Crosstalk of Brain and Bone—Clinical Observations and Their Molecular Bases


 and
and
Abstract
1. Introduction
2. Clinical Observations
2.1. Efferent ‘Brain-Bone’
2.2. Afferent ‘Bone-Brain’
2.3. Trauma Affecting Brain and Bone
3. Molecular Bases of Brain-Bone Crosstalk
3.1. Brain- and Nerve-Derived Mediators Affecting Bone Cell Function
3.1.1. Central Regulation
3.1.2. SNS and PSNS
3.1.3. Sensory Innervation
3.2. Bone-Derived Mediators Acting on the Central Nervous System
3.3. Mediators Affecting Both Brain and Bone Function
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chamouni, A.; Schreiweis, C.; Oury, F. Bone, brain & beyond. Rev. Endocr. Metab. Disord. 2015, 16, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Rousseaud, A.; Moriceau, S.; Ramos-Brossier, M.; Oury, F. Bone-brain crosstalk and potential associated diseases. Horm. Mol. Biol. Clin. Investig. 2016, 28, 69–83. [Google Scholar] [CrossRef]
- Dimitri, P.; Rosen, C. The Central Nervous System and Bone Metabolism: An Evolving Story. Calcif. Tissue Int. 2017, 100, 476–485. [Google Scholar] [CrossRef]
- Idelevich, A.; Baron, R. Brain to bone: What is the contribution of the brain to skeletal homeostasis? Bone 2018, 115, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Takeishi, S.; Frenette, P.S. Neural regulation of bone and bone marrow. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Obri, A.; Khrimian, L.; Karsenty, G.; Oury, F. Osteocalcin in the brain: From embryonic development to age-related decline in cognition. Nat. Rev. Endocrinol. 2018, 14, 174–182. [Google Scholar] [CrossRef]
- Frame, G.; Bretland, K.A.; Dengler-Crish, C.M. Mechanistic complexities of bone loss in Alzheimer’s disease: A review. Connect. Tissue Res. 2020, 61, 4–18. [Google Scholar] [CrossRef]
- Yuan, J.; Meloni, B.P.; Shi, T.; Bonser, A.; Papadimitriou, J.M.; Mastaglia, F.L.; Zhang, C.; Zheng, M.; Gao, J. The potential influence of bone-derived modulators on the progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 69, 59–70. [Google Scholar] [CrossRef]
- Tatangelo, G.; Watts, J.; Lim, K.; Connaughton, C.; Abimanyi-Ochom, J.; Borgström, F.; Nicholson, G.C.; Shore-Lorenti, C.; Stuart, A.L.; Iuliano-Burns, S.; et al. The Cost of Osteoporosis, Osteopenia, and Associated Fractures in Australia in 2017. J. Bone Miner. Res. 2019, 34, 616–625. [Google Scholar] [CrossRef]
- Siris, E.S.; Adler, R.; Bilezikian, J.; Bolognese, M.; Dawson-Hughes, B.; Favus, M.J.; Harris, S.T.; Jan De Beur, S.M.; Khosla, S.; Lane, N.E.; et al. The clinical diagnosis of osteoporosis: A position statement from the National Bone Health Alliance Working Group. Osteoporos. Int. 2014, 25, 1439–1443. [Google Scholar] [CrossRef]
- Petty, S.J.; Wilding, H.; Wark, J.D. Osteoporosis Associated with Epilepsy and the Use of Anti-Epileptics—A Review. Curr. Osteoporos. Rep. 2016, 14, 54–65. [Google Scholar] [CrossRef]
- Kishimoto, T.; De Hert, M.; Carlson, H.E.; Manu, P.; Correll, C.U. Osteoporosis and fracture risk in people with schizophrenia. Curr. Opin. Psychiatry 2012, 25, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Bukowska-Damska, A.; Skowronska-Jozwiak, E.; Peplonska, B. Night shift work and osteoporosis: Evidence and hypothesis. Chronobiol. Int. 2019, 36, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.S.; Hsu, J.W.; Huang, K.L.; Bai, Y.M.; Su, T.P.; Li, C.T.; Lin, W.C.; Chen, T.J.; Tsai, S.J.; Liou, Y.J.; et al. Post-traumatic stress disorder and risk of osteoporosis: A nationwide longitudinal study. Stress Heal. 2018, 34, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Aloumanis, K.; Mavroudis, K. The “depressive” face of osteoporosis and the “osteoporotic” face of depression. Hormones 2013, 12, 350–362. [Google Scholar] [CrossRef]
- Carda, S.; Cisari, C.; Invernizzi, M.; Bevilacqua, M. Osteoporosis after stroke: A review of the causes and potential treatments. Cerebrovasc. Dis. 2009, 28, 191–200. [Google Scholar] [CrossRef]
- Chen, Y.H.; Lo, R.Y. Alzheimer’s disease and osteoporosis. Tzu Chi Med. J. 2017, 29, 138–142. [Google Scholar]
- Invernizzi, M.; Carda, S.; Viscontini, G.S.; Cisari, C. Osteoporosis in Parkinson’s disease. Park. Relat. Disord. 2009, 15, 339–346. [Google Scholar] [CrossRef]
- Smith, E.; Comiskey, C.; Carroll, A. Prevalence of and risk factors for osteoporosis in adults with acquired brain injury. Ir. J. Med. Sci. 2016, 185, 473–481. [Google Scholar] [CrossRef]
- Wang, L.; Yao, X.; Xiao, L.; Tang, X.; Ding, H.; Zhang, H.; Yuan, J. The effects of spinal cord injury on bone healing in patients with femoral fractures. J. Spinal Cord Med. 2014, 37, 414–419. [Google Scholar] [CrossRef]
- Borchers, A.T.; Gershwin, M.E. Complex regional pain syndrome: A comprehensive and critical review. Autoimmun. Rev. 2014, 13, 242–265. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.J.; Sun, M.; Agoston, D.V.; Shultz, S.R. The effect of concomitant peripheral injury on traumatic brain injury pathobiology and outcome. J. Neuroinflammation 2016, 13. [Google Scholar] [CrossRef] [PubMed]
- Dennison, E.M.; Compston, J.E.; Flahive, J.; Siris, E.S.; Gehlbach, S.H.; Adachi, J.D.; Boonen, S.; Chapurlat, R.; Díez-Pérez, A.; Anderson, F.A.; et al. Effect of co-morbidities on fracture risk: Findings from the Global Longitudinal Study of Osteoporosis in Women (GLOW). Bone 2012, 50, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Ellman, R.; Spatz, J.; Cloutier, A.; Palme, R.; Christiansen, B.A.; Bouxsein, M.L. Partial reductions in mechanical loading yield proportional changes in bone density, bone architecture, and muscle mass. J. Bone Miner. Res. 2013, 28, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Feskanich, D.; Hankinson, S.E.; Schernhammer, E.S. Nightshift work and fracture risk: The Nurses’ Health Study. Osteoporos. Int. 2009, 20, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Quevedo, I.; Zuniga, A.M. Low bone mineral density in rotating-shift workers. J. Clin. Densitom. 2010, 13, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Weibel, L.; Brandenberger, G. Disturbances in Hormonal Profiles of Night Workers during Their Usual Sleep and Work Times. J. Biol. Rhythms 1998, 13, 202–208. [Google Scholar] [CrossRef]
- Dumont, M.; Lanctt, V.; Cadieux-Viau, R.; Paquet, J. Melatonin production and light exposure of rotating night workers. Chronobiol. Int. 2012, 29, 203–210. [Google Scholar] [CrossRef]
- Ulhôa, M.A.; Marqueze, E.C.; Burgos, L.G.A.; Moreno, C.R.C. Shift work and endocrine disorders. Int. J. Endocrinol. 2015, 2015. [Google Scholar] [CrossRef]
- Schernhammer, E.S.; Rosner, B.; Willet, W.C.; Laden, F.; Colditz, G.A.; Hankinson, S.E. Epidemiology of urinary melatonin in women and its relation to other hormones and night work. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 936–943. [Google Scholar]
- Manenschijn, L.; Van Kruysbergen, R.G.P.M.; De Jong, F.H.; Koper, J.W.; Van Rossum, E.F.C. Shift work at young age is associated with elevated long-term cortisol levels and body mass index. J. Clin. Endocrinol. Metab. 2011, 96, 1862–1865. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, A.M.; Kac, G.; Campos e Souza, R.R.; Maria de Barros Almeida Ferreira, L.; Maria de Fátima Silqueira, S. Night-shift work and cardiovascular risk among employees of a public university. Rev. da Assoc. Médica Bras. (English Ed.) 2012, 58, 168–177. [Google Scholar] [CrossRef]
- Strohmaier, S.; Devore, E.E.; Zhang, Y.; Schernhammer, E.S. A Review of Data of Findings on Night Shift Work and the Development of DM and CVD Events: A Synthesis of the Proposed Molecular Mechanisms. Curr. Diab. Rep. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.; Schernhammer, E.S.; Sun, Q.; Hu, F.B. Rotating night shift work and risk of type 2 diabetes: Two prospective cohort studies in women. PLoS Med. 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Shao, P.; Ohtsuka-Isoya, M.; Shinoda, H. Circadian rhythms in serum bone markers and their relation to the effect of etidronate in rats. Chronobiol. Int. 2003, 20, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Schibler, U.; Sassone-Corsi, P. A web of circadian pacemakers. Cell 2002, 111, 919–922. [Google Scholar] [CrossRef]
- Maronde, E.; Schilling, A.F.; Seitz, S.; Schinke, T.; Schmutz, I.; van der Horst, G.; Amling, M.; Albrecht, U. The clock genes Period 2 and Cryptochrome 2 differentially balance bone formation. PLoS ONE 2010, 5, 1–8. [Google Scholar] [CrossRef]
- Everson, C.A.; Folley, A.E.; Toth, J.M. Chronically inadequate sleep results in abnormal bone formation and abnormal bone marrow in rats. Exp. Biol. Med. 2012, 237, 1101–1109. [Google Scholar] [CrossRef]
- Swanson, C.M.; Kohrt, W.M.; Buxton, O.M.; Everson, C.A.; Wright, K.P.; Orwoll, E.S.; Shea, S.A. The importance of the circadian system & sleep for bone health. Metabolism 2018, 84, 28–43. [Google Scholar] [CrossRef]
- Swanson, C.M.; Shea, S.A.; Wolfe, P.; Cain, S.W.; Munch, M.; Vujović, N.; Czeisler, C.A.; Buxton, O.M.; Orwoll, E.S. Bone turnover markers after sleep restriction and circadian disruption: A mechanism for sleep-related bone loss in humans. J. Clin. Endocrinol. Metab. 2017, 102, 3722–3730. [Google Scholar] [CrossRef]
- El-Gabalawy, R.; Blaney, C.; Tsai, J.; Sumner, J.A.; Pietrzak, R.H. Physical health conditions associated with full and subthreshold PTSD in U.S. military veterans: Results from the National Health and Resilience in Veterans Study. J. Affect. Disord. 2018, 227, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Toft, H.; Bramness, J.G.; Lien, L.; Abebe, D.S.; Wampold, B.E.; Tilden, T.; Hestad, K.; Neupane, S.P. PTSD patients show increasing cytokine levels during treatment despite reduced psychological distress. Neuropsychiatr. Dis. Treat. 2018, 14, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Pfeilschifter, J.; Chenu, C.; Bird, A.; Mundy, G.R.; Roodman, D.G. Interleukin-1 and tumor necrosis factor stimulate the formation of human osteoclastlike cells in vitro. J. Bone Miner. Res. 1989, 4, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Daskalakis, N.P.; Lehrner, A.; Yehuda, R. Endocrine Aspects of Post-traumatic Stress Disorder and Implications for Diagnosis and Treatment. Endocrinol. Metab. Clin. N. Am. 2013, 42, 503–513. [Google Scholar] [CrossRef]
- Hasan, K.M.M.; Rahman, M.S.; Arif, K.M.T.; Sobhani, M.E. Psychological stress and aging: Role of glucocorticoids (GCs). Age 2012, 34, 1421–1433. [Google Scholar] [CrossRef]
- Weinstein, R.S.; Jilka, R.L.; Michael Parfitt, A.; Manolagas, S.C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts end osteocytes by glucocorticoids potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998, 102, 274–282. [Google Scholar] [CrossRef]
- Foertsch, S.; Haffner-Luntzer, M.; Kroner, J.; Gross, F.; Kaiser, K.; Erber, M.; Reber, S.O.; Ignatius, A. Chronic psychosocial stress disturbs long-bone growth in adolescent mice. DMM Dis. Model. Mech. 2017, 10, 1399–1409. [Google Scholar] [CrossRef]
- Haffner-Luntzer, M.; Foertsch, S.; Fischer, V.; Prystaz, K.; Tschaffon, M.; Mödinger, Y.; Bahney, C.S.; Marcucio, R.S.; Miclau, T.; Ignatius, A.; et al. Chronic psychosocial stress compromises the immune response and endochondral ossification during bone fracture healing via β-AR signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 8615–8622. [Google Scholar] [CrossRef]
