Signaling of Tumor-Derived sEV Impacts Melanoma Progression




Abstract
1. Introduction
2. General Characteristics of sEV
3. General Features of Melanoma
4. The Molecular Cargo of MTEX
4.1. The Proteome of MTEX
4.2. Micro RNA Component of MTEX
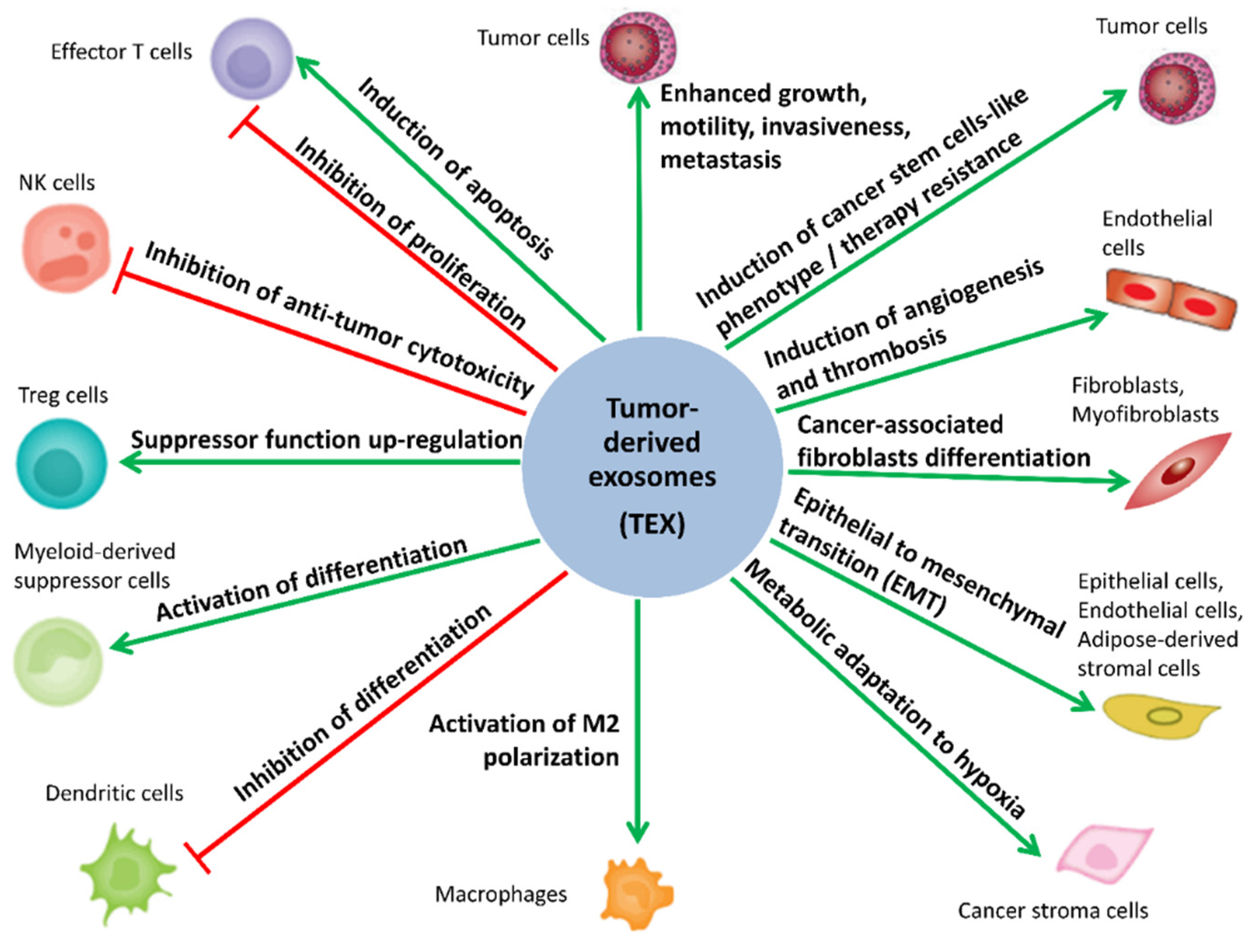
5. Biological Activity of MTEX
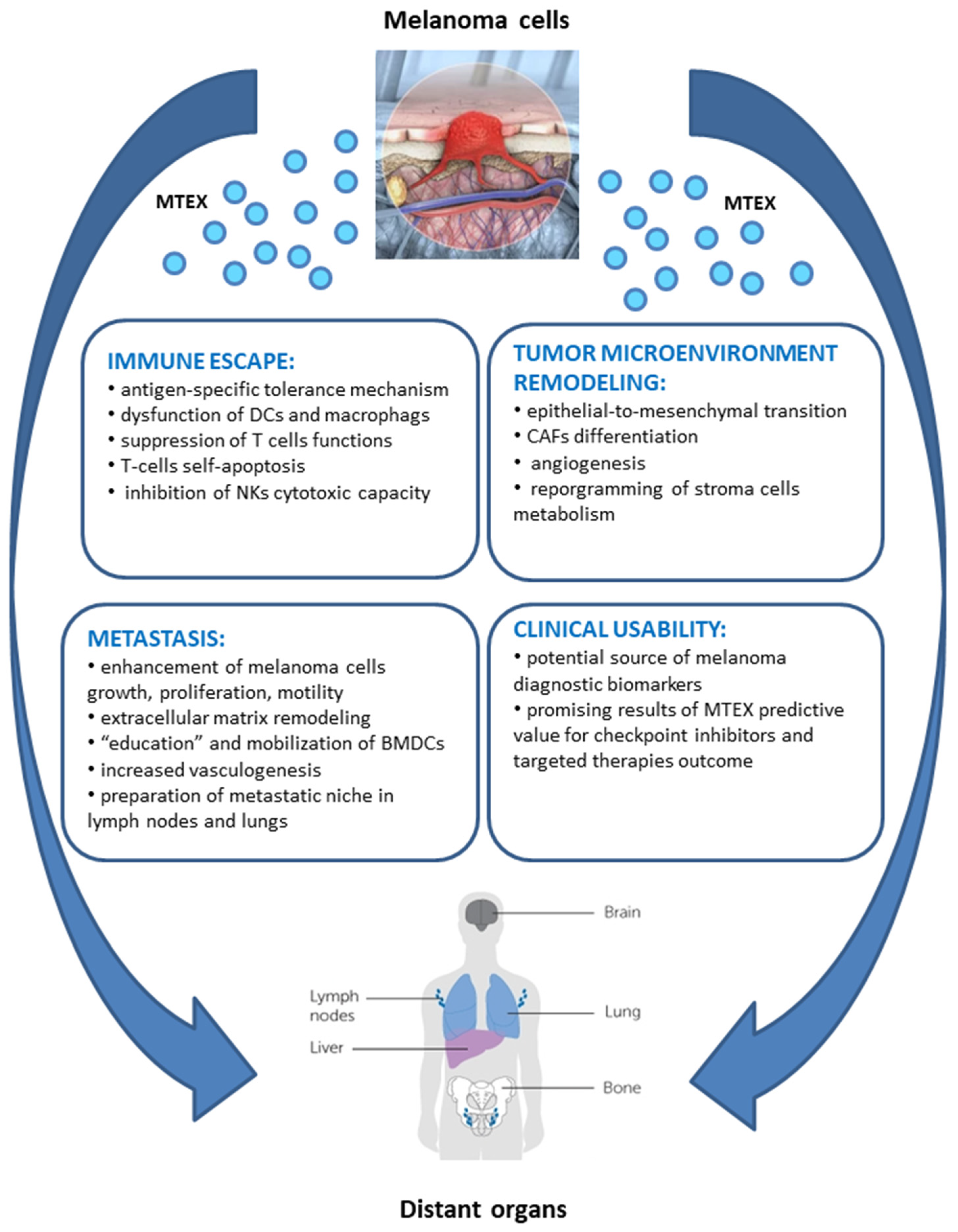
5.1. MTEX Participate in the Reprogramming of Immune Cells
5.2. MTEX Participate in the Reprogramming of TME
5.3. MTEX Can Modulate Tumor Progression and Invasiveness
6. MTEX as Potential Clinical Biomarkers
7. Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AB | apoptotic bodies |
| APC | antigen presenting cells |
| BM | bone marrow |
| BMDC | bone marrow derived cells |
| CAF | cancer-associated fibroblasts |
| CEM | cryo-electron microscopy |
| CM | confocal microscopy |
| DC | differential centrifugation |
| DC | dendritic cells |
| DLS | dynamic light scattering |
| EC | endothelial cells |
| EMT | epithelial-to-mesenchymal transition |
| EV | extracellular vesicles |
| EX | exosomes |
| HADF | human adult dermal fibroblasts |
| IA-FCM | immune-affinity flow cytometry |
| KO | knockdown |
| MAA | melanoma associated antigens |
| mAb | monoclonal antibodies |
| MSC | myeloid stem cells |
| MTEX | melanoma cell-derived exosomes |
| MV | microvesicles |
| MVB | multivesicular bodies |
| NTA | nanoparticle tracking analysis |
| SEC | size-exclusion chromatography |
| SEM | scanning electron microscopy |
| sEV | small extracellular vesicles |
| TEM | transmission electron microscopy |
| TEX | tumor-derived exosomes |
| TME | tumor microenvironment |
| UC | ultracentrifugation |
| UF | ultrafiltration |
| WB | western blotting |
References
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular Vesicles in Cancer: Exosomes, Microvesicles and the Emerging Role of Large Oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriansitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2019, 8, 1535750. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, P.J.; Primdal-Bengtson, B.; Dingli, F.; Leow, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, 968–977. [Google Scholar] [CrossRef]
- Abramowicz, A.; Widlak, P.; Pietrowska, M. Proteomic analysis of exosomal cargo: The challenge of high purity vesicle isolation. Mol. Biosyst. 2016, 12, 1407–1419. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef]
- Zebrowska, A.; Skowronek, A.; Wojakowska, A.; Widlak, P.; Pietrowska, M. Metabolome of Exosomes: Focus on Vesicles Released by Cancer Cells and Present in Human Body Fluids. Int. J. Mol. Sci. 2019, 20, 3461. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Dixon, C.L.; Sheller-Miller, S.; Saade, G.R.; Fortunato, S.J.; Lai, A.; Palma, C.; Guanzon, D.; Salomon, C.; Menon, R. Amniotic Fluid Exosome Proteomic Profile Exhibits Unique Pathways of Term and Preterm Labor. Endocrinology 2018, 159, 2229–2240. [Google Scholar] [CrossRef]
- Yu, J.; Lin, Y.; Xiong, X.; Li, K.; Yao, Z.; Dong, H.; Jiang, Z.; Yu, D.; Yeung, S.J.; Zhang, H. Detection of Exosomal PD-L1 RNA in Saliva of Patients With Periodontitis. Front. Genet. 2019, 10, 202. [Google Scholar] [CrossRef]
- Grigor’eva, A.E.; Tamkovich, S.N.; Eremina, A.V.; Tupikin, A.E.; Kabilov, M.R.; Chernykh, V.V.; Vlassov, V.V.; Laktionov, P.P.; Ryabchikova, E.I. Exosomes in tears of healthy individuals: Isolation, identification, and characterization. Biochem. Moscow Suppl. Ser. B 2016, 10, 165. [Google Scholar] [CrossRef]
- Wu, G.; Yang, G.; Zhang, R. Altered microRNA Expression Profiles of Extracellular Vesicles in Nasal Mucus From Patients With Allergic Rhinitis. Allergy Asthma Immunol. Res. 2015, 7, 449–457. [Google Scholar] [CrossRef]
- Saunderson, S.C.; Dunn, A.C.; Crocker, P.R.; McLellan, A.D. CD169 mediates the capture of exosomes in spleen and lymph node. Blood 2014, 123, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Chiasserini, D.; van Weering, J.R.; Piersma, S.R.; Pham, T.V.; Malekzadeh, A.; Teunissen, C.E.; de Wit, H.; Jiménez, C.R. Proteomic analysis of cerebrospinal fluid extracellular vesicles: A comprehensive dataset. J. Proteom. 2014, 106, 191–204. [Google Scholar] [CrossRef]
