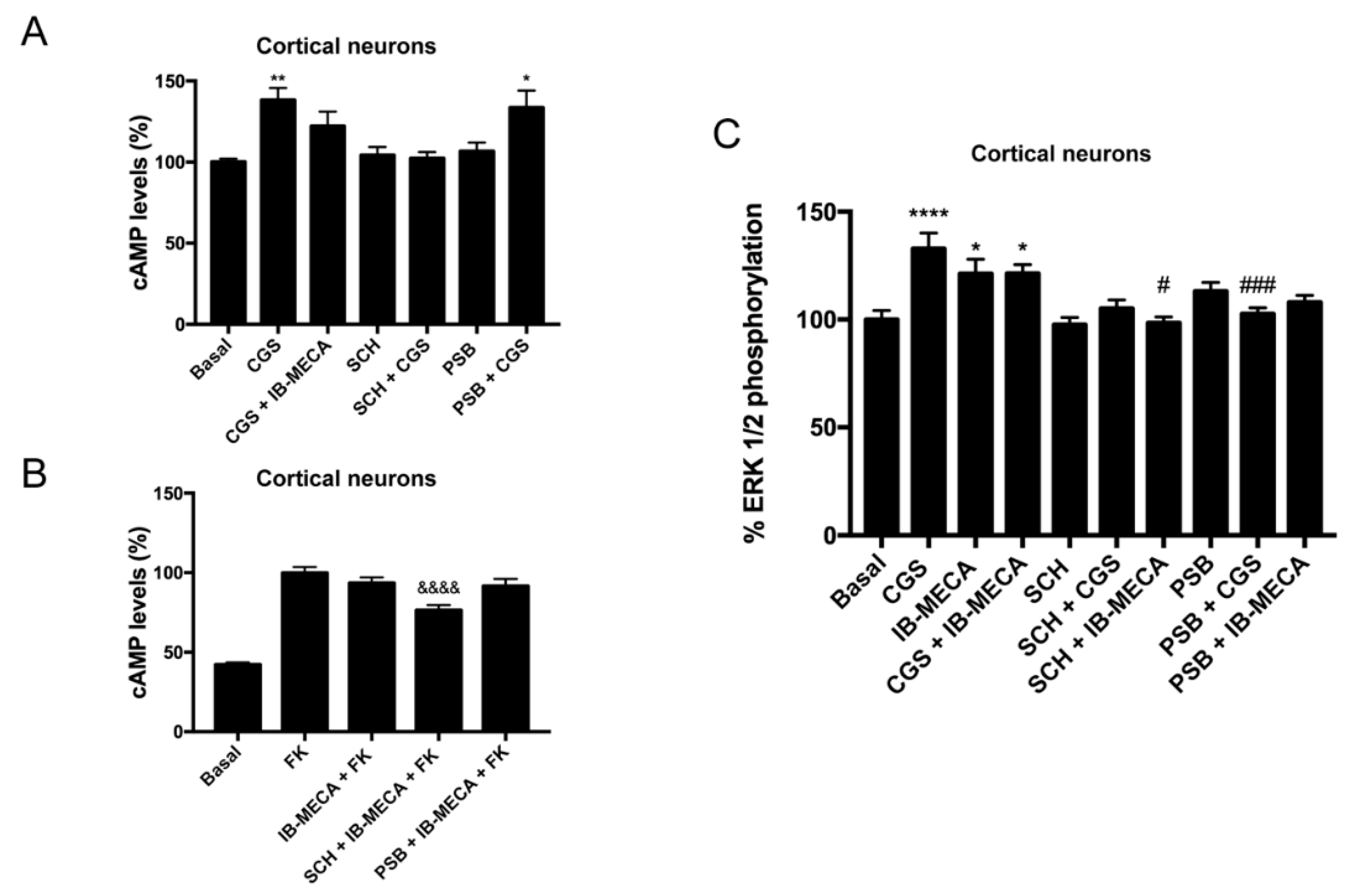
2.4. Discovery of A2AR/A3R-Hets in Primary Cultures of Cortical Neurons
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Neuronal Primary Cultures
4.3. Cell Culture and Transient Transfection
4.4. Fusion Proteins and Expression Vectors
4.5. Bioluminescence Resonance Energy Transfer (BRET) Assays
4.6. Immunocytochemistry
4.7. Homogeneous Competition Binding Assays in Living Cells
4.8. cAMP Determination
4.9. ERK Phosphorylation Assays
4.10. Dynamic Mass Redistribution (DMR) Assays
4.11. β-arrestin 2 Recruitment
4.12. Data Handling and Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A1R | A1 receptor |
| A2AR | A2A receptor |
| A2AR/A3R-Het | A2A–A3 receptor heteromer |
| A2BR | A2B receptor |
| A3R | A3 receptor |
| BRET | Bioluminescence resonance energy transfer |
| DMR | Dynamic mass redistribution |
| DMSO | Dimethyl sulfoxide |
| FBS | Fetal bovine serum |
| FRET | Fluorescence resonance energy transfer |
| GPCRs | G-protein-coupled receptors |
| HBSS | Hank’s balanced salt solution |
| HTRF | Homogeneous time resolved fluorescence |
| TLB | Tag-lite buffer |
| YFP | Yellow fluorescent protein |
References
- Jordan, B.A.; Devi, L.A. G-Protein-Coupled Receptor Heterodimerization Modulates Receptor Function. Nature 1999, 399, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Jordan, B.A.; Gupta, A.; Trapaidze, N.; Nagy, V.; Devi, L.A. Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J. Neurosci. 2000, 20, RC110. [Google Scholar] [CrossRef]
- Gomes, I.; Gupta, A.; Filipovska, J.; Szeto, H.H.; Pintar, J.E.; Devi, L.A. A Role for Heterodimerization of Mu and Delta Opiate Receptors in Enhancing Morphine Analgesia. Proc. Natl. Acad. Sci. USA 2004, 101, 5135–5139. [Google Scholar] [CrossRef]
- Gomes, I.; Filipovska, J.; Jordan, B.A.; Devi, L.A. Oligomerization of opioid receptors. Methods 2002, 27, 358–365. [Google Scholar] [CrossRef]
- Rashid, A.J.; So, C.H.; Kong, M.M.C.; Furtak, T.; El-Ghundi, M.; Cheng, R.; O’Dowd, B.F.; George, S.R. D1-D2 Dopamine Receptor Heterooligomers with Unique Pharmacology are Coupled to Rapid Activation of Gq/11 in the Striatum. Proc. Natl. Acad. Sci. USA 2007, 104, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Hasbi, A.; O’Dowd, B.F.; George, S.R. Dopamine D1-D2 Receptor Heteromer-Mediated Calcium Release is Desensitized by D1 Receptor Occupancy with or without Signal Activation: Dual Functional Regulation by G Protein-Coupled Receptor Kinase 2. J. Biol. Chem. 2010, 285, 35092–35103. [Google Scholar] [CrossRef]
- Lee, S.P.; So, C.H.; Rashid, A.J.; Varghese, G.; Cheng, R.; Lança, A.J.; O’Dowd, B.F.; George, S.R. Dopamine D1 and D2 Receptor Co-Activation Generates a Novel Phospholipase C-Mediated Calcium Signal. J. Biol. Chem. 2004, 279, 35671–35678. [Google Scholar] [CrossRef]
- George, S.R.S.R.; Kern, A.; Smith, R.G.R.G.; Franco, R. Dopamine Receptor Heteromeric Complexes and Their Emerging Functions. Prog. Brain Res. 2014, 211, 183–200. [Google Scholar] [CrossRef]