- Schweiger, J.U.; Schweiger, U.; Hüppe, M.; Kahl, K.G.; Greggersen, W.; Fassbinder, E. Bone density and depressive disorder: A meta-analysis. Brain Behav. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Cizza, G. Major depressive disorder is a risk factor for low bone mass, central obesity, and other medical conditions. Dialogues Clin. Neurosci. 2011, 13, 73–87. [Google Scholar]
- Cizza, G.; Primma, S.; Coyle, M.; Gourgiotis, L.; Csako, G. Depression and osteoporosis: A research synthesis with meta-analysis. Horm. Metab. Res. 2010, 42, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Yirmiya, R.; Bab, I. Major Depression Is a Risk Factor for Low Bone Mineral Density: A Meta-Analysis. Biol. Psychiatry 2009, 66, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.; Gomez, R.; Williams, G.; Lembke, A.; Lazzeroni, L.; Murphy, G.M.; Schatzberg, A.F. HPA axis in major depression: Cortisol, clinical symptomatology and genetic variation predict cognition. Mol. Psychiatry 2017, 22, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Yirmiya, R.; Goshen, I.; Bajayo, A.; Kreisel, T.; Feldman, S.; Tam, J.; Trembovler, V.; Csernus, V.; Shohami, E.; Bab, I. Depression induces bone loss through stimulation of the sympathetic nervous system. Proc. Natl. Acad. Sci. USA 2006, 103, 16876–16881. [Google Scholar] [CrossRef]
- Mezuk, B.; Eaton, W.W.; Golden, S.H. Depression and osteoporosis: Epidemiology and potential mediating pathways. Osteoporos. Int. 2008, 19, 1–12. [Google Scholar] [CrossRef]
- Schweiger, U.; Weber, B.; Deuschle, M.; Heuser, I. Lumbar bone mineral density in patients with major depression: Evidence of increased bone loss at follow-up. Am. J. Psychiatry 2000, 157, 118–120. [Google Scholar] [CrossRef]
- Nie, C.; Wang, Z.; Liu, X. The effect of depression on fracture healing and osteoblast differentiation in rats. Neuropsychiatr. Dis. Treat. 2018, 14, 1705–1713. [Google Scholar] [CrossRef]
- Roth, G.A.; Feigin, V.L.; Nguyen, G.; Cercy, K.; Johnson, C.O.; Alam, T.; Parmar, P.G.; Abajobir, A.A.; Abate, K.H.; Abd-Allah, F.; et al. Global, regional, and country-specific lifetime risks of stroke, 1990 and 2016. N. Engl. J. Med. 2018, 379, 2429–2437. [Google Scholar] [CrossRef]
- Mughal, N.; Inderjeeth, A.J.; Inderjeeth, C.A. Osteoporosis in patients with dementia is associated with high morbidity and mortality: Findings from a single orthogeriatric unit. Aust. J. Gen. Pract. 2019, 48, 53–58. [Google Scholar] [CrossRef]
- Posada, I.J.; Benito-León, J.; Louis, E.D.; Trincado, R.; Villarejo, A.; Medrano, M.J.; Bermejo-Pareja, F. Mortality from Parkinson’s disease: A population-based prospective study (NEDICES). Mov. Disord. 2011, 26, 2522–2529. [Google Scholar] [CrossRef]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef] [PubMed]
- Ramnemark, A.; Nyberg, L.; Borssén, B.; Olsson, T.; Gustafson, Y. Fractures after stroke. Osteoporos. Int. 1998, 8, 92–95. [Google Scholar] [CrossRef]
- Wei, M.; Lyu, H.; Huo, K.; Su, H. Impact of bone fracture on ischemic stroke recovery. Int. J. Mol. Sci. 2018, 19, 1533. [Google Scholar] [CrossRef]
- Sato, Y.; Kuno, H.; Kaji, M.; Etoh, K.; Oizumi, K. Influence of immobilization upon calcium metabolism in the week following hemiplegic stroke. J. Neurol. Sci. 2000, 175, 135–139. [Google Scholar] [CrossRef]
- He, X.W.; Wang, E.; Bao, Y.Y.; Wang, F.; Zhu, M.; Hu, X.F.; Jin, X.P. High serum levels of sclerostin and Dickkopf-1 are associated with acute ischaemic stroke. Atherosclerosis 2016, 253, 22–28. [Google Scholar] [CrossRef]
- Zhu, Z.; Guo, D.; Zhong, C.; Wang, A.; Xie, X.; Xu, T.; Chen, C.S.; Peng, Y.; Peng, H.; Li, Q.; et al. Serum Dkk-1 (Dickkopf-1) Is a Potential Biomarker in the Prediction of Clinical Outcomes among Patients with Acute Ischemic Stroke. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 285–293. [Google Scholar] [CrossRef]
- Mathold, K.; Wanby, P.; Brudin, L.; Von, S.P.; Carlsson, M. Alterations in bone turnover markers in patients with noncardio-embolic ischemic stroke. PLoS ONE 2018, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Beaupre, G.S.; Lew, H.L. Bone-density changes after stroke. Am. J. Phys. Med. Rehabil. 2006, 85, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Wheatley, B.M.; Cholok, D.; Agarwal, S.; Yu, P.B.; Levi, B.; Davis, T.A. The traumatic bone: Trauma-induced heterotopic ossification. Transl. Res. 2017, 186, 95–111. [Google Scholar] [CrossRef]
- Gerber, H.P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628. [Google Scholar] [CrossRef]
- Street, J.; Bao, M.; DeGuzman, L.; Bunting, S.; Peale, F.V.; Ferrara, N.; Steinmetz, H.; Hoeffel, J.; Cleland, J.L.; Daugherty, A.; et al. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc. Natl. Acad. Sci. USA 2002, 99, 9656–9661. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kang, S.; Zou, D.; Zhan, L.; Li, Z.; Zhu, W.; Su, H. Bone fracture pre-ischemic stroke exacerbates ischemic cerebral injury in mice. PLoS ONE 2016, 11, e0153835. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.L.; Cook, M.N.; Arrighi, H.M.; Bullock, R. Hip fracture risk and subsequent mortality among Alzheimer’s disease patients in the United Kingdom, 1988–2007. Age Ageing 2011, 40, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shen, L.; Ji, H.F. Alzheimer’s disease and risk of hip fracture: A meta-analysis study. Sci. World J. 2012, 2012. [Google Scholar] [CrossRef]
- Wang, H.K.; Hung, C.M.; Lin, S.H.; Tai, Y.C.; Lu, K.; Liliang, P.C.; Lin, C.W.; Lee, Y.C.; Fang, P.H.; Chang, L.C.; et al. Increased risk of hip fractures in patients with dementia: A nationwide population-based study. BMC Neurol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Loskutova, N.; Honea, R.A.; Vidoni, E.D.; Brooks, W.M.; Burns, J.M. Bone density and brain atrophy in early Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 18, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yin, X.S.; Guo, H.; Han, R.K.; He, R.D.; Chi, L.J. Elevated osteopontin levels in mild cognitive impairment and Alzheimer’s disease. Mediators Inflamm. 2013, 2013. [Google Scholar] [CrossRef]
- Comi, C.; Carecchio, M.; Chiocchetti, A.; Nicola, S.; Galimberti, D.; Fenoglio, C.; Cappellano, G.; Monaco, F.; Scarpini, E.; Dianzani, U. Osteopontin is increased in the cerebrospinal fluid of patients with Alzheimer’s disease and its levels correlate with cognitive decline. J. Alzheimer’s Dis. 2010, 19, 1143–1148. [Google Scholar] [CrossRef]
- Fodor, D.; Bondor, C.; Albu, A.; Simon, S.P.; Craciun, A.; Muntean, L. The value of osteopontin in the assessment of bone mineral density status in postmenopausal women. J. Investig. Med. 2013, 61, 15–21. [Google Scholar] [CrossRef]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Li, S.; Yang, B.; Teguh, D.; Zhou, L.; Xu, J.; Rong, L. Amyloid β peptide enhances RANKL-induced osteoclast activation through NF-κB, ERK, and calcium oscillation signaling. Int. J. Mol. Sci. 2016, 17, 1683. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Gerlach, M.; Youdim, M.B.H.; Double, K.L.; Zecca, L.; Riederer, P.; Becker, G. Brain iron pathways and their relevance to Parkinson’s disease. J. Neurochem. 2001, 79, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L. The aggregation and fibrillation of α-synuclein. Acc. Chem. Res. 2006, 39, 628–634. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Definition of a molecular pathway mediating α-synuclein neurotoxicity. J. Neurosci. 2015, 35, 5221–5232. [Google Scholar] [CrossRef]
- Kapral, M.K.; Fang, J.; Alibhai, S.M.H.; Cram, P.; Cheung, A.M.; Casaubon, L.K.; Prager, M.; Stamplecoski, M.; Rashkovan, B.; Austin, P.C. Risk of fractures after stroke: Results from the Ontario Stroke Registry. Neurology 2017, 88, 57–64. [Google Scholar] [CrossRef]
- Van Den Bos, F.; Speelman, A.D.; Van Nimwegen, M.; Van Der Schouw, Y.T.; Backx, F.J.G.; Bloem, B.R.; Munneke, M.; Verhaar, H.J.J. Bone mineral density and vitamin D status in Parkinson’s disease patients. J. Neurol. 2013, 260, 754–760. [Google Scholar] [CrossRef]
- Torsney, K.M.; Noyce, A.J.; Doherty, K.M.; Bestwick, J.P.; Dobson, R.; Lees, A.J. Bone health in Parkinson’s disease: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1159–1166. [Google Scholar] [CrossRef]
- Handa, K.; Kiyohara, S.; Yamakawa, T.; Ishikawa, K.; Hosonuma, M.; Sakai, N.; Karakawa, A.; Chatani, M.; Tsuji, M.; Inagaki, K.; et al. Bone loss caused by dopaminergic degeneration and levodopa treatment in Parkinson’s disease model mice. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Mundlos, S. Cleidocranial dysplasia: Clinical and molecular genetics. J. Med. Genet. 1999, 36, 177–182. [Google Scholar] [CrossRef]
- Izumi, K.; Yahagi, N.; Fujii, Y.; Higuchi, M.; Kosaki, R.; Naito, Y.; Nishimura, G.; Hosokai, N.; Takahashi, T.; Kosaki, K. Cleidocranial dysplasia plus vascular anomalies with 6p21.2 microdeletion spanning RUNX2 and VEGF (4). Am. J. Med. Genet. 2006, 140 A, 398–401. [Google Scholar] [CrossRef]
- Takenouchi, T.; Sato, W.; Torii, C.; Kosaki, K. Progressive cognitive decline in an adult patient with cleidocranial dysplasia. Eur. J. Med. Genet. 2014, 57, 319–321. [Google Scholar] [CrossRef]
- Trivier, E.; De Cesare, D.; Jacquot, S.; Pannetier, S.; Zackai, E.; Young, I.; Mandel, J.L.; Sassone-Corsi, P.; Hanauer, A. Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome. Nature 1996, 384, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Marques Pereira, P.; Schneider, A.; Pannetier, S.; Heron, D.; Hanauer, A. Coffin-Lowry syndrome. Eur. J. Hum. Genet. 2010, 18, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Poirier, R.; Jacquot, S.; Vaillend, C.; Soutthiphong, A.A.; Libbey, M.; Davis, S.; Laroche, S.; Hanauer, A.; Welzl, H.; Lipp, H.P.; et al. Deletion of the Coffin-Lowry syndrome gene Rsk2 in mice is associated with impaired spatial learning and reduced control of exploratory behavior. Behav. Genet. 2007, 37, 31–50. [Google Scholar] [CrossRef]
- Kandel, E.R.; Dudai, Y.; Mayford, M.R. The molecular and systems biology of memory. Cell 2014, 157, 163–186. [Google Scholar] [CrossRef]
- Yang, X.; Matsuda, K.; Bialek, P.; Jacquot, S.; Masuoka, H.C.; Schinke, T.; Li, L.; Brancorsini, S.; Sassone-Corsi, P.; Townes, T.M.; et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology: Implication for Coffin-Lowry syndrome. Cell 2004, 117, 387–398. [Google Scholar] [CrossRef]
- Xiao, G.; Jiang, D.; Ge, C.; Zhao, Z.; Lai, Y.; Boules, H.; Phimphilai, M.; Yang, X.; Karsenty, G.; Franceschi, R.T. Cooperative interactions between activating transcription factor 4 and Runx2/Cbfa1 stimulate osteoblast-specific osteocalcin gene expression. J. Biol. Chem. 2005, 280, 30689–30696. [Google Scholar] [CrossRef] [PubMed]
- Balemans, W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.M.; De Vera, M.A.; Heslip, T.R.; Casey, B. Evaluation of the anatomic burden of patients with hereditary multiple exostoses. Clin. Orthop. Relat. Res. 2007, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yamagata, T.; Mori, M.; Momoi, M.Y. Association of autism in two patients with hereditary multiple exostoses caused by novel deletion mutations of EXT1. J. Hum. Genet. 2002, 47, 262–265. [Google Scholar] [CrossRef]
- Narvid, J.; Gorno-Tempini, M.L.; Slavotinek, A.; DeArmond, S.J.; Cha, Y.H.; Miller, B.L.; Rankin, K. Of brain and bone: The unusual case of Dr. A. Neurocase 2009, 15, 190–205. [Google Scholar] [CrossRef]