- Romancino, D.P.; Paterniti, G.; Campos, Y.; De Luca, A.; Di Felice, V.; d’Azzo, A.; Bongiovanni, A. Identification and characterization of the nano-sized vesicles released by muscle cells. FEBS Lett. 2013, 587, 1379–1384. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Huang, B.Q.; Ward, C.J.; Gradilone, S.A.; Banales, J.M.; Masyuk, T.V.; Radtke, B.; Splinter, P.L.; LaRusso, N.F. Biliary exosomes influence cholangiocyte regulatory mechanisms and proliferation through interaction with primary cilia. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G990–G999. [Google Scholar] [CrossRef] [PubMed]
- Runz, S.; Keller, S.; Rupp, C.; Stoeck, A.; Issa, Y.; Koensgen, D.; Mustea, A.; Sehouli, J.; Kristiansen, G.; Altevogt, P. Malignant ascites-derived exosomes of ovarian carcinoma patients contain CD24 and EpCAM. Gynecol. Oncol. 2007, 107, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Admyre, C.; Johansson, S.M.; Qazi, K.R.; Filén, J.J.; Lahesmaa, R.; Norman, M.; Neve, E.P.; Scheynius, A.; Gabrielsson, S. Exosomes with immune modulatory features are present in human breast milk. J. Immunol. 2007, 179, 1969–1978. [Google Scholar] [CrossRef]
- Andreau, Z.; Yanez-Mo, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-Derived Microvesicles Promote Regulatory T Cell Expansion and Induce Apoptosis in Tumor-Reactive Activated CD8+ T Lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whietside, T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020, 10, 92. [Google Scholar] [CrossRef]
- Ferrone, S.; Whiteside, T.L. Targeting CSPG4 for isolation of melanoma cell-derived exosomes from body fluids. HNO 2020, 68, 100–105. [Google Scholar] [CrossRef]
- Sharma, P.; Ludwig, S.; Muller, L.; Hong, C.S.; Kirkwood, J.M.; Ferrone, S.; Whiteside, T.L. Immunoaffinity-based isolation of melanoma cell-derived exosomes from plasma of patients with melanoma. J. Extracell. Vesicles 2018, 7, 1435138. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef]
- Mizutani, K.; Terazawa, R.; Kameyama, K.; Kato, T.; Horie, K.; Tsuchiya, T.; Seike, K.; Ehara, H.; Fujita, Y.; Kawakami, K.; et al. Isolation of prostate cancer-related exosomes. Anticancer Res. 2014, 34, 3419–3423. [Google Scholar]
- Hong, C.S.; Muller, L.; Boyiadzis, M.; Whiteside, T.L. Isolation and characterization of CD34+ blast-derived exosomes in acute myeloid leukemia. PLoS ONE 2014, 9, e103310. [Google Scholar] [CrossRef]
- Raimondo, S.; Pucci, M.; Alessandro, R.; Fontana, S. Extracellular Vesicles and Tumor-Immune Escape: Biological Functions and Clinical Perspectives. Int. J. Mol. Sci. 2020, 21, 2286. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Exosomes in Cancer: Another Mechanism of Tumor-Induced Immune Suppression. In Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy; Kalinski, P., Ed.; Springer: Cham, Switzerland, 2017; Volume 1036, pp. 81–89. [Google Scholar] [CrossRef]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef]
- Ludwig, A.K.; Giebel, B. Exosomes: Small vesicles participating in intercellular communication. Int. J. Biochem. Cell Biol. 2012, 44, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Alegre, E.; Zubiri, L.; Perez-Gracia, J.L.; González-Cao, M.; Soria, L.; Martín-Algarra, S.; González, A. Circulating melanoma exosomes as diagnostic and prognosis biomarkers. Clin. Chim. Acta 2016, 454, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The potential of tumor-derived exosomes for noninvasive cancer monitoring. Expert Rev. Mol. Diagn. 2015, 15, 1293–1310. [Google Scholar] [CrossRef] [PubMed]
- Duijvesz, D.; Versluis, C.Y.; van der Fels, C.A.; Vredenbregt-van den Berg, M.S.; Leivo, J.; Peltola, M.T.; Bangma, C.H.; Pettersson, K.S.; Jenster, G. Immuno-based detection of extracellular vesicles in urine as diagnostic marker for prostate cancer. Int. J. Cancer 2015, 137, 2869–2878. [Google Scholar] [CrossRef]
- Szajnik, M.; Derbis, M.; Lach, M.; Patalas, P.; Michalak, M.; Drzewiecka, H.; Szpurek, D.; Nowakowski, A.; Spaczynski, M.; Baranowski, W.; et al. Exosomes in Plasma of Patients with Ovarian Carcinoma: Potential Biomarkers of Tumor Progression and Response to Therapy. Gynecol. Obstet. 2013, 4, 003. [Google Scholar] [CrossRef]