- Franco, R.; Martínez-Pinilla, E.; Lanciego, J.L.; Navarro, G. Basic Pharmacological and Structural Evidence for Class A G-Protein-Coupled Receptor Heteromerization. Front. Pharm. 2016, 7, 76. [Google Scholar] [CrossRef]
- Ferré, S.; Baler, R.; Bouvier, M.; Caron, M.G.; Devi, L.A.; Durroux, T.; Fuxe, K.; George, S.R.; Javitch, J.A.; Lohse, M.J.; et al. Building a New Conceptual Framework for Receptor Heteromers. Nat. Chem. Biol. 2009, 5, 131–134. [Google Scholar] [CrossRef]
- Franco, N.; Franco, R. Understanding the Added Value of g-Protein-Coupled Receptor Heteromers. Scientifica 2014, 2014, 362937. [Google Scholar] [CrossRef]
- Ciruela, F.; Casadó, V.; Rodrigues, R.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.; Mallol, J.; et al. Presynaptic Control of Striatal Glutamatergic Neurotransmission by Adenosine A1-A2A Receptor Heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, A.I.; Castro, E.; Mirabet, M.; Franco, R.; Delicado, E.G.; Miras-Portugal, M.T. Potentiation of ATP Calcium Responses by A(2B) Receptor Stimulation and Other Signals Coupled to G(S) Proteins in Type-1 Cerebellar Astrocytes. Glia 1999, 26, 119–128. [Google Scholar] [CrossRef]
- Bernareggi, A.; Luin, E.; Pavan, B.; Parato, G.; Sciancalepore, M.; Urbani, R.; Lorenzon, P. Adenosine Enhances Acetylcholine Receptor Channel Openings and Intracellular Calcium “Spiking” in mouse Skeletal Myotubes. Acta Physiol. 2015, 214, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Matta, C.; Fodor, J.; Miosge, N.; Takács, R.; Juhász, T.; Rybaltovszki, H.; Tóth, A.; Csernoch, L.; Zákány, R. Purinergic signalling is required for calcium oscillations in migratory chondrogenic progenitor cells. Pflug. Arch. Eur. J. Physiol. 2014, 467, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Cordomí, A.; Zelman-Femiak, M.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Perez-Benito, L.; Cortés, A.; Casadó, V.; Mallol, J.; et al. Quaternary Structure of a G-protein-Coupled Receptor Heterotetramer in Complex with Gi and Gs. BMC Biol. 2016, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Cordomí, A.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Pérez-Benito, L.; Ferre, S.; Cortés, A.; Casadó, V.; Mallol, J.; et al. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018, 16, 1–15. [Google Scholar] [CrossRef]
- Cristóvão-Ferreira, S.; Navarro, G.; Brugarolas, M.; Pérez-Capote, K.; Vaz, S.H.; Fattorini, G.; Conti, F.; Lluis, C.; Ribeiro, J.A.; McCormick, P.J.; et al. A1R-A2AR Heteromers Coupled to Gs and G i/o proteins Modulate GABA Transport into Astrocytes. Purinergic Signal. 2013, 9, 433–449. [Google Scholar] [CrossRef]
- Hinz, S.; Navarro, G.; Borroto-Escuela, D.; Seibt, B.F.; Ammon, C.; Filippo, E.; De Danish, A.; Lacher, S.K.; Červinková, B.; Rafehi, M. Adenosine A2A Receptor Ligand Recognition and Signaling is Blocked by A2B Receptors. Oncotarget 2018, 9, 13593–13611. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.D.O.; Hinz, S.; Navarro, G.; Franco, R.; Muller, C.; Fuxe, K.; Müller, C.E.; Fuxe, K. Understanding the Role of Adenosine A2AR Heteroreceptor Complexes in Neurodegeneration and Neuroinflammation. Front. Neurosci. 2018, 12, 43. [Google Scholar] [CrossRef]