- Marinus, J.; Moseley, L.; Birklein, F.; Baron, R.; Maihöfner, C.; Kingery, W.S.; Hilten, J.J. Van Syndrome–current state of the art. Lancet Neurol. 2011, 10, 637–648. [Google Scholar] [CrossRef]
- Lohnberg, J.A.; Altmaier, E.M. A review of psychosocial factors in complex regional pain syndrome. J. Clin. Psychol. Med. Settings 2013, 20, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Bazika-Gerasch, B.; Maier, C.; Kumowski, N.; Fiege, C.; Kaisler, M.; Vollert, J.; Dietrich, J.W. Compared to limb pain of other origin, ultrasonographic osteodensitometry reveals loss of bone density in complex regional pain syndrome. Pain 2019, 160, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Krämer, H.H.; Hofbauer, L.C.; Szalay, G.; Breimhorst, M.; Eberle, T.; Zieschang, K.; Rauner, M.; Schlereth, T.; Schreckenberger, M.; Birklein, F. Osteoprotegerin: A new biomarker for impaired bone metabolism in complex regional pain syndrome? Pain 2014, 155, 889–895. [Google Scholar] [CrossRef]
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I. Position statement: Definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640. [Google Scholar] [CrossRef]
- Lu, J.; Marmarou, A.; Choi, S.; Maas, A.; Murray, G.; Steyerberg, E.W. Mortality from traumatic brain injury. Acta Neurochir. Suppl. 2005, 281–285. [Google Scholar] [CrossRef]
- Blennow, K.; Hardy, J.; Zetterberg, H. The Neuropathology and Neurobiology of Traumatic Brain Injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef]
- Barshikar, S.; Bell, K.R. Sleep Disturbance After TBI. Curr. Neurol. Neurosci. Rep. 2017, 17. [Google Scholar] [CrossRef]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef]
- Strbian, D.; Durukan, A.; Pitkonen, M.; Marinkovic, I.; Tatlisumak, E.; Pedrono, E.; Abo-Ramadan, U.; Tatlisumak, T. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience 2008, 153, 175–181. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.J.; Sharkey, J.M.; Sun, M.; Kaukas, L.M.; Shultz, S.R.; Turner, R.J.; Leonard, A.V.; Brady, R.D.; Corrigan, F. Beyond the Brain: Peripheral Interactions after Traumatic Brain Injury. J. Neurotrauma 2020, 37, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Trentz, O.A.; Handschin, A.E.; Bestmann, L.; Hoerstrup, S.P.; Trentz, O.L.; Platz, A. Influence of brain injury on early posttraumatic bone metabolism. Crit. Care Med. 2005, 33, 399–406. [Google Scholar] [CrossRef]
- Banham-Hall, N.; Kothwal, K.; Pipkin, J.; Bentley, J.; Dickens, G.L. Prevalence of low bone mineral density in inpatients with traumatic brain injury receiving neurobehavioural rehabilitation: A postoperative, observational study. Physiother 2013, 99, 328–334. [Google Scholar] [CrossRef]
- Yu, H.; Watt, H.; Mohan, S. The negative impact of traumatic brain injury (TBI) on bone in a mouse model. Brain Inj. 2014, 28, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.D.; Shultz, S.R.; Sun, M.; Romano, T.; van der Poel, C.; Wright, D.K.; Wark, J.D.; O’Brien, T.J.; Grills, B.L.; McDonald, S.J. Experimental Traumatic Brain Injury Induces Bone Loss in Rats. J. Neurotrauma 2016, 33, 2154–2160. [Google Scholar] [CrossRef]
- Bajwa, N.M.; Kesavan, C.; Mohan, S. Long-term consequences of Traumatic brain injury in bone metabolism. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef]
- Singleton, Q.; Vaibhav, K.; Braun, M.; Patel, C.; Khayrullin, A.; Mendhe, B.; Lee, B.; Kolhe, R.; Kaiser, H.; Awad, M.; et al. Bone Marrow Derived Extracellular Vesicles Activate Osteoclast Differentiation in Traumatic Brain Injury Induced Bone Loss. Cells 2019, 8, 63. [Google Scholar] [CrossRef]
- Simonsen, L.L.; Sonne-Holm, S.; Krasheninnikoff, M.; Engberg, A.W. Symptomatic heterotopic ossification after very severe traumatic brain injury in 114 patients: Incidence and risk factors. Injury 2007, 38, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Almangour, W.; Schnitzler, A.; Salga, M.; Debaud, C.; Denormandie, P.; Genêt, F. Recurrence of heterotopic ossification after removal in patients with traumatic brain injury: A systematic review. Ann. Phys. Rehabil. Med. 2016, 59, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Cheng, W.X.; Hu, Y.P.; Chen, J.H.; Zheng, Z.T.; Zhang, P. Relationship between heterotopic ossification and traumatic brain injury: Why severe traumatic brain injury increases the risk of heterotopic ossification. J. Orthop. Transl. 2018, 12, 16–25. [Google Scholar] [CrossRef]
- Brady, R.D.; Shultz, S.R.; McDonald, S.J.; O’Brien, T.J. Neurological heterotopic ossification: Current understanding and future directions. Bone 2018, 109, 35–42. [Google Scholar] [CrossRef]
- Davis, E.L.; Davis, A.R.; Gugala, Z.; Olmsted-Davis, E.A. Is heterotopic ossification getting nervous?: The role of the peripheral nervous system in heterotopic ossification. Bone 2018, 109, 22–27. [Google Scholar] [CrossRef]
- Anthonissen, J.; Steffen, C.T.; Alessandri, B.; Baranowski, A.; Rommens, P.M.; Victor, J.; Hofmann, A. Traumatic brain injury enhances the formation of heterotopic ossification around the hip: An animal model study. Arch. Orthop. Trauma Surg. 2019. [Google Scholar] [CrossRef]
- Brady, R.D.; Zhao, M.Z.; Wong, K.R.; Casilla-Espinosa, P.M.; Yamakawa, G.R.; Wortman, R.C.; Sun, M.; Grills, B.L.; Mychasiuk, R.; O’Brien, T.J.; et al. A novel rat model of heterotopic ossification after polytrauma with traumatic brain injury. Bone 2020, 133. [Google Scholar] [CrossRef]
- Newman, R.J.; Stone, M.H.; Mukherjee, S.K. Accelerated fracture union in association with severe head injury. Injury 1987, 18, 241–246. [Google Scholar] [CrossRef]
- Perkins, R.; Skirving, A.P. Callus formation and the rate of healing of femoral fractures in patients with head injuries. J. Bone Jt. Surg. Ser. B 1987, 69, 521–524. [Google Scholar] [CrossRef]
- Spencer, R.F. The effect of head injury on fracture healing. A quantitative assessment. J. Bone Jt. Surg. Ser. B 1987, 69, 525–528. [Google Scholar] [CrossRef]
- Giannoudis, P.V.; Mushtaq, S.; Harwood, P.; Kambhampati, S.; Dimoutsos, M.; Stavrou, Z.; Pape, H.C. Accelerated bone healing and excessive callus formation in patients with femoral fracture and head injury. Injury 2006, 37. [Google Scholar] [CrossRef] [PubMed]
- Cadosch, D.; Gautschi, O.P.; Thyer, M.; Song, S.; Skirving, A.P.; Filgueira, L.; Zellweger, R. Humoral factors enhance fracture-healing and callus formation in patients with traumatic brain injury. J. Bone Jt. Surg. Ser. A 2009, 91, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, Z.; Li, Z.; Yang, R. Does traumatic brain injury result in accelerated mandibular fracture healing? J. Oral Maxillofac. Surg. 2012, 70, 2135–2142. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.Y.; Wang, T.C.; Tsai, Y.H.; Huang, K.C. The effects of an injury to the brain on bone healing and callus formation in young adults with fractures of the femoral shaft. J. Bone Jt. Surg. Ser. B 2012, 94 B, 227–230. [Google Scholar] [CrossRef]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef]
- Schell, H.; Duda, G.N.; Peters, A.; Tsitsilonis, S.; Johnson, K.A.; Schmidt-Bleek, K. The haematoma and its role in bone healing. J. Exp. Orthop. 2017, 4. [Google Scholar] [CrossRef]
- Hofman, M.; Koopmans, G.; Kobbe, P.; Poeze, M.; Andruszkow, H.; Brink, P.R.G.; Pape, H.C. Improved fracture healing in patients with concomitant traumatic brain injury: Proven or not? Mediators Inflamm. 2015, 2015. [Google Scholar] [CrossRef]
- Toffoli, A.M.; Gautschi, O.P.; Frey, S.P.; Filgueira, L.; Zellweger, R. From brain to bone: Evidence for the release of osteogenic humoral factors after traumatic brain injury. Brain Inj. 2008, 22, 511–518. [Google Scholar] [CrossRef]
- Bidner, S.M.; Rubins, I.M.; Desjardins, J.V.; Zukor, D.J.; Goltzman, D. Evidence for a humoral mechanism for enhanced osteogenesis after head injury. J. Bone Jt. Surg. Ser. A 1990, 72, 1144–1149. [Google Scholar] [CrossRef]
- Gautschi, O.P.; Toffoli, A.M.; Joesbury, K.A.; Skirving, A.P.; Filgueira, L.; Zellweger, R. Osteoinductive effect of cerebrospinal fluid from brain-injured patients. J. Neurotrauma 2007, 24, 154–162. [Google Scholar] [CrossRef]
- Tsitsilonis, S.; Seemann, R.; Misch, M.; Wichlas, F.; Haas, N.P.; Schmidt-Bleek, K.; Kleber, C.; Schaser, K.D. The effect of traumatic brain injury on bone healing: An experimental study in a novel in vivo animal model. Injury 2015, 46, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Locher, R.J.; Lünnemann, T.; Garbe, A.; Schaser, K.D.; Schmidt-Bleek, K.; Duda, G.; Tsitsilonis, S. Traumatic brain injury and bone healing: Radiographic and biomechanical analyses of bone formation and stability in a combined murine trauma model. J. Musculoskelet. Neuronal Interact. 2015, 15, 309–315. [Google Scholar] [PubMed]
- Morley, J.; Marsh, S.; Drakoulakis, E.; Pape, H.C.; Giannoudis, P.V. Does traumatic brain injury result in accelerated fracture healing? Injury 2005, 36, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Morioka, K.; Marmor, Y.; Sacramento, J.A.; Lin, A.; Shao, T.; Miclau, K.R.; Clark, D.R.; Beattie, M.S.; Marcucio, R.S.; Miclau, T.; et al. Differential fracture response to traumatic brain injury suggests dominance of neuroinflammatory response in polytrauma. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Jiang, S.D.; Dai, L.Y.; Jiang, L.S. Osteoporosis after spinal cord injury. Osteoporos. Int. 2006, 17, 180–192. [Google Scholar] [CrossRef]
- Dauty, M.; Perrouin Verbe, B.; Maugars, Y.; Dubois, C.; Mathe, J.F. Supralesional and sublesional bone mineral density in spinal cord-injured patients. Bone 2000, 27, 305–309. [Google Scholar] [CrossRef]
- BIERING-SØRENSEN, F.; BOHR, H.H.; SCHAADT, O.P. Longitudinal study of bone mineral content in the lumbar spine, the forearm and the lower extremities after spinal cord injury. Eur. J. Clin. Investig. 1990, 20, 330–335. [Google Scholar] [CrossRef]
- Frey-Rindova, P.; De Bruin, E.D.; Stüssi, E.; Dambacher, M.A.; Dietz, V. Bone mineral density in upper and lower extremities during 12 months after spinal cord injury measured by peripheral quantitative computed tomography. Spinal Cord 2000, 38, 26–32. [Google Scholar] [CrossRef]
- de Bruin, E.D.; Dietz, V.; Dambacher, M.A.; Stüssi, E. Longitudinal changes in bone in men with spinal cord injury. Clin. Rehabil. 2000, 14, 145–152. [Google Scholar] [CrossRef]
- Sullivan, M.P.; Torres, S.J.; Mehta, S.; Ahn, J. Heterotopic ossification after central nervous system trauma. Bone Joint Res. 2013, 2, 51–57. [Google Scholar] [CrossRef]
- Albrecht, J.S.; Al Kibria, G.; Gruber-Baldini, A.; Magaziner, J. Risk of Mortality in Individuals with Hip Fracture and Traumatic Brain Injury. J. Am. Geriatr. Soc. 2019, 67, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Hensler, T.; Sauerland, S.; Bouillon, B.; Raum, M.; Rixen, D.; Helling, H.J.; Andermahr, J.; Neugebauer, E.A.M. Association between injury pattern of patients with multiple injuries and circulating levels of soluble tumor necrosis factor receptors, interleukin-6 and interleukin-10, and polymorphonuclear neutrophil elastase. J. Trauma 2002, 52, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain. Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.G.; Diamond, M.L.; Boles, J.A.; Berger, R.P.; Tisherman, S.A.; Kochanek, P.M.; Wagner, A.K. Acute CSF interleukin-6 trajectories after TBI: Associations with neuroinflammation, polytrauma, and outcome. Brain. Behav. Immun. 2015, 45, 253–262. [Google Scholar] [CrossRef]