- Silva, J.; Garcia, V.; Rodriguez, M.; Compte, M.; Cisneros, E.; Veguillas, P.; Garcia, J.M.; Dominguez, G.; Campos-Martin, Y.; Cuevas, J.; et al. Analysis of exosome release and its prognostic value in human colorectal cancer. Genes Chromosomes Cancer 2012, 51, 409–418. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Baran, J.; Baj-Krzyworzeka, M.; Weglarczyk, K.; Szatanek, R.; Zembala, M.; Barbasz, J.; Czupryna, A.; Szczepanik, A.; Zembala, M. Circulating tumour-derived microvesicles in plasma of gastric cancer patients. Cancer Immunol. Immunother. 2010, 59, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef]
- Tung, K.H.; Ernstoff, M.S.; Allen, C.; Shu, S. A Review of Exosomes and their Role in The Tumor Microenvironment and Host-Tumor “Macroenvironment”. J. Immunol. Sci. 2019, 3, 4–8. [Google Scholar] [CrossRef]
- Wang, T.; Nasser, M.I.; Shen, J.; Qu, S.; He, Q.; Zhao, M. Functions of Exosomes in the Triangular Relationship between the Tumor, Inflammation, and Immunity in the Tumor Microenvironment. J. Immunol. Res. 2019, 2019, 4197829. [Google Scholar] [CrossRef]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef]
- Whiteside, T.L. The role of tumor-derived exosomes in epithelial mesenchymal transition (EMT). Transl. Cancer Res. 2017, 6, S90–S92. [Google Scholar] [CrossRef]
- Ludwig, N.; Yerneni, S.S.; Razzo, B.M.; Whiteside, T.L. Exosomes from HNSCC Promote Angiogenesis through Reprogramming of Endothelial Cells. Mol. Cancer Res. 2018, 16, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.M.; Carneiro, F.; Machado, J.C.; Melo, S.A. Exosomes and Immune Response in Cancer: Friends or Foes? Front. Immunol. 2018, 9, 730. [Google Scholar] [CrossRef] [PubMed]
- Czernek, L.; Düchler, M. Functions of Cancer-Derived Extracellular Vesicles in Immunosuppression. Arch. Immunol. Ther. Exp. 2017, 65, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEX (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Mannavola, F.; Passarelli, A.; Stucci, L.S.; Cives, M.; Silvestris, F. Exosomes in melanoma: A role in tumor progression, metastasis and impaired immune system activity. Oncotarget 2018, 9, 20826–20837. [Google Scholar] [CrossRef]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019, 9, 1148. [Google Scholar] [CrossRef]
- Isola, A.L.; Eddy, K.; Chen, S. Biology, Therapy and Implications of Tumor Exosomes in the Progression of Melanoma. Cancers 2016, 8, 110. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Osella-Abate, S.; Ribero, S.; Sanlorenzo, M.; Maule, M.M.; Richiardi, L.; Merletti, F.; Tomasini, C.; Marra, E.; Macripò, G.; Fierro, M.T.; et al. Risk factors related to late metastases in 1,372 melanoma patients disease free more than 10 years. Int. J. Cancer 2015, 136, 2453–2457. [Google Scholar] [CrossRef]
- Rutkowski, P.; Wysocki, P.J.; Nasierowska-Guttmejer, A.; Jeziorski, A.; Wysocki, W.M.; Kalinka-Warzocha, E.; Świtaj, T.; Kozak, K.; Kamińska-Winciorek, G.; Czarnecka, A.M.; et al. Cutaneous melanomas. Oncol. Clin. Pract. 2019, 15, 1–19. [Google Scholar] [CrossRef]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Clin. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef]
- Fujimura, T.; Fujisawa, Y.; Kambayashi, Y.; Aiba, S. Significance of BRAF Kinase Inhibitors for Melanoma Treatment: From Bench to Bedside. Cancers 2019, 11, 1342. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, S.; Hadjinicolaou, A.V.; Chiarion Sileni, V.; Rossi, C.R.; Mocellin, S. Systemic treatments for metastatic cutaneous melanoma. Cochrane Database Syst. Rev. 2018, 2, CD011123. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huo, C.; Qiao, Z.; Shang, Z.; Uzzaman, A.; Liu, S.; Jiang, X.; Fan, L.Y.; Ji, L.; Guan, X.; et al. Comparative Proteomic Analysis of Exosomes and Microvesicles in Human Saliva for Lung Cancer. J. Proteome Res. 2018, 17, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Zhan, S.; Xia, W.; Huang, L.; Ge, W.; Wang, T. Proteomics study of serum exosomes from papillary thyroid cancer patients. Endocr. Relat. Cancer 2018, 25, 879–891. [Google Scholar] [CrossRef]