- Gnad, T.; Navarro, G.; Lahesmaa, M.; Reverte-Salisa, L.; Copperi, F.; Cordomi, A.; Naumann, J.; Hochhäuser, A.; Haufs-Brusberg, S.; Wenzel, D.; et al. Adenosine/A2B Receptor Signaling Ameliorates the Effects of Aging and Counteracts Obesity. Cell Metab. 2020, 32, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J.; May, L.T.; Kellam, B.; Woolard, J. Allosteric Interactions at Adenosine A(1) and A(3) Receptors: New Insights into the Role of Small Molecules and Receptor Dimerization. Br. J. Pharm. 2014, 171, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Otah, M.E.; Stiles, G.L. Adenosine Receptors. Annu. Rev. Physiol. 1992, 54, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.M.; Stiles, G.L. Adenosine receptors. Neuropharmacology 1995, 271, 15272–15278. [Google Scholar] [CrossRef]
- Grundmann, M.; Kostenis, E. Holistic Methods for the Analysis of cNMP Effects. In Handbook of Experimental Pharmacology; Flockerzi, V., Frohman, M.A., Geppeti, P., Eds.; Springer: Cham, Switzerland, 2015; pp. 339–357. [Google Scholar] [CrossRef]
- Corset, V.; Nguyen-Ba-Charvet, K.T.; Forcet, C.; Moyse, E.; Chédotal, A.; Mehlen, P. Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2b receptor. Nature 2000, 407, 747–750. [Google Scholar] [CrossRef]
- De Filippo, E.; Hinz, S.; Pellizzari, V.; Deganutti, G.; El-Tayeb, A.; Navarro, G.; Franco, R.; Moro, S.; Schiedel, A.C.; Müller, C.E. A2A and A2B Adenosine Receptors: The Extracellular Loop 2 Determines High (A2A) or Low Affinity (A2B) for Adenosine. Biochem. Pharm. 2020, 172, 113718. [Google Scholar] [CrossRef]
- Martínez-Pinilla, E.; Rodríguez-Pérez, A.I.I.; Navarro, G.; Aguinaga, D.; Moreno, E.; Lanciego, J.L.L.; Labandeira-García, J.L.L.; Franco, R. Dopamine D2 and Angiotensin II Type 1 Receptors form Functional Heteromers in Rat Striatum. Biochem. Pharm. 2015, 96, 131–142. [Google Scholar] [CrossRef]
- Rodríguez-Ruiz, M.; Moreno, E.; Moreno-Delgado, D.; Navarro, G.; Mallol, J.; Cortés, A.; Lluís, C.; Canela, E.I.; Casadó, V.; McCormick, P.J.; et al. Heteroreceptor Complexes Formed by Dopamine D1, Histamine H3, and N-Methyl-D-Aspartate Glutamate Receptors as Targets to Prevent Neuronal Death in Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 4537–4550. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Hauser, R.; Morelli, M.; Fredholm, B.B.; Chen, J.F. Adenosine, adenosine A 2A antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 406–413. [Google Scholar] [CrossRef]
- Jenner, P. An Overview of Adenosine A2A Receptor Antagonists in Parkinson’s Disease. Int. Rev. Neurobiol. 2014, 119, 71–86. [Google Scholar] [CrossRef]
- Mizuno, Y.; Kondo, T. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 2013, 28, 1138–1141. [Google Scholar] [CrossRef] [PubMed]