- Shultz, S.R.; Sun, M.; Wright, D.K.; Brady, R.D.; Liu, S.; Beynon, S.; Schmidt, S.F.; Kaye, A.H.; Hamilton, J.A.; O’Brien, T.J.; et al. Tibial fracture exacerbates traumatic brain injury outcomes and neuroinflammation in a novel mouse model of multitrauma. J. Cereb. Blood Flow Metab. 2015, 35, 1339–1347. [Google Scholar] [CrossRef]
- Yang, L.; Guo, Y.; Wen, D.; Yang, L.; Chen, Y.; Zhang, G.; Fan, Z. Bone Fracture Enhances Trauma Brain Injury. Scand. J. Immunol. 2016, 83, 26–32. [Google Scholar] [CrossRef]
- Elabd, C.; Cousin, W.; Upadhyayula, P.; Chen, R.Y.; Chooljian, M.S.; Li, J.; Kung, S.; Jiang, K.P.; Conboy, I.M. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Sanchez-Barcelo, E.; Mediavilla, M.D.; Reiter, R.J. Significance of High Levels of Endogenous Melatonin in Mammalian Cerebrospinal Fluid and in the Central Nervous System. Curr. Neuropharmacol. 2010, 8, 162–167. [Google Scholar] [CrossRef]
- Li, T.; Jiang, S.; Lu, C.; Yang, W.; Yang, Z.; Hu, W.; Xin, Z.; Yang, Y. Melatonin: Another avenue for treating osteoporosis? J. Pineal Res. 2019, 66, 1–12. [Google Scholar] [CrossRef]
- Schweiger, U.; Deuschle, M.; Körner, A.; Lammers, C.H.; Schmider, J.; Gotthardt, U.; Holsboer, F.; Heuser, I. Low lumbar bone mineral density in patients with major depression. Am. J. Psychiatry 1994, 151, 1691–1693. [Google Scholar] [CrossRef]
- Ramnemark, A.; Nilsson, M.; Borssén, B.; Gustafson, Y. Stroke, a major and increasing risk factor for femoral neck fracture. Stroke 2000, 31, 1572–1577. [Google Scholar] [CrossRef]
- Pouwels, S.; Lalmohamed, A.; Leufkens, B.; De Boer, A.; Cooper, C.; Van Staa, T.; De Vries, F. Risk of hip/femur fracture after stroke: A population-based case-control study. Stroke 2009, 40, 3281–3285. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wan, W.; Xia, S.; Kalionis, B.; Li, Y. Dysfunctional Wnt/β-catenin signaling contributes to blood-brain barrier breakdown in Alzheimer’s disease. Neurochem. Int. 2014, 75, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Genêt, F.; Jourdan, C.; Schnitzler, A.; Lautridou, C.; Guillemot, D.; Judet, T.; Poiraudeau, S.; Denormandie, P. Troublesome Heterotopic ossification after central nervous system damage: A survey of 570 surgeries. PLoS ONE 2011, 6, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Perumal, R.; SP, S.; Shankar, V.; Gaddam, S.R.; Jayaramaraju, D.; Rajasekaran, S. Does accelerated bone healing associated with traumatic brain injury (TBI) occur in open fractures. Int. J. Orthop. Sci. 2018, 4, 677–681. [Google Scholar] [CrossRef][Green Version]
- Cipriano, C.A.; Pill, S.G.; Keenan, M.A. Heterotopic ossification following traumatic brain injury and spinal cord injury. J. Am. Acad. Orthop. Surg. 2009, 17, 689–697. [Google Scholar] [CrossRef]
- van Bezooijen, R.L.; Papapoulos, S.E.; Hamdy, N.A.; ten Dijke, P.; Löwik, C.W. Control of bone formation by osteocytes? lessons from the rare skeletal disorders sclerosteosis and van Buchem disease. BoneKEy-Osteovision 2005, 2, 33–38. [Google Scholar] [CrossRef]
- Tsai, C.H.; Chuang, C.S.; Hung, C.H.; Lin, C.L.; Sung, F.C.; Tang, C.H.; Hsu, H.C.; Chung, C.J. Fracture as an independent risk factor of dementia: A nationwide population-based cohort study. Medicine 2014, 93, 1–7. [Google Scholar] [CrossRef]
- Jodoin, M.; Rouleau, D.M.; Charlebois-Plante, C.; Benoit, B.; Leduc, S.; Laflamme, G.Y.; Gosselin, N.; Larson-Dupuis, C.; De Beaumont, L. Incidence rate of mild traumatic brain injury among patients who have suffered from an isolated limb fracture: Upper limb fracture patients are more at risk. Injury 2016, 47, 1835–1840. [Google Scholar] [CrossRef]
- Zaidi, M.; Yuen, T.; Sun, L.; Rosen, C.J. Regulation of skeletal homeostasis. Endocr. Rev. 2018, 39, 701–718. [Google Scholar] [CrossRef]
- Zaidi, M.; Lizneva, D.; Kim, S.M.; Sun, L.; Iqbal, J.; New, M.I.; Rosen, C.J.; Yuen, T. FSH, Bone Mass, Body Fat, and Biological Aging. Endocrinology 2018, 159, 3503–3514. [Google Scholar] [CrossRef] [PubMed]
- Lizneva, D.; Rahimova, A.; Kim, S.M.; Atabiekov, I.; Javaid, S.; Alamoush, B.; Taneja, C.; Khan, A.; Sun, L.; Azziz, R.; et al. FSH beyond fertility. Front. Endocrinol. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Coss, D. Commentary on the Recent FSH Collection: Known Knowns and Known Unknowns. Endocrinology 2020, 161, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Seibel, M.J.; Dunstan, C.R.; Zhou, H.; Allan, C.M.; Handelsman, D.J. Sex Steroids, Not FSH, Influence Bone Mass. Cell 2006, 127, 1079. [Google Scholar] [CrossRef] [PubMed]
- Prior, J.C. FSH and bone—Important physiology or not? Trends Mol. Med. 2007, 13, 1–3. [Google Scholar] [CrossRef]
- García-Martín, A.; Reyes-García, R.; García-Castro, J.M.; Rozas-Moreno, P.; Escobar-Jiménez, F.; Muñoz-Torres, M. Role of serum FSH measurement on bone resorption in postmenopausal women. Endocrine 2012, 41, 302–308. [Google Scholar] [CrossRef]
- Sun, L.; Peng, Y.; Sharrow, A.C.; Iqbal, J.; Zhang, Z.; Papachristou, D.J.; Zaidi, S.; Zhu, L.L.; Yaroslavskiy, B.B.; Zhou, H.; et al. FSH Directly Regulates Bone Mass. Cell 2006, 125, 247–260. [Google Scholar] [CrossRef]
- Iqbal, J.; Blair, H.C.; Zallone, A.; Sun, L.; Zaidi, M. Further evidence that FSH causes bone loss independently of low estrogen. Endocrine 2012, 41, 171–175. [Google Scholar] [CrossRef]
- Zaidi, S.; Zhu, L.L.; Mali, R.; Iqbal, J.; Yang, G.; Zaidi, M.; Sun, L. Regulation of FSH receptor promoter activation in the osteoclast. Biochem. Biophys. Res. Commun. 2007, 361, 910–915. [Google Scholar] [CrossRef]
- Ji, Y.; Liu, P.; Yuen, T.; Haider, S.; He, J.; Romero, R.; Chen, H.; Bloch, M.; Kim, S.M.; Lizneva, D.; et al. Epitope-specific monoclonal antibodies to FSHβ increase bone mass. Proc. Natl. Acad. Sci. USA 2018, 115, 2192–2197. [Google Scholar] [CrossRef]
- Abe, E.; Marians, R.C.; Yu, W.; Wu, X.B.; Ando, T.; Li, Y.; Iqbal, J.; Eldeiry, L.; Rajendren, G.; Blair, H.C.; et al. TSH is a negative regulator of skeletal remodeling. Cell 2003, 115, 151–162. [Google Scholar] [CrossRef]
- Ma, R.; Morshed, S.; Latif, R.; Zaidi, M.; Davies, T.F. The influence of thyroid-stimulating hormone and thyroid-stimulating hormone receptor antibodies on osteoclastogenesis. Thyroid 2011, 21, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Hase, H.; Ando, T.; Eldeiry, L.; Brebene, A.; Peng, Y.; Liu, L.; Amano, H.; Davies, T.F.; Sun, L.; Zaidi, M.; et al. TNFα mediates the skeletal effects of thyroid-stimulating hormone. Proc. Natl. Acad. Sci. USA 2006, 103, 12849–12854. [Google Scholar] [CrossRef] [PubMed]
- Sampath, T.K.; Simic, P.; Sendak, R.; Draca, N.; Bowe, A.E.; O’Brien, S.; Schiavi, S.C.; McPherson, J.M.; Vukicevic, S. Thyroid-stimulating hormone restores bone volume, microarchitecture, and strength in aged ovariectomized rats. J. Bone Miner. Res. 2007, 22, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Baliram, R.; Latif, R.; Berkowitz, J.; Frid, S.; Colaianni, G.; Sun, L.; Zaidi, M.; Davies, T.F. Thyroid-stimulating hormone induces a Wnt-dependent, feed-forward loop for osteoblastogenesis in embryonic stem cell cultures. Proc. Natl. Acad. Sci. USA 2011, 108, 16277–16282. [Google Scholar] [CrossRef]
- Bernard, V.; Young, J.; Binart, N. Prolactin—A pleiotropic factor in health and disease. Nat. Rev. Endocrinol. 2019, 15. [Google Scholar] [CrossRef]
- Clément-Lacroix, P.; Ormandy, C.; Lepescheux, L.; Ammann, P.; Damotte, D.; Goffin, V.; Bouchard, B.; Amling, M.; Gaillard-Kelly, M.; Binart, N.; et al. Osteoblasts are a new target for prolactin: Analysis of bone formation in prolactin receptor knockout mice. Endocrinology 1999, 140, 96–105. [Google Scholar] [CrossRef]
- Chiloiro, S.; Giampietro, A.; Bianchi, A.; De Marinis, L. Prolactinoma and Bone. Curr. Opin. Endocr. Metab. Res. 2018, 3, 21–24. [Google Scholar] [CrossRef]
- Coss, D.; Yang, L.; Kuo, C.B.; Xu, X.; Luben, R.A.; Walker, A.M. Effects of prolactin on osteoblast alkaline phosphatase and bone formation in the developing rat. Am. J. Physiol. Endocrinol. Metab. 2000, 279, 1216–1225. [Google Scholar] [CrossRef]
- Seriwatanachai, D.; Thongchote, K.; Charoenphandhu, N.; Pandaranandaka, J.; Tudpor, K.; Teerapornpuntakit, J.; Suthiphongchai, T.; Krishnamra, N. Prolactin directly enhances bone turnover by raising osteoblast-expressed receptor activator of nuclear factor κB ligand/osteoprotegerin ratio. Bone 2008, 42, 535–546. [Google Scholar] [CrossRef]
- Seriwatanachai, D.; Charoenphandhu, N.; Suthiphongchai, T.; Krishnamra, N. Prolactin decreases the expression ratio of receptor activator of nuclear factor κB ligand/osteoprotegerin in human fetal osteoblast cells. Cell Biol. Int. 2008, 32, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Seriwatanachai, D.; Krishnamra, N.; Van Leeuwen, J.P.T.M. Evidence for direct effects of prolactin on human osteoblasts: Inhibition of cell growth and mineralization. J. Cell. Biochem. 2009, 107, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Krishnamra, N.; Seemoung, J. Effects of acute and long-term administration of prolactin on bone 45Ca uptake, calcium deposit, and calcium resorption in weaned, young, and mature rats. Can. J. Physiol. Pharmacol. 1996, 74, 1157–1165. [Google Scholar] [CrossRef]
- Zaidi, M.; Sun, L.; Liu, P.; Davies, T.F.; New, M.; Zallone, A.; Yuen, T. Pituitary-bone connection in skeletal regulation. Horm. Mol. Biol. Clin. Investig. 2016, 28, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Isales, C.M.; Zaidi, M.; Blair, H.C. ACTH is a novel regulator of bone mass. Ann. N. Y. Acad. Sci. 2010, 1192, 110–116. [Google Scholar] [CrossRef]
- Weinstein, R.S. Glucocorticoid-induced osteonecrosis. Endocrine 2012, 41, 183–190. [Google Scholar] [CrossRef]
- Pállinger, É.; Csaba, G. A hormone map of human immune cells showing the presence of adrenocorticotropic hormone, triiodothyronine and endorphin in immunophenotyped white blood cells. Immunology 2008, 123, 584–589. [Google Scholar] [CrossRef]
- Ohlsson, C.; Bengtsson, B.-Å.; Isaksson, O.G.P.; Andreassen, T.T.; Slootweg, M.C. Growth Hormone and Bone. Endocr. Rev. 1998, 19, 55–79. [Google Scholar]
- Giustina, A.; Mazziotti, G.; Canalis, E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr. Rev. 2008, 29, 535–559. [Google Scholar] [CrossRef]
- Tamma, R.; Sun, L.; Cuscito, C.; Lu, P.; Corcelli, M.; Li, J.; Colaianni, G.; Moonga, S.S.; Di Benedetto, A.; Grano, M.; et al. Regulation of bone remodeling by vasopressin explains the bone loss in hyponatremia. Proc. Natl. Acad. Sci. USA 2013, 110, 18644–18649. [Google Scholar] [CrossRef]
- Mavani, G.P.; DeVita, M.V.; Michelis, M.F. A review of the nonpressor and nonantidiuretic actions of the hormone vasopressin. Front. Med. 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, S.; Wray, S. Oxytocin: Its mechanism of action and receptor signalling in the myometrium. J. Neuroendocrinol. 2014, 26, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Tamma, R.; Yuen, T.; Colaianni, G.; Ji, Y.; Cuscito, C.; Bailey, J.; Dhawan, S.; Lu, P.; Calvano, C.D.; et al. Functions of vasopressin and oxytocin in bone mass regulation. Proc. Natl. Acad. Sci. USA 2016, 113, 164–169. [Google Scholar] [CrossRef]