- Gangoda, L.; Liem, M.; Ang, C.S.; Keerthikumar, S.; Adda, C.G.; Parker, B.S.; Mathivanan, S. Proteomic Profiling of Exosomes Secreted by Breast Cancer Cells with Varying Metastatic Potential. Proteomics 2017, 17, 23–24. [Google Scholar] [CrossRef]
- Ludwig, S.; Marczak, L.; Sharma, P.; Abramowicz, A.; Gawin, M.; Widlak, P.; Whiteside, T.L.; Pietrowska, M. Proteomes of exosomes from HPV(+) or HPV(−) head and neck cancer cells: Differential enrichment in immunoregulatory proteins. Oncoimmunology 2019, 8, 1593808. [Google Scholar] [CrossRef]
- Mears, R.; Craven, R.A.; Hanrahan, S.; Totty, N.; Upton, C.; Young, S.L.; Patel, P.; Selby, P.J.; Banks, R.E. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics 2004, 4, 4019–4031. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Ohlendorf, J.; Chen, Y.; Taylor, D.D.; Rai, S.N.; Waigel, S.; Zacharias, W.; Hao, H.; McMasters, K.M. Identifying mRNA, microRNA and protein profiles of melanoma exosomes. PLoS ONE 2012, 7, e46874. [Google Scholar] [CrossRef]
- Lazar, I.; Clement, E.; Ducoux-Petit, M.; Denat, L.; Soldan, V.; Dauvillier, S.; Balor, S.; Burlet-Schiltz, O.; Larue, L.; Muller, C.; et al. Proteome characterization of melanoma exosomes reveals a specific signature for metastatic cell lines. Pigment Cell Melanoma Res. 2015, 28, 464–475. [Google Scholar] [CrossRef]
- Muhsin-Sharafaldine, M.R.; Saunderson, S.C.; Dunn, A.C.; Faed, J.M.; Kleffmann, T.; McLellan, A.D. Procoagulant and immunogenic properties of melanoma exosomes, microvesicles and apoptotic vesicles. Oncotarget 2016, 7, 56279–56294. [Google Scholar] [CrossRef]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.; Sadik, M.; Alaarg, A.; Smith, C.I.; Lehtiö, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef] [PubMed]
- Boussadia, Z.; Lamberti, J.; Mattei, F.; Pizzi, E.; Puglisi, R.; Zanetti, C.; Pasquini, L.; Fratini, F.; Fantozzi, L.; Felicetti, F.; et al. Acidic microenvironment plays a key role in human melanoma progression through a sustained exosome mediated transfer of clinically relevant metastatic molecules. J. Exp. Clin. Cancer Res. 2018, 37, 245. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Surman, M.; Stępień, E.; Przybyło, M. Melanoma-Derived Extracellular Vesicles: Focus on Their Proteome. Proteomes 2019, 7, 21. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Tsitsiou, E.; Lindsay, M.A. microRNAs and the immune response. Trends Immunol. 2008, 29, 343–351. [Google Scholar] [CrossRef]
- Salehi, M.; Sharifi, M. Exosomal miRNAs as novel cancer biomarkers: Challenges and opportunities. J. Cell. Physiol. 2018, 233, 6370–6380. [Google Scholar] [CrossRef]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Cancer 2018, 17, 147. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Phuyal, S.; Brech, A.; Sandvig, K.; Llorente, A. Profiling of microRNAs in exosomes released from PC-3 prostate cancer cells. Biochim. Biophys. Acta 2012, 1819, 1154–1163. [Google Scholar] [CrossRef]
- Gajos-Michniewicz, A.; Duechler, M.; Czyz, M. MiRNA in melanoma-derived exosomes. Cancer Lett. 2014, 347, 29–37. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Wang, S.; Li, P.; Zheng, C.; Zhou, X.; Tao, Y.; Chen, X.; Sun, L.; Wang, A.; et al. Blockage of transferred exosome-shuttled miR-494 inhibits melanoma growth and metastasis. J. Cell. Physiol. 2019, 234, 15763–15774. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.; Peczek, L.; Czernek, L.; Düchler, M. Analysis of the miRNA Profiles of Melanoma Exosomes Derived Under Normoxic and Hypoxic Culture Conditions. Anticancer Res. 2017, 37, 6779–6789. [Google Scholar] [CrossRef] [PubMed]
- Bland, C.L.; Byrne-Hoffman, C.N.; Fernandez, A.; Rellick, S.L.; Deng, W.; Klinke, D.J., 2nd. Exosomes derived from B16F0 melanoma cells alter the transcriptome of cytotoxic T cells that impacts mitochondrial respiration. FEBS J. 2018, 285, 1033–1050. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, Y.; Wang, W.; Zhang, Y.; Chen, Z.; Hao, C.; Zhang, J. Melanoma-released exosomes directly activate the mitochondrial apoptotic pathway of CD4+ T cells through their microRNA cargo. Exp. Cell Res. 2018, 371, 364–371. [Google Scholar] [CrossRef]