- Saki, M.; Yamada, K.; Koshimura, E.; Sasaki, K.; Kanda, T. In Vitro Pharmacological Profile of the A2A Receptor Antagonist Istradefylline. Naunyn. Schmiedebergs. Arch. Pharm. 2013, 386, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Mizuno, Y.; Japanese Istradefylline Study Group. A Long-Term Study of Istradefylline Safety and Efficacy in Patients with Parkinson Disease. Clin. Neuropharmacol. 2015, 38, 41–46. [Google Scholar] [CrossRef]
- Hradsky, J.; Mikhaylova, M.; Karpova, A.; Kreutz, M.R.; Zuschratter, W. Super-Resolution Microscopy of the Neuronal Calcium-Binding Proteins Calneuron-1 and Caldendrin. Methods Mol. Biol. 2013, 963, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Medrano, M.; Aguinaga, D.; Reyes-Resina, I.; Canela, E.I.; Mallol, J.; Navarro, G.; Franco, R. Orexin A/Hypocretin Modulates Leptin Receptor-Mediated Signaling by Allosteric Modulations Mediated by the Ghrelin GHS-R1A Receptor in Hypothalamic Neurons. Mol. Neurobiol. 2018, 55, 4718–4730. [Google Scholar] [CrossRef]
- Martinez-Pinilla, E.; Rabal, O.; Reyes-Resina, I.; Zamarbide, M.; Navarro, G.; Sanchez-Arias, J.A.; de Miguel, I.; Lanciego, J.L.; Oyarzabal, J.; Franco, R. Two Affinity Sites of the Cannabinoid Subtype 2 Receptor Identified by a Novel Homogeneous Binding Assay. J. Pharm. Exp. 2016, 358, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Ferré, S.; Cordomi, A.; Moreno, E.; Mallol, J.; Casadó, V.; Cortés, A.; Hoffmann, H.; Ortiz, J.; Canela, E.I.; et al. Interactions between intracellular domains as key determinants of the quaternary structure and function of receptor heteromers. J. Biol. Chem. 2010, 285, 27346–27359. [Google Scholar] [CrossRef]
- Cheng, H.C. The power issue: Determination of KB or Ki from IC50—A closer look at the Cheng-Prusoff Equation, the Schild Plot and Related Power Equations. J. Pharm. Toxicol. Methods 2001, 46, 61–71. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lillo, A.; Martínez-Pinilla, E.; Reyes-Resina, I.; Navarro, G.; Franco, R. Adenosine A2A and A3 Receptors Are Able to Interact with Each Other. A Further Piece in the Puzzle of Adenosine Receptor-Mediated Signaling. Int. J. Mol. Sci. 2020, 21, 5070. https://doi.org/10.3390/ijms21145070
Lillo A, Martínez-Pinilla E, Reyes-Resina I, Navarro G, Franco R. Adenosine A2A and A3 Receptors Are Able to Interact with Each Other. A Further Piece in the Puzzle of Adenosine Receptor-Mediated Signaling. International Journal of Molecular Sciences. 2020; 21(14):5070. https://doi.org/10.3390/ijms21145070
Chicago/Turabian StyleLillo, Alejandro, Eva Martínez-Pinilla, Irene Reyes-Resina, Gemma Navarro, and Rafael Franco. 2020. "Adenosine A2A and A3 Receptors Are Able to Interact with Each Other. A Further Piece in the Puzzle of Adenosine Receptor-Mediated Signaling" International Journal of Molecular Sciences 21, no. 14: 5070. https://doi.org/10.3390/ijms21145070
APA StyleLillo, A., Martínez-Pinilla, E., Reyes-Resina, I., Navarro, G., & Franco, R. (2020). Adenosine A2A and A3 Receptors Are Able to Interact with Each Other. A Further Piece in the Puzzle of Adenosine Receptor-Mediated Signaling. International Journal of Molecular Sciences, 21(14), 5070. https://doi.org/10.3390/ijms21145070