- Colaianni, G.; Sun, L.; Zaidi, M.; Zallone, A. Oxytocin and bone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R970–R977. [Google Scholar] [CrossRef]
- Tamma, R.; Colaianni, G.; Zhu, L.L.; DiBenedetto, A.; Greco, G.; Montemurro, G.; Patano, N.; Strippoli, M.; Vergari, R.; Mancini, L.; et al. Oxytocin is an anabolic bone hormone. Proc. Natl. Acad. Sci. USA 2009, 106, 7149–7154. [Google Scholar] [CrossRef] [PubMed]
- Copland, J.A.; Ives, K.L.; Simmons, D.J.; Soloff, M.S. Functional oxytocin receptors discovered in human osteoblasts. Endocrinology 1999, 140, 4371–4374. [Google Scholar] [CrossRef] [PubMed]
- Colucci, S.; Colaianni, G.; Mori, G.; Grano, M.; Zallone, A. Human osteoclasts express oxytocin receptor. Biochem. Biophys. Res. Commun. 2002, 297, 442–445. [Google Scholar] [CrossRef]
- Colaianni, G.; Sun, L.; Di Benedetto, A.; Tamma, R.; Zhu, L.L.; Cao, J.; Grano, M.; Yuen, T.; Colucci, S.; Cuscito, C.; et al. Bone marrow oxytocin mediates the anabolic action of estrogen on the skeleton. J. Biol. Chem. 2012, 287, 29159–29167. [Google Scholar] [CrossRef]
- Leston, J.; Harthé, C.; Brun, J.; Mottolese, C.; Mertens, P.; Sindou, M.; Claustrat, B. Melatonin is released in the third ventricle in humans. A study in movement disorders. Neurosci. Lett. 2010, 469, 294–297. [Google Scholar] [CrossRef]
- Slominski, R.M.; Reiter, R.J.; Schlabritz-Loutsevitch, N.; Ostrom, R.S.; Slominski, A.T. Melatonin membrane receptors in peripheral tissues: Distribution and functions. Mol. Cell. Endocrinol. 2012, 351, 152–166. [Google Scholar] [CrossRef]
- Roth, J.A.; Kim, B.G.; Lin, W.L.; Cho, M. Il Melatonin promotes osteoblast differentiation and bone formation. J. Biol. Chem. 1999, 274, 22041–22047. [Google Scholar] [CrossRef]
- Nakade, O.; Koyama, H.; Ariji, H.; Yajima, A.; Kaku, T. Melatonin stimulates proliferation and type I collagen synthesis in human bone cells in vitro. J. Pineal Res. 1999, 27, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Su, P.; Xu, C.; Chen, C.; Liang, A.; Du, K.; Peng, Y.; Huang, D. Melatonin inhibits adipogenesis and enhances osteogenesis of human mesenchymal stem cells by suppressing PPARγ expression and enhancing Runx2 expression. J. Pineal Res. 2010, 49, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Amstrup, A.K.; Sikjaer, T.; Heickendorff, L.; Mosekilde, L.; Rejnmark, L. Melatonin improves bone mineral density at the femoral neck in postmenopausal women with osteopenia: A randomized controlled trial. J. Pineal Res. 2015, 59, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Barcelo, E.J.; Mediavilla, M.D.; Tan, D.X.; Reiter, R.J. Clinical Uses of Melatonin: Evaluation of Human Trials. Curr. Med. Chem. 2010, 17, 2070–2095. [Google Scholar] [CrossRef]
- Koch, B.C.P.; Nagtegaal, J.E.; Hagen, E.C.; Van Der Westerlaken, M.M.L.; Boringa, J.B.S.; Kerkhof, G.A.; Ter Wee, P.M. The effects of melatonin on sleep-wake rhythm of daytime haemodialysis patients: A randomized, placebo-controlled, cross-over study (EMSCAP study). Br. J. Clin. Pharmacol. 2009, 67, 68–75. [Google Scholar] [CrossRef]
- Driessler, F.; Baldock, P.A. Hypothalamic regulation of bone. J. Mol. Endocrinol. 2010, 45, 175–181. [Google Scholar] [CrossRef]
- Gehlert, D.R. Role of hypothalamic neuropeptide Y in feeding and obesity. Neuropeptides 1999, 33, 329–338. [Google Scholar] [CrossRef]
- Teixeira, L.; Sousa, D.M.; Nunes, A.F.; Sousa, M.M.; Herzog, H.; Lamghari, M. NPY revealed as a critical modulator of osteoblast function in vitro: New insights into the role of Y1 and Y2 receptors. J. Cell. Biochem. 2009, 107, 908–916. [Google Scholar] [CrossRef]
- Baldock, P.A.; Allison, S.J.; Lundberg, P.; Lee, N.J.; Slack, K.; Lin, E.J.D.; Enriquez, R.F.; McDonald, M.M.; Zhang, L.; During, M.J.; et al. Novel role of Y1 receptors in the coordinated regulation of bone and energy homeostasis. J. Biol. Chem. 2007, 282, 19092–19102. [Google Scholar] [CrossRef]
- Elefteriou, F. Impact of the autonomic nervous system on the skeleton. Physiol. Rev. 2018, 98, 1083–1112. [Google Scholar] [CrossRef]
- Horsnell, H.; Baldock, P.A. Osteoblastic Actions of the Neuropeptide Y System to Regulate Bone and Energy Homeostasis. Curr. Osteoporos. Rep. 2016, 14, 26–31. [Google Scholar] [CrossRef]
- Zhang, W.; Cline, M.A.; Gilbert, E.R. Hypothalamus-adipose tissue crosstalk: Neuropeptide y and the regulation of energy metabolism. Nutr. Metab. 2014, 11, 1–12. [Google Scholar] [CrossRef]
- Baldock, P.; Lin, S.; Zhang, L.; Karl, T.; Shi, Y.; Driessler, F.; Zengin, A.; Hörmer, B.; Lee, N.; Wong, I.; et al. Neuropeptide Y Attenuates Stress-Induced Bone Loss Through Suppression of Noradrenaline Circuits. J. Bone Miner. Res. 2014, 29, 2238–2249. [Google Scholar] [CrossRef]
- Ilnytska, O.; Argyropoulos, G. The role of the Agouti-related protein in energy balance regulation. Cell. Mol. Life Sci. 2008, 65, 2721–2731. [Google Scholar] [CrossRef]
- Deng, J.; Yuan, F.; Guo, Y.; Xiao, Y.; Niu, Y.; Deng, Y.; Han, X.; Guan, Y.; Chen, S.; Guo, F. Deletion of ATF4 in AgRP neurons promotes fat loss mainly via increasing energy expenditure. Diabetes 2017, 66, 640–650. [Google Scholar] [CrossRef]
- Kim, J.G.; Sun, B.H.; Dietrich, M.O.; Koch, M.; Yao, G.Q.; Diano, S.; Insogna, K.; Horvath, T.L. AgRP Neurons Regulate Bone Mass. Cell Rep. 2015, 13, 8–14. [Google Scholar] [CrossRef]
- Swart, I.; Jahng, J.W.; Overton, J.M.; Houpt, T.A. Hypothalamic NPY, AGRP, and POMC mRNA responses to leptin and refeeding in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, 1020–1026. [Google Scholar] [CrossRef]
- Kuperman, Y.; Weiss, M.; Dine, J.; Staikin, K.; Golani, O.; Ramot, A.; Nahum, T.; Kühne, C.; Shemesh, Y.; Wurst, W.; et al. CRFR1 in AgRP Neurons Modulates Sympathetic Nervous System Activity to Adapt to Cold Stress and Fasting. Cell Metab. 2016, 23, 1185–1199. [Google Scholar] [CrossRef]
- Shi, Z.; Madden, C.J.; Brooks, V.L. Arcuate neuropeptide y inhibits sympathetic nerve activity via multiple neuropathways. J. Clin. Investig. 2017, 127, 2868–2880. [Google Scholar] [CrossRef]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef]
- Maletínská, L.; Maixnerová, J.; Matyšková, R.; Haugvicová, R.; Pirník, Z.; Kiss, A.; Železná, B. Synergistic effect of CART (cocaine- and amphetamine-regulated transcript) peptide and cholecystokinin on food intake regulation in lean mice. BMC Neurosci. 2008, 9. [Google Scholar] [CrossRef]
- Elefteriou, F.; Ahn, J.D.; Takeda, S.; Starbuck, M.; Yang, X.; Liu, X.; Kondo, H.; Richards, W.G.; Bannon, T.W.; Noda, M.; et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 2005, 434, 514–520. [Google Scholar] [CrossRef]
- Singh, M.K.; Elefteriou, F.; Karsenty, G. Cocaine and amphetamine-regulated transcript may regulate bone remodeling as a circulating molecule. Endocrinology 2008, 149, 3933–3941. [Google Scholar] [CrossRef]
- Yamamoto, H.; Yamane, T.; Iguchi, K.; Tanaka, K.; Iddamalgoda, A.; Unno, K.; Hoshino, M.; Takeda, A. Melanin production through novel processing of proopiomelanocortin in the extracellular compartment of the auricular skin of C57BL/6 mice after UV-irradiation. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Böhm, M.; Grässel, S. Role of proopiomelanocortin-derived peptides and their receptors in the osteoarticular system: From basic to translational research. Endocr. Rev. 2012, 33, 623–651. [Google Scholar] [CrossRef]
- Cornish, J.; Callon, K.E.; Mountjoy, K.G.; Bava, U.; Lin, J.M.; Myers, D.E.; Naot, D.; Reid, I.R. A-Melanocyte-Stimulating Hormone Is a Novel Regulator of Bone. Am. J. Physiol. Endocrinol. Metab. 2003, 284. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Yeo, G.S.H.; Keogh, J.M.; Aminian, S.; Jebb, S.A.; Butler, G.; Cheetham, T.; O’Rahilly, S. Dominant and recessive inheritance of morbid obesity associate with melanocortin 4 receptor deficiency. J. Clin. Investig. 2000, 106, 271–279. [Google Scholar] [CrossRef]
- Garg, G.; Kumar, J.; McGuigan, F.E.; Ridderstråle, M.; Gerdhem, P.; Luthman, H.; Åkesson, K. Variation in the MC4R gene is associated with bone phenotypes in elderly Swedish women. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Grässel, S.; Muschter, D. Do neuroendocrine peptides and their receptors qualify as novel therapeutic targets in osteoarthritis? Int. J. Mol. Sci. 2018, 19, 367. [Google Scholar] [CrossRef]
- Sudo, Y.; Ezura, Y.; Kajita, M.; Yoshida, H.; Suzuki, T.; Hosoi, T.; Inoue, S.; Shiraki, M.; Ito, H.; Emi, M. Association of single nucleotide polymorphisms in the promoter region of the pro-opiomelanocortin gene (POMC) with low bone mineral density in adult women. J. Hum. Genet. 2005, 50, 235–240. [Google Scholar] [CrossRef][Green Version]
- Farman, H.H.; Windahl, S.H.; Westberg, L.; Isaksson, H.; Egecioglu, E.; Schele, E.; Ryberg, H.; Jansson, J.O.; Tuukkanen, J.; Koskela, A.; et al. Female mice lacking estrogen receptor-α in hypothalamic proopiomelanocortin (POMC) neurons display enhanced estrogenic response on cortical bone mass. Endocrinology 2016, 157, 3242–3252. [Google Scholar] [CrossRef]
- Fujii, R.; Hosoya, M.; Fukusumi, S.; Kawamata, Y.; Habata, Y.; Hinuma, S.; Onda, H.; Nishimura, O.; Fujino, M. Identification of neuromedin U as the cognate ligand of the orphan G protein-coupled receptor FM-3. J. Biol. Chem. 2000, 275, 21068–21074. [Google Scholar] [CrossRef] [PubMed]
- Brighton, P.J.; Szekeres, P.G.; Willars, G.B. Neuromedin U and its receptors: Structure, function, and physiological roles. Pharmacol. Rev. 2004, 56, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Hanada, R.; Kimura, A.; Abe, T.; Matsumoto, T.; Iwasaki, M.; Inose, H.; Ida, T.; Mieda, M.; Takeuchi, Y.; et al. Central control of bone remodeling by neuromedin U. Nat. Med. 2007, 13, 1234–1240. [Google Scholar] [CrossRef]
- Hanada, T.; Date, Y.; Shimbara, T.; Sakihara, S.; Murakami, N.; Hayashi, Y.; Kanai, Y.; Suda, T.; Kangawa, K.; Nakazato, M. Central actions of neuromedin U via corticotropin-releasing hormone. Biochem. Biophys. Res. Commun. 2003, 311, 954–958. [Google Scholar] [CrossRef]
- Wren, A.M.; Small, C.J.; Abbott, C.R.; Jethwa, P.H.; Kennedy, A.R.; Murphy, K.G.; Stanley, S.A.; Zollner, A.N.; Ghatei, M.A.; Bloom, S.R. Hypothalamic actions of neuromedin U. Endocrinology 2002, 143, 4227–4234. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T.; Nagai, H.; Asakawa, T.; Suzuki, N.; Fujita, H.; Matsumiya, K.; Nishizawa, N.; Kanematsu-Yamaki, Y.; Dote, K.; Sakamoto, J.I.; et al. Effects of peripheral administration of a Neuromedin U receptor 2-selective agonist on food intake and body weight in obese mice. Int. J. Obes. 2017, 41, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Sampson, C.M.; Kasper, J.M.; Felsing, D.E.; Raval, S.R.; Ye, N.; Wang, P.; Patrikeev, I.; Rytting, E.; Zhou, J.; Allen, J.A.; et al. Small-Molecule Neuromedin U Receptor 2 Agonists Suppress Food Intake and Decrease Visceral Fat in Animal Models. Pharmacol. Res. Perspect. 2018, 6, 1–11. [Google Scholar] [CrossRef]
- Jones, K.B.; Mollano, A.V.; Morcuende, J.A.; Cooper, R.R.; Saltzman, C.L. Bone and brain: A review of neural, hormonal, and musculoskeletal connections. Iowa Orthop. J. 2004, 24, 123–132. [Google Scholar]
- Hohmann, E.; Elde, R.; Rysavy, J.; Einzig, S.; Gebhard, R. Innervation of periosteum and bone by sympathetic vasoactive intestinal peptide-containing nerve fibers. Science 1986, 232, 868–871. [Google Scholar] [CrossRef]