- Zhou, X.; Yan, T.; Huang, C.; Xu, Z.; Wang, L.; Jiang, E.; Wang, H.; Chen, Y.; Liu, K.; Shao, Z.; et al. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 2018, 37, 242. [Google Scholar] [CrossRef]
- Felicetti, F.; De Feo, A.; Coscia, C.; Puglisi, R.; Pedini, F.; Pasquini, L.; Bellenghi, M.; Errico, M.C.; Pagani, E.; Carè, A. Exosome-mediated transfer of miR-222 is sufficient to increase tumor malignancy in melanoma. J. Transl. Med. 2016, 1, 56. [Google Scholar] [CrossRef]
- Chen, L.; Karisma, V.W.; Liu, H.; Zhong, L. MicroRNA-300: A Transcellular Mediator in Exosome Regulates Melanoma Progression. Front. Oncol. 2019, 9, 1005. [Google Scholar] [CrossRef]
- Shu, S.L.; Yang, Y.; Allen, C.L.; Maguire, O.; Minderman, H.; Sen, A.; Ciesielski, M.J.; Collins, K.A.; Bush, P.J.; Singh, P.; et al. Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci. Rep. 2018, 8, 12905. [Google Scholar] [CrossRef]
- Vignard, V.; Labbé, M.; Marec, N.; André-Grégoire, G.; Jouand, N.; Fonteneau, J.F.; Labarrière, N.; Fradin, D. MicroRNAs in Tumor Exosomes Drive Immune Escape in Melanoma. Cancer Immunol. Res. 2020, 8, 255–267. [Google Scholar] [CrossRef]
- Pfeffer, S.R.; Grossmann, K.F.; Cassidy, P.B.; Yang, C.H.; Fan, M.; Kopelovich, L.; Leachman, S.A.; Pfeffer, L.M. Detection of Exosomal miRNAs in the Plasma of Melanoma Patients. J. Clin. Med. 2015, 4, 2012–2027. [Google Scholar] [CrossRef]
- Alegre, E.; Sanmamed, M.F.; Rodriguez, C.; Carranza, O.; Martín-Algarra, S.; González, A. Study of circulating microRNA-125b levels in serum exosomes in advanced melanoma. Arch. Pathol. Lab. Med. 2014, 138, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, E.; Hershkovitz, L.; Itzhaki, O.; Hajdu, S.; Nemlich, Y.; Ortenberg, R.; Gefen, N.; Edry, L.; Modai, S.; Keisari, Y.; et al. Regulation of cancer aggressive features in melanoma cells by microRNAs. PLoS ONE 2011, 6, e18936. [Google Scholar] [CrossRef] [PubMed]
- Rao, E.; Jiang, C.; Ji, M.; Huang, X.; Iqbal, J.; Lenz, G.; Wright, G.; Staudt, L.M.; Zhao, Y.; McKeithan, T.W.; et al. The miRNA-17 approximately 92 cluster mediates chemoresistance and enhances tumor growth in mantle cell lymphoma via pi3k/akt pathway activation. Leukemia 2012, 26, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Iliopoulos, D.; Janas, M.M.; Schubert, S.; Pinner, S.; Chen, P.H.; Li, S.; Fletcher, A.L.; Yokoyama, S.; et al. Intronic miR-211 assumes the tumor suppressive function of its host gene in melanoma. Mol. Cell 2010, 40, 841–849. [Google Scholar] [CrossRef]
- Si, M.L.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y.Y. MiR-21-mediated tumor growth. Oncogene 2007, 26, 2799–2803. [Google Scholar] [CrossRef]
- Zhu, S.; Wu, H.; Wu, F.; Nie, D.; Sheng, S.; Mo, Y.Y. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008, 18, 350–359. [Google Scholar] [CrossRef]
- Yang, C.H.; Yue, J.; Pfeffer, S.R.; Handorf, C.R.; Pfeffer, L.M. MicroRNA miR-21 regulates the metastatic behavior of b16 melanoma cells. J. Biol. Chem. 2011, 286, 39172–39178. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Yue, J.; Fan, M.; Pfeffer, L.M. IFN induces miR-21 through a signal transducer and activator of transcription 3-dependent pathway as a suppressive negative feedback on IFN-induced apoptosis. Cancer Res. 2010, 70, 8108–8116. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Hannafon, B.N.; Khatri, U.; Gin, A.; Ding, W.Q. The origin of exosomal miR-1246 in human cancer cells. RNA Biol. 2019, 16, 770–784. [Google Scholar] [CrossRef]
- Bhagirath, D.; Yang, T.L.; Bucay, N.; Sekhon, K.; Majid, S.; Shahryari, V.; Dahiya, R.; Tanaka, Y.; Saini, S. microRNA-1246 Is an Exosomal Biomarker for Aggressive Prostate Cancer. Cancer Res. 2018, 78, 1833–1844. [Google Scholar] [CrossRef]
- Felicetti, F.; Errico, M.C.; Bottero, L.; Segnalini, P.; Stoppacciaro, A.; Biffoni, M.; Felli, N.; Mattia, G.; Petrini, M.; Colombo, M.P.; et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008, 68, 2745–2754. [Google Scholar] [CrossRef]
- Müller, D.; Bosserhoff, A. Integrin β3 expression is regulated by let-7a miRNA in malignant melanoma. Oncogene 2008, 27, 6698–6706. [Google Scholar] [CrossRef]
- Xiao, D.; Barry, S.; Kmetz, D.; Egger, M.; Pan, J.; Rai, S.N.; Qu, J.; McMasters, K.M.; Hao, H. Melanoma cell-derived exosomes promote epithelial-mesenchymal transition in primary melanocytes through paracrine/autocrine signaling in the tumor microenvironment. Cancer Lett. 2016, 376, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.; Rafferty, M.; Hegarty, S.; O’Leary, P.; Faller, W.; Gremel, G.; Bergqvist, M.; Agnarsdottir, M.; Strömberg, S.; Kampf, C.; et al. Topoisomerase I amplification in melanoma is associated with more advanced tumours and poor prognosis. Pigment Cell Melanoma Res. 2010, 23, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tang, L.; Lin, J.; Shen, Z.; Yao, Y.; Wang, W.; Tao, S.; Gu, C.; Ma, J.; Xie, Y.; et al. ABCB5 promotes melanoma metastasis through enhancing NF-κB p65 protein stability. Biochem. Biophys. Res. Commun. 2017, 492, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Suryo Rahmanto, Y.; Dunn, L.; Richardson, D. The melanoma tumor antigen, melanotransferrin (p97): A 25-year hallmark—From iron metabolism to tumorigenesis. Oncogene 2007, 26, 6113–6124. [Google Scholar] [CrossRef] [PubMed]
- Soengas, M.; Hernando, E. TYRP1 mRNA goes fishing for miRNAs in melanoma. Nat. Cell Biol. 2017, 19, 1311–1312. [Google Scholar] [CrossRef]
- Gilot, D.; Migault, M.; Bachelot, L.; Journé, F.; Rogiers, A.; Donnou-Fournet, E.; Mogha, A.; Mouchet, N.; Pinel-Marie, M.L.; Mari, B.; et al. A non-coding function of TYRP1 mRNA promotes melanoma growth. Nat. Cell Biol. 2017, 19, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Wang, Q.; Liu, Y.; Bao, W.; Wu, S. Exosomes in cancer: Small transporters with big functions. Cancer Lett. 2018, 435, 55–65. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Kaiser, J. Malignant messengers. Science 2016, 352, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Muller, L.; Mitsuhashi, M.; Simms, P.; Gooding, W.E.; Whiteside, T.L. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 2016, 6, 20254. [Google Scholar] [CrossRef]
- Bergmann, C.; Strauss, L.; Wang, Y.; Szczepanski, M.J.; Lang, S.; Johnson, J.T.; Whiteside, T.L. T regulatory Type 1 cells (Tr1) in squamous cell carcinoma of the head and neck: Mechanisms of suppression and expansion in advanced disease. Clin. Cancer Res. 2008, 14, 3706–3715. [Google Scholar] [CrossRef]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef] [PubMed]
- Düchler, M.; Czernek, L.; Peczek, L.; Cypryk, W.; Sztiller-Sikorska, M.; Czyz, M. Melanoma-Derived Extracellular Vesicles Bear the Potential for the Induction of Antigen-Specific Tolerance. Cells 2019, 8, 665. [Google Scholar] [CrossRef]
- Gyukity-Sebestyén, E.; Harmati, M.; Dobra, G.; Németh, I.B.; Mihály, J.; Zvara, Á.; Hunyadi-Gulyás, É.; Katona, R.; Nagy, I.; Horváth, P.; et al. Melanoma-Derived Exosomes Induce PD-1 Overexpression and Tumor Progression via Mesenchymal Stem Cell Oncogenic Reprogramming. Front. Immunol. 2019, 10, 2459. [Google Scholar] [CrossRef]
- Martínez-Lorenzo, M.J.; Anel, A.; Alava, M.A.; Piñeiro, A.; Naval, J.; Lasierra, P.; Larrad, L. The human melanoma cell line MelJuSo secretes bioactive FasL and APO2L/TRAIL on the surface of microvesicles. Possible contribution to tumor counterattack. Exp. Cell Res. 2004, 295, 315–329. [Google Scholar] [CrossRef]
- Wu, Y.; Deng, W.; McGinley, E.C.; Klinke, D.J., 2nd. Melanoma exosomes deliver a complex biological payload that upregulates PTPN11 to suppress T lymphocyte function. Pigment Cell Melanoma Res. 2017, 30, 203–218. [Google Scholar] [CrossRef]
- Kulkarni, Y.M.; Chambers, E.; McGray, A.J.R.; Ware, J.S.; Bramson, J.L.; Klinke, D.J. A quantitative systems approach to identify paracrine mechanisms that locally suppress immune response to Interleukin-12 in the B16 melanoma model. Integr. Biol. 2012, 4, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Cottonham, C.L.; Kaneko, S.; Xu, L. miR-21 and miR-31 converge on TIAM1 to regulate migration and invasion of colon carcinoma cells. J. Biol. Chem. 2010, 285, 35293–35302. [Google Scholar] [CrossRef]