- Bjurholm, A.; Kreicbergs, A.; Terenius, L.; Goldstein, M.; Schultzberg, M. Neuropeptide Y-, tyrosine hydroxylase- and vasoactive intestinal polypeptide-immunoreactive nerves in bone and surrounding tissues. J. Auton. Nerv. Syst. 1988, 25, 119–125. [Google Scholar] [CrossRef]
- Hill, E.L.; Elde, R. Distribution of CGRP-, VIP-, DβH-, SP-, and NPY-immunoreactive nerves in the periosteum of the rat. Cell Tissue Res. 1991, 264, 469–480. [Google Scholar] [CrossRef]
- Togari, A.; Arai, M.; Mizutani, S.; Mizutani, S.; Koshihara, Y.; Nagatsu, T. Expression of mRNAs for neuropeptide receptors and β-adrenergic receptors in human osteoblasts and human osteogenic sarcoma cells. Neurosci. Lett. 1997, 233, 125–128. [Google Scholar] [CrossRef]
- Ransjö, M.; Lie, A.; Mukohyama, H.; Lundberg, P.; Lerner, U.H. Microisolated mouse osteoclasts express VIP-1 and PACAP receptors. Biochem. Biophys. Res. Commun. 2000, 274, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Mukohyama, H.; Ransjö, M.; Taniguchi, H.; Ohyama, T.; Lerner, U.H. The inhibitory effects of vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide on osteoclast formation are associated with upregulation of osteoprotegerin and downregulation of RANKL and RANK. Biochem. Biophys. Res. Commun. 2000, 271, 158–163. [Google Scholar] [CrossRef]
- Juarranz, Y.; Abad, C.; Martinez, C.; Arranz, A.; Gutierrez-Cañas, I.; Rosignoli, F.; Gomariz, R.P.; Leceta, J. Protective effect of vasoactive intestinal peptide on bone destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R1034. [Google Scholar] [CrossRef]
- de Vernejoul, M.-C.; Collet, C.; Chabbi-Achengli, Y. Serotonin: Good or bad for bone. Bonekey Rep. 2012, 1. [Google Scholar] [CrossRef]
- Yadav, V.K.; Ryu, J.H.; Suda, N.; Tanaka, K.F.; Gingrich, J.A.; Schütz, G.; Glorieux, F.H.; Chiang, C.Y.; Zajac, J.D.; Insogna, K.L.; et al. Lrp5 Controls Bone Formation by Inhibiting Serotonin Synthesis in the Duodenum. Cell 2008, 135, 825–837. [Google Scholar] [CrossRef]
- Park, K.R.; Kim, E.C.; Hong, J.T.; Yun, H.M. Dysregulation of 5-hydroxytryptamine 6 receptor accelerates maturation of bone-resorbing osteoclasts and induces bone loss. Theranostics 2018, 8, 3087–3098. [Google Scholar] [CrossRef]
- Cui, Y.; Niziolek, P.J.; MacDonald, B.T.; Zylstra, C.R.; Alenina, N.; Robinson, D.R.; Zhong, Z.; Matthes, S.; Jacobsen, C.M.; Conlon, R.A.; et al. Lrp5 functions in bone to regulate bone mass. Nat. Med. 2011, 17, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Karsenty, G. The two faces of serotonin in bone biology. J. Cell Biol. 2010, 191, 7–13. [Google Scholar] [CrossRef]
- Sheu, Y.H.; Lanteigne, A.; Stürmer, T.; Pate, V.; Azrael, D.; Miller, M. SSRI use and risk of fractures among perimenopausal women without mental disorders. Inj. Prev. 2015, 21, 397–403. [Google Scholar] [CrossRef]
- Feuer, A.J.; Demmer, R.T.; Thai, A.; Vogiatzi, M.G. Use of selective serotonin reuptake inhibitors and bone mass in adolescents: An NHANES study. Bone 2015, 78, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Rauma, P.H.; Honkanen, R.J.; Williams, L.J.; Tuppurainen, M.T.; Kröger, H.P.; Koivumaa-Honkanen, H. Effects of antidepressants on postmenopausal bone loss—A 5-year longitudinal study from the OSTPRE cohort. Bone 2016, 89, 25–31. [Google Scholar] [CrossRef]
- Wang, C.X.; Ge, X.Y.; Wang, M.Y.; Ma, T.; Zhang, Y.; Lin, Y. Dopamine D1 receptor-mediated activation of the ERK signaling pathway is involved in the osteogenic differentiation of bone mesenchymal stem cells. Stem Cell Res. Ther. 2020, 11, 12. [Google Scholar] [CrossRef]
- Cheong, P.; Ma, T.; Zheng, Y.; Ge, X.; Zhang, Y.; Lin, Y. Dopamine receptor expression on primary osteoblasts and bone marrow mesenchymal stem cells of rats. Int. J. Clin. Exp. Med. 2018, 11, 1765–1771. [Google Scholar]
- Lee, D.J.; Tseng, H.C.; Wong, S.W.; Wang, Z.; Deng, M.; Ko, C.C. Dopaminergic effects on in vitro osteogenesis. Bone Res. 2015, 3. [Google Scholar] [CrossRef]
- Hanami, K.; Nakano, K.; Saito, K.; Okada, Y.; Yamaoka, K.; Kubo, S.; Kondo, M.; Tanaka, Y. Dopamine D2-like receptor signaling suppresses human osteoclastogenesis. Bone 2013, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Motyl, K.J.; Beauchemin, M.; Barlow, D.; Le, P.T.; Nagano, K.; Treyball, A.; Contractor, A.; Baron, R.; Rosen, C.J.; Houseknecht, K.L. A novel role for dopamine signaling in the pathogenesis of bone loss from the atypical antipsychotic drug risperidone in female mice. Bone 2017, 103, 168–176. [Google Scholar] [CrossRef]
- Chen, C.Y.; Lane, H.Y.; Lin, C.H. Effects of Antipsychotics on Bone Mineral Density in Patients with Schizophrenia: Gender Differences. Clin. Psychopharmacol. Neurosci. 2016, 14, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Genever, P.G.; Skerry, T.M.; Publicover, S.J. The NMDA type glutamate receptors expressed by primary rat osteoblasts have the same electrophysiological characteristics as neuronal receptors. Calcif. Tissue Int. 2002, 70, 194–203. [Google Scholar] [CrossRef]
- Chenu, C.; Serre, C.M.; Raynal, C.; Burt-Pichat, B.; Delmas, P.D. Glutamate receptors are expressed by bone cells and are involved in bone resorption. Bone 1998, 22, 295–299. [Google Scholar] [CrossRef]
- Merle, B.; Itzstein, C.; Delmas, P.D.; Chenu, C. NMDA glutamate receptors are expressed by osteoclast precursors and involved in the regulation of osteoclastogenesis. J. Cell. Biochem. 2003, 90, 424–436. [Google Scholar] [CrossRef]
- Morimoto, R.; Uehara, S.; Yatsushiro, S.; Juge, N.; Hua, Z.; Senoh, S.; Echigo, N.; Hayashi, M.; Mizoguchi, T.; Ninomiya, T.; et al. Secretion of L-glutamate from osteoclasts through transcytosis. EMBO J. 2006, 25, 4175–4186. [Google Scholar] [CrossRef]
- Taylor, A.F. Osteoblastic glutamate receptor function regulates bone formation and resorption. J. Musculoskelet. Neuronal Interact. 2002, 2, 285–290. [Google Scholar]
- Serre, C.M.; Farlay, D.; Delmas, P.D.; Chenu, C. Evidence for a dense and intimate innervation of the bone tissue, including glutamate-containing fibers. Bone 1999, 25, 623–629. [Google Scholar] [CrossRef]
- Bhangu, P.S.; Genever, P.G.; Spencer, G.J.; Grewal, T.S.; Skerry, T.M. Evidence for targeted vesicular glutamate exocytosis in osteoblasts. Bone 2001, 29, 16–23. [Google Scholar] [CrossRef]
- Olkku, A.; Mahonen, A. Wnt and steroid pathways control glutamate signalling by regulating glutamine synthetase activity in osteoblastic cells. Bone 2008, 43, 483–493. [Google Scholar] [CrossRef]
- Idris, A.I.; Ralston, S.H. Role of cannabinoids in the regulation of bone remodeling. Front. Endocrinol. 2012, 3, 1–8. [Google Scholar] [CrossRef]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef]
- Kalsbeek, A.; Palm, I.F.; La Fleur, S.E.; Scheer, F.A.J.L.; Perreau-Lenz, S.; Ruiter, M.; Kreier, F.; Cailotto, C.; Buijs, R.M. SCN outputs and the hypothalamic balance of life. J. Biol. Rhythms 2006, 21, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Bartness, T.J.; Song, C.K.; Demas, G.E. SCN efferents to peripheral tissues: Implications for biological rhythms. J. Biol. Rhythms 2001, 16, 196–204. [Google Scholar] [CrossRef]
- Buijs, R.M.; Escobar, C.; Swaab, D.F. The circadian system and the balance of the autonomic nervous system. Handb. Clin. Neurol. 2013, 117, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Buijs, F.N.; León-Mercado, L.; Guzmán-Ruiz, M.; Guerrero-Vargas, N.N.; Romo-Nava, F.; Buijs, R.M. The circadian system: A regulatory feedback network of periphery and brain. Physiology 2016, 31, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Takarada, T.; Xu, C.; Ochi, H.; Nakazato, R.; Yamada, D.; Nakamura, S.; Kodama, A.; Shimba, S.; Mieda, M.; Fukasawa, K.; et al. Bone Resorption Is Regulated by Circadian Clock in Osteoblasts. J. Bone Miner. Res. 2017, 32, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.E.; Walker, W.V.; Fenster, R.J.; Simmons, D.J. In Vitro Evaluation of Circadian Patterns of Bone Collagen Formation. Proc. Soc. Exp. Biol. Med. 1985, 180, 375–381. [Google Scholar] [CrossRef] [PubMed]
- McElderry, J.D.P.; Zhao, G.; Khmaladze, A.; Wilson, C.G.; Franceschi, R.T.; Morris, M.D. Tracking circadian rhythms of bone mineral deposition in murine calvarial organ cultures. J. Bone Miner. Res. 2013, 28, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ochi, H.; Fukuda, T.; Sato, S.; Sunamura, S.; Takarada, T.; Hinoi, E.; Okawa, A.; Takeda, S. Circadian Clock Regulates Bone Resorption in Mice. J. Bone Miner. Res. 2016, 31, 1344–1355. [Google Scholar] [CrossRef]
- Kawai, M.; Kinoshita, S.; Shimba, S.; Ozono, K.; Michigami, T. Sympathetic activation induces skeletal Fgf23 expression in a circadian rhythm-dependent manner. J. Biol. Chem. 2014, 289, 1457–1466. [Google Scholar] [CrossRef]
- Boucher, H.; Vanneaux, V.; Domet, T.; Parouchev, A.; Larghero, J. Circadian clock genes modulate human bone marrow mesenchymal stem cell differentiation, migration and cell cycle. PLoS ONE 2016, 11, 1–16. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; De Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Ishac, E.J.N.; Jiang, L.; Lake, K.D.; Varga, K.; Abood, M.E.; Kunos, G. Inhibition of exocytotic noradrenaline release by presynaptic cannabinoid CB1 receptors on peripheral sympathetic nerves. Br. J. Pharmacol. 1996, 118, 2023–2028. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002, 68–69, 619–631. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Ross, R.A. Cannabinoid receptors and their ligands. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 101–121. [Google Scholar] [CrossRef]
- Mackie, K. Signaling via CNS cannabinoid receptors. Mol. Cell. Endocrinol. 2008, 286. [Google Scholar] [CrossRef] [PubMed]
- Bab, I.; Zimmer, A.; Melamed, E. Cannabinoids and the skeleton: From marijuana to reversal of bone loss. Ann. Med. 2009, 41, 560–567. [Google Scholar] [CrossRef]
- Karsak, M.; Malkin, I.; Tolia, M.R.; Kubisch, C.; Nürnberg, P.; Zimmer, A.; Livshits, G. The cannabinoid receptor type 2 (CNR2) gene is associated with hand bone strength phenotypes in an ethnically homogeneous family sample. Hum. Genet. 2009, 126, 629–636. [Google Scholar] [CrossRef]
- Tam, J.; Trembovler, V.; Di Marzo, V.; Petrosino, S.; Leo, G.; Alexandrovich, A.; Regev, E.; Casap, N.; Shteyer, A.; Ledent, C.; et al. The cannabinoid CB1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J. 2008, 22, 285–294. [Google Scholar] [CrossRef]
- Deis, S.; Srivastava, R.K.; de Azua, I.R.; Bindila, L.; Baraghithy, S.; Lutz, B.; Bab, I.; Tam, J. Age-related regulation of bone formation by the sympathetic cannabinoid CB1 receptor. Bone 2018, 108, 34–42. [Google Scholar] [CrossRef]
- Apostu, D.; Lucaciu, O.; Mester, A.; Benea, H.; Oltean-Dan, D.; Onisor, F.; Baciut, M.; Bran, S. Cannabinoids and bone regeneration. Drug Metab. Rev. 2019, 51, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Kogan, N.M.; Melamed, E.; Wasserman, E.; Raphael, B.; Breuer, A.; Stok, K.S.; Sondergaard, R.; Escudero, A.V.V.; Baraghithy, S.; Attar-Namdar, M.; et al. Cannabidiol, a major non-psychotropic cannabis constituent enhances fracture healing and stimulates lysyl hydroxylase activity in osteoblasts. J. Bone Miner. Res. 2015, 30, 1905–1913. [Google Scholar] [CrossRef]