- Yasuda, M.; Schmid, T.; Rubsamen, D.; Colburn, N.H.; Irie, K.; Murakami, A. Downregulation of programmed cell death 4 by inflammatory conditions contributes to the generation of the tumor promoting microenvironment. Mol. Carcinog. 2010, 49, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Diao, J.; Gu, H.; Khatri, I.; Zhao, J.; Cattral, M.S. Toll-like Receptor 2 Activation Promotes Tumor Dendritic Cell Dysfunction by Regulating IL-6 and IL-10 Receptor Signaling. Cell Rep. 2015, 13, 2851–2864. [Google Scholar] [CrossRef]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ji, X.; Liu, J.; Fan, D.; Zhou, Q.; Chen, C.; Wang, W.; Wang, G.; Wang, H.; Yuan, W.; et al. Effects of exosomes on pre-metastatic niche formation in tumors. Mol. Cancer 2019, 18, 39. [Google Scholar] [CrossRef]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell. 2013, 24, 466–480. [Google Scholar] [CrossRef]
- Hu, T.; Hu, J. Melanoma-derived exosomes induce reprogramming fibroblasts into cancer-associated fibroblasts via Gm26809 delivery. Cell Cycle 2019, 18, 3085–3094. [Google Scholar] [CrossRef]
- Zhao, X.-P.; Wang, M.; Song, Y.; Song, K.; Yan, T.L.; Wang, L.; Li, K.; Shang, Z.J. Membrane microvesicles as mediators for melanoma-fibroblasts communication: Roles of the VCAM-1/VLA-4 axis and the ERK1/2 signal pathway. Cancer Lett. 2015, 360, 125–133. [Google Scholar] [CrossRef]
- Hood, J.; Pan, H.; Lanza, G.; Wickline, S.A. Paracrine induction of endothelium by tumor exosomes. Lab. Investig. 2009, 89, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Wu, X.; Jiang, Z.; Kasman, I.; Yao, J.; Guan, Y.; Oeh, J.; Modrusan, Z.; Bais, C.; Sampath, D.; et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J. 2012, 31, 3513–3523. [Google Scholar] [CrossRef] [PubMed]
- Ekström, E.J.; Bergenfelz, C.; von Bülow, V.; Serifler, F.; Carlemalm, E.; Jönsson, G.; Andersson, T.; Leandersson, K. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol. Cancer 2014, 13, 88. [Google Scholar] [CrossRef] [PubMed]
- Sento, S.; Sasabe, E.; Yamamoto, T. Application of a persistent heparin treatment inhibits the malignant potential of oral squamous carcinoma cells induced by tumor cell-derived exosomes. PLoS ONE 2016, 11, e0148454. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Rosenblum, D.; Weinstein, S.; Bairey, O.; Raanani, P.; Peer, D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015, 364, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Morishita, M.; Charoenviriyakul, C.; Saji, H.; Takakura, Y. Accelerated growth of B16BL6 tumor in mice through efficient uptake of their own exosomes by B16BL6 cells. Cancer Sci. 2017, 108, 1803–1810. [Google Scholar] [CrossRef]
- Isola, A.L.; Eddy, K.; Zembrzuski, K.; Goydos, J.S.; Chen, S. Exosomes released by metabotropic glutamate receptor 1 (GRM1) expressing melanoma cells increase cell migration and invasiveness. Oncotarget 2018, 9, 1187–1199. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2009, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Lui, G.Y.L.; Lai, S.L.; Wilmott, J.S.; Tikoo, S.; Jackett, L.A.; Quek, C.; Brown, D.L.; Sharp, D.M.; Kwan, R.Y.Q.; et al. RAB27A promotes melanoma cell invasion and metastasis via regulation of pro-invasive exosomes. Int. J. Cancer 2019, 144, 3070–3085. [Google Scholar] [CrossRef]
- Mannavola, F.; Tucci, M.; Felici, C.; Passarelli, A.; D’Oronzo, S.; Silvestris, F. Tumor-derived exosomes promote the in vitro osteotropism of melanoma cells by activating the SDF-1/CXCR4/CXCR7 axis. J. Transl. Med. 2019, 17, 230. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [PubMed]
- Angi, M.; Kalirai, H.; Prendergast, S.; Simpson, D.; Hammond, D.E.; Madigan, M.C.; Beynon, R.J.; Coupland, S.E. In-depth proteomic profiling of the uveal melanoma secretome. Oncotarget 2016, 7, 49623–49635. [Google Scholar] [CrossRef] [PubMed]