- Elefteriou, F.; Campbell, P.; Ma, Y. Control of bone remodeling by the peripheral sympathetic nervous system. Calcif. Tissue Int. 2014, 94, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Bajayo, A.; Bar, A.; Denes, A.; Bachar, M.; Kram, V.; Attar-Namdar, M.; Zallone, A.; Kovács, K.J.; Yirmiya, R.; Bab, I. Skeletal parasympathetic innervation communicates central IL-1 signals regulating bone mass accrual. Proc. Natl. Acad. Sci. USA 2012, 109, 15455–15460. [Google Scholar] [CrossRef] [PubMed]
- Eimar, H.; Tamimi, I.; Murshed, M.; Tamimi, F. Cholinergic regulation of bone. J. Musculoskelet. Neuronal Interact. 2013, 13, 124–132. [Google Scholar]
- Shi, Y.; Oury, F.; Yadav, V.K.; Wess, J.; Liu, X.S.; Guo, X.E.; Murshed, M.; Karsenty, G. Signaling through the M3 Muscarinic Receptor Favors Bone Mass Accrual by Decreasing Sympathetic Activity. Cell Metab. 2010, 11, 231–238. [Google Scholar] [CrossRef]
- Kondo, H.; Togari, A. Continuous treatment with a low-dose β-agonist reduces bone mass by increasing bone resorption without suppressing bone formation. Calcif. Tissue Int. 2011, 88, 23–32. [Google Scholar] [CrossRef]
- Ducy, P.; Amling, M.; Takeda, S.; Priemel, M.; Schilling, A.F.; Beil, F.T.; Shen, J.; Vinson, C.; Rueger, J.M.; Karsenty, G. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell 2000, 100, 197–207. [Google Scholar] [CrossRef]
- Takeda, S.; Elefteriou, F.; Levasseur, R.; Liu, X.; Zhao, L.; Parker, K.L.; Armstrong, D.; Ducy, P.; Karsenty, G. Leptin regulates bone formation via the sympathetic nervous system. Cell 2002, 111, 305–317. [Google Scholar] [CrossRef]
- Hamrick, M.W.; Ferrari, S.L. Leptin and the sympathetic connection of fat to bone. Osteoporos. Int. 2008, 19, 905–912. [Google Scholar] [CrossRef]
- Martin, A.; David, V.; Malaval, L.; Lafage-Proust, M.H.; Vico, L.; Thomas, T. Opposite effects of leptin on bone metabolism: A dose-dependent balance related to energy intake and insulin-like growth factor-I pathway. Endocrinology 2007, 148, 3419–3425. [Google Scholar] [CrossRef][Green Version]
- Mach, D.B.; Rogers, S.D.; Sabino, M.C.; Luger, N.M.; Schwei, M.J.; Pomonis, J.D.; Keyser, C.P.; Clohisy, D.R.; Adams, D.J.; O’leary, P.; et al. Origins of skeletal pain: Sensory and sympathetic innervation of the mouse femur. Neuroscience 2002, 113, 155–166. [Google Scholar] [CrossRef]
- Chartier, S.R.; Mitchell, S.A.T.; Majuta, L.A.; Mantyh, P.W. The Changing Sensory and Sympathetic Innervation of the Young, Adult and Aging Mouse Femur. Neuroscience 2018, 387, 178–190. [Google Scholar] [CrossRef]
- Offley, S.C.; Guo, T.Z.; Wei, T.; Clark, J.D.; Vogel, H.; Lindsey, D.P.; Jacobs, C.R.; Yao, W.; Lane, N.E.; Kingery, W.S. Capsaicin-sensitive sensory neurons contribute to the maintenance of trabecular bone integrity. J. Bone Miner. Res. 2005, 20, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Arai, M.; Kondo, H.; Togari, A. Effects of capsaicin-induced sensory denervation on bone metabolism in adult rats. Bone 2010, 46, 1591–1596. [Google Scholar] [CrossRef] [PubMed]
- Russell, F.A.; King, R.; Smillie, S.J.; Kodji, X.; Brain, S.D. Calcitonin gene-related peptide: Physiology and pathophysiology. Physiol. Rev. 2014, 94, 1099–1142. [Google Scholar] [CrossRef]
- Wang, L.; Shi, X.; Zhao, R.; Halloran, B.P.; Clark, D.J.; Jacobs, C.R.; Kingery, W.S. Calcitonin-gene-related peptide stimulates stromal cell osteogenic differentiation and inhibits RANKL induced NF-κB activation, osteoclastogenesis and bone resorption. Bone 2010, 46, 1369–1379. [Google Scholar] [CrossRef]
- Zhou, R.; Yuan, Z.; Liiu, J.; Liu, J. Calcitonin gene-related peptide promotes the expression of osteoblastic genes and activates the WNT signal transduction pathway in bone marrow stromal stem cells. Mol. Med. Rep. 2016, 13, 4689–4696. [Google Scholar] [CrossRef]
- Ishizuka, K.; Hirukawa, K.; Nakamura, H.; Togari, A. Inhibitory effect of CGRP on osteoclast formation by mouse bone marrow cells treated with isoproterenol. Neurosci. Lett. 2005, 379, 47–51. [Google Scholar] [CrossRef]
- Onuoha, G.N.; Alpar, E.K. Elevation of plasma CGRP and SP levels in orthopedic patients with fracture neck of femur. Neuropeptides 2000, 34, 116–120. [Google Scholar] [CrossRef]
- Xu, Y.Q.; Qin, M.L.; Feng, S.Y.; Huang, Y.; Jia, Z. Expressions and significance of calcitonin gene-related peptide and nerve growth factor in rabbit model of traumatic brain injury complicated with tibial fracture: Preliminary results. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5040–5050. [Google Scholar] [PubMed]
- Harrison, S.; Geppetti, P. Substance P. Int. J. Biochem. Cell Biol. 2001, 33, 555–576. [Google Scholar] [CrossRef]
- Cook, N.L.; Vink, R.; Donkin, J.J.; van den Heuvel, C. Validation of reference genes for normalization of real-time quantitative RT-PCR data in traumatic brain injury. J. Neurosci. Res. 2009, 87, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Donkin, J.J.; Nimmo, A.J.; Cernak, I.; Blumbergs, P.C.; Vink, R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J. Cereb. Blood Flow Metab. 2009, 29, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, R.; Shi, X.; Wei, T.; Halloran, B.P.; Clark, D.J.; Jacobs, C.R.; Kingery, W.S. Substance P stimulates bone marrow stromal cell osteogenic activity, osteoclast differentiation, and resorption activity in vitro. Bone 2009, 45, 309–320. [Google Scholar] [CrossRef]
- Li, F.X.Z.; Xu, F.; Lin, X.; Wu, F.; Zhong, J.Y.; Wang, Y.; Guo, B.; Zheng, M.H.; Shan, S.K.; Yuan, L.Q. The Role of Substance P in the Regulation of Bone and Cartilage Metabolic Activity. Front. Endocrinol. 2020, 11. [Google Scholar] [CrossRef]
- Niedermair, T.; Schirner, S.; Seebröker, R.; Straub, R.H.; Grässel, S. Substance P modulates bone remodeling properties of murine osteoblasts and osteoclasts. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Ding, W.G.; Zhang, Z.M.; Zhang, Y.H.; Jiang, S.D.; Jiang, L.S.; Dai, L.Y. Changes of substance P during fracture healing in ovariectomized mice. Regul. Pept. 2010, 159, 28–34. [Google Scholar] [CrossRef]
- Hofman, M.; Rabenschlag, F.; Andruszkow, H.; Andruszkow, J.; Möckel, D.; Lammers, T.; Kolejewska, A.; Kobbe, P.; Greven, J.; Teuben, M.P.J.; et al. Effect of neurokinin-1-receptor blockage on fracture healing in rats. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Zheng, X.F.; Zhao, E.D.; He, J.Y.; Zhang, Y.H.; Jiang, S.D.; Jiang, L.S. Inhibition of substance P signaling aggravates the bone loss in ovariectomy-induced osteoporosis. Prog. Biophys. Mol. Biol. 2016, 122, 112–121. [Google Scholar] [CrossRef]
- Tran, T.S.; Kolodkin, A.L.; Bharadwaj, R. Semaphorin Regulation of Cellular Morphology. Annu. Rev. Cell Dev. Biol. 2007, 23, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Wee, N.K.Y.; Kulkarni, R.N.; Horsnell, H.; Baldock, P.A. The brain in bone and fuel metabolism. Bone 2016, 82, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, Z.; Liu, Y.; Gao, D.; Zhang, X.; Hao, J.; Yang, F. Neural regulation of bone remodeling: Identifying novel neural molecules and pathways between brain and bone. J. Cell. Physiol. 2019, 234, 5466–5477. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Nakashima, T.; Taniguchi, M.; Kodama, T.; Kumanogoh, A.; Takayanagi, H. Osteoprotection by semaphorin 3A. Nature 2012, 485, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Takeda, S.; Xu, R.; Ochi, H.; Sunamura, S.; Sato, T.; Shibata, S.; Yoshida, Y.; Gu, Z.; Kimura, A.; et al. Sema3A regulates bone-mass accrual through sensory innervations. Nature 2013, 497, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Desbois, C.; Boyce, B.; Pinero, G.; Story, B.; Dunstan, C.; Smith, E.; Bonadio, J.; Goldstein, S.; Gundberg, C.; et al. Increased bone formation in osteocalcin-deficient mice. Nature 1996, 382, 448–452. [Google Scholar] [CrossRef]
- Diaz-Franco, M.C.; Franco-Diaz de Leon, R.; Villafan-Bernal, J.R. Osteocalcin-GPRC6A: An update of its clinical and biological multi-organic interactions (Review). Mol. Med. Rep. 2019, 19, 15–22. [Google Scholar] [CrossRef]
- Oury, F.; Khrimian, L.; Denny, C.A.; Gardin, A.; Chamouni, A.; Goeden, N.; Huang, Y.Y.; Lee, H.; Srinivas, P.; Gao, X.B.; et al. XMaternal and offspring pools of osteocalcin influence brain development and functions. Cell 2013, 155, 228. [Google Scholar] [CrossRef]
- Bradburn, S.; Mcphee, J.S.; Bagley, L.; Sipila, S.; Stenroth, L.; Narici, M.V.; Pääsuke, M.; Gapeyeva, H.; Osborne, G.; Sassano, L.; et al. Association between osteocalcin and cognitive performance in healthy older adults. Age Ageing 2016, 45, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Ghosh, A.; Guo, X.Z.; Wang, S.M.; Hou, Y.F.; Li, S.T.; Liu, J.M. Roles for osteocalcin in brain signalling: Implications in cognition- and motor-related disorders. Mol. Brain 2019, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moser, S.C.; van der Eerden, B.C.J. Osteocalcin — A versatile bone-derived hormone. Front. Endocrinol. 2019, 10, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Diegel, C.R.; Hann, S.; Ayturk, U.M.; Hu, J.C.W.; Lim, K.E.; Droscha, C.J.; Madaj, Z.B.; Foxa, G.E.; Izaguirre, I.; Transgenics Core, V.V.A.; et al. An osteocalcin-deficient mouse strain without endocrine abnormalities. PLoS Genet. 2020, 16, e1008361. [Google Scholar] [CrossRef]
- Moriishi, T.; Ozasa, R.; Ishimoto, T.; Nakano, T.; Hasegawa, T.; Miyazaki, T.; Liu, W.; Fukuyama, R.; Wang, Y.; Komori, H.; et al. Osteocalcin is necessary for the alignment of apatite crystallites, but not glucose metabolism, testosterone synthesis, or muscle mass. PLoS Genet. 2020, 16, e1008586. [Google Scholar] [CrossRef] [PubMed]
- Mera, P.; Ferron, M.; Mosialou, I. Regulation of energy metabolism by bone-derived hormones. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Mosialou, I.; Shikhel, S.; Liu, J.M.; Maurizi, A.; Luo, N.; He, Z.; Huang, Y.; Zong, H.; Friedman, R.A.; Barasch, J.; et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 2017, 543, 385–390. [Google Scholar] [CrossRef]
- Koide, M.; Kobayashi, Y. Regulatory mechanisms of sclerostin expression during bone remodeling. J. Bone Miner. Metab. 2019, 37, 9–17. [Google Scholar] [CrossRef]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Shah, A.D.; Shoback, D.; Michael Lewiecki, E. Sclerostin inhibition: A novel therapeutic approach in the treatment of osteoporosis. Int. J. Womens. Health 2015, 7, 565–580. [Google Scholar]
- McClung, M.R.; Grauer, A.; Boonen, S.; Bolognese, M.A.; Brown, J.P.; Diez-Perez, A.; Langdahl, B.L.; Reginster, J.Y.; Zanchetta, J.R.; Wasserman, S.M.; et al. Romosozumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2014, 370, 412–420. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Ferrari, S.; Khan, A.; Lane, N.E.; Lippuner, K.; Matsumoto, T.; Milmont, C.E.; Libanati, C.; Grauer, A. FRAME Study: The Foundation Effect of Building Bone With 1 Year of Romosozumab Leads to Continued Lower Fracture Risk After Transition to Denosumab. J. Bone Miner. Res. 2018, 33, 1219–1226. [Google Scholar] [CrossRef]
- Graeff, C.; Campbell, G.M.; Peña, J.; Borggrefe, J.; Padhi, D.; Kaufman, A.; Chang, S.; Libanati, C.; Glüer, C.C. Administration of romosozumab improves vertebral trabecular and cortical bone as assessed with quantitative computed tomography and finite element analysis. Bone 2015, 81, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N. Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Youssef, S.J.; Oursler, M.J. Sclerostin expression and functions beyond the osteocyte. Bone 2017, 96, 45–50. [Google Scholar] [CrossRef]