- Tengda, L.; Shuping, L.; Mingli, G.; Jie, G.; Yun, L.; Weiwei, Z.; Anmei, D. Serum exosomal microRNAs as potent circulating biomarkers for melanoma. Melanoma Res. 2018, 28, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Lunavat, T.R.; Cheng, L.; Einarsdottir, B.O.; Olofsson Bagge, R.; Veppil Muralidharan, S.; Sharples, R.A.; Lässer, C.; Gho, Y.S.; Hill, A.F.; Nilsson, J.A.; et al. BRAFV600 inhibition alters the microRNA cargo in the vesicular secretome of malignant melanoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E5930–E5939. [Google Scholar] [CrossRef]
- Svedman, F.C.; Lohcharoenkal, W.; Bottai, M.; Brage, S.E.; Sonkoly, E.; Hansson, J.; Pivarcsi, A.; Eriksson, H. Extracellular microvesicle microRNAs as predictive biomarkers for targeted therapy in metastastic cutaneous malignant melanoma. PLoS ONE 2018, 13, e0206942. [Google Scholar] [CrossRef]
- Segura, M.F.; Belitskaya-Lévy, I.; Rose, A.E.; Zakrzewski, J.; Gaziel, A.; Hanniford, D.; Darvishian, F.; Berman, R.S.; Shapiro, R.L.; Pavlick, A.C.; et al. Melanoma MicroRNA signature predicts post-recurrence survival. Clin. Cancer Res. 2010, 16, 1577–1586. [Google Scholar] [CrossRef]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.I.; Weber, J.S.; et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Cordonnier, M.; Nardin, C.; Chanteloup, G.; Derangere, V.; Algros, M.P.; Arnould, L.; Garrido, C.; Aubin, F.; Gobbo, J. Tracking the evolution of circulating exosomal-PD-L1 to monitor melanoma patients. J. Extracell. Vesicles 2020, 9, 1. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Line | Method of MTEX Purification and Characterization | MS Approach | Major Findings | Ref. |
|---|---|---|---|---|
| MeWo, SK-MEL-28 (human) | UC/TEM, WB, 1D/2D SDS-PAGE | MALDI-TOF MS/MS | A few proteins identified in MTEX for the first time: prostaglandin regulatory-like protein (PGRL), p120 catenin, syntaxin-binding proteins 1 and 2, septin 2 (Nedd5), ezrin, radixin, tryptophan/aspartic acid (WD) repeat-containing protein 1 | [68] |
| A375 (human) | UC/TEM, NTA, WB | LC-MS/MS | Different sets of proteins present in MTEX and melanocyte-derived EV, including annexin A1, HAPLN1, GRP78, endoplasmin precursor (gp 96), TUBA1B, PYGB), ferritin, heavy polypeptide 1 (MTEX-upregulated), annexin A2, syntenin-1, MFGE8, OXCT (MTEX-downregulated) | [69] |
| MNT-1, G1, 501 mel, SKMEL28, Daju, A375M, 1205Lu (human) | UC+SEC/WB, TEM, NTA | nanoLC-MS/MS | Different sets of proteins present in MTEX from nontumorigenic, tumorigenic, and metastatic cell lines, including EGFR, PTK2/FAK1, EPHB2, SRC, LGALS1/LEG1, LGALS3/LEG3, NT5E/5NTD-CD73, NRAS, KIT, MCAM/MUC18, MET specific for metastatic cell lines | [70] |
| B16-F1 (murine) | UC+SEC/CEM, DLS, IA-FCM | uHPLC-MS | 10 most abundant proteins: CD81, CD9, histones (H2A, H2B, H3.1, H4), heat shock proteins (HSPA5/GRP78, HSC71), syntetin-1 | [71] |
| B16-F10 (murine) | UC+SEC, UF+SEC/TEM, NTA, WB | nanoLC-MS/MS | Different sets of proteins identified in low- and high-density MTEX, including ACTN4 and CCNY enriched in LD-MTEX and EPHA2 enriched in HD-MTEX | [72] |
| Mel501 (human) | UC+SEC/WB, CM | RPLC-MS/MS | Different sets of proteins identified in MTEX released in neutral and acidic environment (pH 6.7 and 6.0, respectively), including HRAS, NRAS, TIMP3, HSP90AB1, HSP90B1, HSPAIL, HSPA5, GANAB, gelsolin, and cofilin upregulated in acidic conditions. | [73] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zebrowska, A.; Widlak, P.; Whiteside, T.; Pietrowska, M. Signaling of Tumor-Derived sEV Impacts Melanoma Progression. Int. J. Mol. Sci. 2020, 21, 5066. https://doi.org/10.3390/ijms21145066
Zebrowska A, Widlak P, Whiteside T, Pietrowska M. Signaling of Tumor-Derived sEV Impacts Melanoma Progression. International Journal of Molecular Sciences. 2020; 21(14):5066. https://doi.org/10.3390/ijms21145066
Chicago/Turabian StyleZebrowska, Aneta, Piotr Widlak, Theresa Whiteside, and Monika Pietrowska. 2020. "Signaling of Tumor-Derived sEV Impacts Melanoma Progression" International Journal of Molecular Sciences 21, no. 14: 5066. https://doi.org/10.3390/ijms21145066
APA StyleZebrowska, A., Widlak, P., Whiteside, T., & Pietrowska, M. (2020). Signaling of Tumor-Derived sEV Impacts Melanoma Progression. International Journal of Molecular Sciences, 21(14), 5066. https://doi.org/10.3390/ijms21145066