- Pinzone, J.J.; Hall, B.M.; Thudi, N.K.; Vonau, M.; Qiang, Y.W.; Rosol, T.J.; Shaughnessy, J.D. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 2009, 113, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sarosi, I.; Cattley, R.C.; Pretorius, J.; Asuncion, F.; Grisanti, M.; Morony, S.; Adamu, S.; Geng, Z.; Qiu, W.; et al. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 2006, 39, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Lieven, O.; Knobloch, J.; Rüther, U. The regulation of Dkk1 expression during embryonic development. Dev. Biol. 2010, 340, 256–268. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, L.; Liu, A. Dickkopf-1: Current knowledge and related diseases. Life Sci. 2018, 209, 249–254. [Google Scholar] [CrossRef]
- Okamoto, M.; Inoue, K.; Iwamura, H.; Terashima, K.; Soya, H.; Asashima, M.; Kuwabara, T. Reduction in paracrine Wnt3 factors during aging causes impaired adult neurogenesis. FASEB J. 2011, 25, 3570–3582. [Google Scholar] [CrossRef]
- Ren, C.; Gu, X.; Li, H.; Lei, S.; Wang, Z.; Wang, J.; Yin, P.; Zhang, C.; Wang, F.; Liu, C. The role of DKK1 in Alzheimer’s disease: A potential intervention point of brain damage prevention? Pharmacol. Res. 2019, 144, 331–335. [Google Scholar] [CrossRef]
- Purro, S.A.; Dickins, E.M.; Salinas, P.C. The secreted Wnt antagonist dickkopf-1 is required for amyloid β-mediated synaptic loss. J. Neurosci. 2012, 32, 3492–3498. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B. Wnt/β-catenin signaling pathway governs a full program for dopaminergic neuron survival, neurorescue and regeneration in the MPTP mouse model of Parkinson’s disease. Int. J. Mol. Sci. 2018, 19, 3743. [Google Scholar] [CrossRef] [PubMed]
- Corrado, A.; Sanpaolo, E.R.; Di Bello, S.; Cantatore, F.P. Osteoblast as a target of anti-osteoporotic treatment. Postgrad. Med. 2017, 129, 858–865. [Google Scholar] [CrossRef]
- Florio, M.; Gunasekaran, K.; Stolina, M.; Li, X.; Liu, L.; Tipton, B.; Salimi-Moosavi, H.; Asuncion, F.J.; Li, C.; Sun, B.; et al. A bispecific antibody targeting sclerostin and DKK-1 promotes bone mass accrual and fracture repair. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Han, K.H.; Arlian, B.M.; Macauley, M.S.; Paulson, J.C.; Lerner, R.A. Migration-based selections of antibodies that convert bone marrow into trafficking microglia-like cells that reduce brain amyloid β. Proc. Natl. Acad. Sci. USA 2018, 115, E372–E381. [Google Scholar] [CrossRef]
- Kawanishi, S.; Takata, K.; Itezono, S.; Nagayama, H.; Konoya, S.; Chisaki, Y.; Toda, Y.; Nakata, S.; Yano, Y.; Kitamura, Y.; et al. Bone-Marrow-Derived Microglia-Like Cells Ameliorate Brain Amyloid Pathology and Cognitive Impairment in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 563–585. [Google Scholar] [CrossRef]
- Kuroda, E.; Takata, K.; Nishimura, K.; Oka, H.; Sueyoshi, M.; Aitani, M.; Kouda, A.; Satake, S.; Shima, C.; Toda, Y.; et al. Peripheral Blood-Derived Microglia-Like Cells Decrease Amyloid-β Burden and Ameliorate Cognitive Impairment in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 413–429. [Google Scholar] [CrossRef]
- Costa-Marques, L.; Arnold, K.; Pardon, M.C.; Leovsky, C.; Swarbrick, S.; Fabian, C.; Stolzing, A. Transplantation of bone marrow derived macrophages reduces markers of neuropathology in an APP/PS1 mouse model. Transl. Neurodegener. 2019, 8, 1–11. [Google Scholar] [CrossRef]
- Lampron, A.; Gosselin, D.; Rivest, S. Targeting the hematopoietic system for the treatment of Alzheimer’s disease. Brain. Behav. Immun. 2011, 25, 71–79. [Google Scholar] [CrossRef]
- Colaianni, G.; Cuscito, C.; Mongelli, T.; Pignataro, P.; Buccoliero, C.; Liu, P.; Lu, P.; Sartini, L.; Comite, M.D.; Mori, G.; et al. The myokine irisin increases cortical bone mass. Proc. Natl. Acad. Sci. USA 2015, 112, 12157–12162. [Google Scholar] [CrossRef] [PubMed]
- Colaianni, G.; Mongelli, T.; Cuscito, C.; Pignataro, P.; Lippo, L.; Spiro, G.; Notarnicola, A.; Severi, I.; Passeri, G.; Mori, G.; et al. Irisin prevents and restores bone loss and muscle atrophy in hind-limb suspended mice. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Arhire, L.I.; Mihalache, L.; Covasa, M. Irisin: A Hope in Understanding and Managing Obesity and Metabolic Syndrome. Front. Endocrinol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Munoz, I.Y.M.; Del Socorro Camarillo Romero, E.; De Jesus Garduno Garcia, J. Irisin a novel metabolic biomarker: Present knowledge and future directions. Int. J. Endocrinol. 2018, 2018. [Google Scholar] [CrossRef]
- Asadi, Y.; Gorjipour, F.; Behrouzifar, S.; Vakili, A. Irisin Peptide Protects Brain Against Ischemic Injury Through Reducing Apoptosis and Enhancing BDNF in a Rodent Model of Stroke. Neurochem. Res. 2018, 43, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Ting, S.-M.; Zhao, X.; Sun, G.; Obertas, L.; Ricote, M.; Aronowski, J. Brain Cleanup as a Potential Target for Poststroke Recovery. Stroke 2020, 958–966. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Gonçalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef]
- Reinholt, F.P.; Hultenby, K.; Oldbergf, A.; Heinegardt, D. Osteopontin-a possible anchor of osteoclasts to bone (bone matrix proteins/cell adhesion/vitronectin receptor). Proc. Nadl. Acad. Sci. USA 1990, 87, 4473–4475. [Google Scholar] [CrossRef]
- Singh, A.; Gill, G.; Kaur, H.; Amhmed, M.; Jakhu, H. Role of osteopontin in bone remodeling and orthodontic tooth movement: A review. Prog. Orthod. 2018, 19. [Google Scholar] [CrossRef]
- Cho, E.H.; Cho, K.H.; Lee, H.A.; Kim, S.W. High serum osteopontin levels are associated with low bone mineral density in postmenopausal women. J. Korean Med. Sci. 2013, 28, 1496–1499. [Google Scholar] [CrossRef]
- Scatena, M.; Liaw, L.; Giachelli, C.M. Osteopontin: A multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Brown, A. Osteopontin: A key link between immunity, inflammation and the central nervous system. Transl. Neurosci. 2012, 3, 288–293. [Google Scholar] [CrossRef][Green Version]
- Gliem, M.; Krammes, K.; Liaw, L.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia 2015, 63, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Carecchio, M.; Comi, C. The role of osteopontin in neurodegenerative diseases. J. Alzheimer’s Dis. 2011, 25, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Hanada, R.; Leibbrandt, A.; Hanada, T.; Kitaoka, S.; Furuyashiki, T.; Fujihara, H.; Trichereau, J.; Paolino, M.; Qadri, F.; Plehm, R.; et al. Central control of fever and female body temperature by RANKL/RANK. Nature 2009, 462, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, X. Receptor activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin system in bone and other tissues (Review). Mol. Med. Rep. 2015, 11, 3212–3218. [Google Scholar] [CrossRef]
- Lacey, D.L.; Boyle, W.J.; Simonet, W.S.; Kostenuik, P.J.; Dougall, W.C.; Sullivan, J.K.; Martin, J.S.; Dansey, R. Bench to bedside: Elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat. Rev. Drug Discov. 2012, 11, 401–419. [Google Scholar] [CrossRef]
- Lipton, A.; Fizazi, K.; Stopeck, A.T.; Henry, D.H.; Brown, J.E.; Yardley, D.A.; Richardson, G.E.; Siena, S.; Maroto, P.; Clemens, M.; et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: A combined analysis of 3 pivotal, randomised, phase 3 trials. Eur. J. Cancer 2012, 48, 3082–3092. [Google Scholar] [CrossRef]
- Zhang, J.; Fujita, Y.; Chang, L.; Pu, Y.; Qu, Y.; Wang, S.; Hashimoto, K. Beneficial effects of anti-RANKL antibody in depression-like phenotype, inflammatory bone markers, and bone mineral density in male susceptible mice after chronic social defeat stress. Behav. Brain Res. 2020, 379. [Google Scholar] [CrossRef]
- Mattson, M.P. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann. N. Y. Acad. Sci. 2008, 1144, 97–112. [Google Scholar]
- Kajiya, M.; Shiba, H.; Fujita, T.; Ouhara, K.; Takeda, K.; Mizuno, N.; Kawaguchi, H.; Kitagawa, M.; Takata, T.; Tsuji, K.; et al. Brain-derived neurotrophic factor stimulates bone/cementum-related protein gene expression in cementoblasts. J. Biol. Chem. 2008, 283, 16259–16267. [Google Scholar] [CrossRef]
- Camerino, C.; Zayzafoon, M.; Rymaszewski, M.; Heiny, J.; Rios, M.; Hauschka, P.V. Central depletion of brain-derived neurotrophic, factorin mice results in high bone mass and metabolic phenotype. Endocrinology 2012, 153, 5394–5405. [Google Scholar] [CrossRef]
- Laviola, L.; Natalicchio, A.; Perrini, S.; Giorgino, F. Abnormalities of IGF-I signaling in the pathogenesis of diseases of the bone, brain, and fetoplacental unit in humans. Am. J. Physiol. Endocrinol. Metab. 2008, 295. [Google Scholar] [CrossRef] [PubMed]
- Wrigley, S.; Arafa, D.; Tropea, D. Insulin-like growth factor 1: At the crossroads of brain development and aging. Front. Cell. Neurosci. 2017, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Boisclair, Y.R.; Leroith, D.; Yakar, S.; Rosen, C.J.; Beamer, W.G.; Ackert-bicknell, C.L.; Wu, Y.; Liu, J.; Ooi, G.T.; et al. Circulating levels of IGF-1 directly regulate bone growth and density. J. Clin. Investig. 2002, 110, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.J. Insulin-like growth factor I and bone mineral density: Experience from animal models and human observational studies. Best Pract. Res. Clin. Endocrinol. Metab. 2004, 18, 423–435. [Google Scholar] [CrossRef]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone Morphogenetic Proteins: A critical review. Cell. Signal. 2011, 23, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Lein, P.J.; Wang, J.Y.; Gash, D.; Hoffer, B.J.; Chiang, Y.H. Expression of bone morphogenetic proteins in the brain during normal aging and in 6-hydroxydopamine-lesioned animals. Brain Res. 2003, 994, 81–90. [Google Scholar] [CrossRef]
- Sato, T.; Mikawa, S.; Sato, K. BMP2 expression in the adult rat brain. J. Comp. Neurol. 2010, 518, 4513–4530. [Google Scholar] [CrossRef]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol. 2012, 72, 1068–1084. [Google Scholar] [CrossRef]
- Chang, J.; Dettman, R.W.; Dizon, M.L.V. Bone morphogenetic protein signaling: A promising target for white matter protection in perinatal brain injury. Neural Regen. Res. 2018, 13, 1183–1184. [Google Scholar] [CrossRef]
- Staal, A.; Frith, J.C.; French, M.H.; Swartz, J.; Güngör, T.; Harrity, T.W.; Tamasi, J.; Rogers, M.J.; Feyen, J.H.M. The ability of statins to inhibit bone resorption is directly related to their inhibitory effect on HMG-CoA reductase activity. J. Bone Miner. Res. 2003, 18, 88–96. [Google Scholar] [CrossRef]
- Mori, M.; Nishikawa, T.; Masuno, K.; Okamura, T.; Tanaka, A.; Shikimori, M. Statins: Candidates for promoting bone formation via BMP-2. Oral Med. Pathol. 2010, 14, 81–87. [Google Scholar] [CrossRef][Green Version]
- Ruan, F.; Zheng, Q.; Wang, J. Mechanisms of bone anabolism regulated by statins. Biosci. Rep. 2012, 32, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Uzzan, B.; Cohen, R.; Nicolas, P.; Cucherat, M.; Perret, G.Y. Effects of statins on bone mineral density: A meta-analysis of clinical studies. Bone 2007, 40, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Morse, L.R.; Coker, J.; Battaglino, R.A. Statins and bone health: A mini review. Actual. osteol. 2018, 14, 31–35. [Google Scholar] [PubMed]
- Samsa, W.E.; Vasanji, A.; Midura, R.J.; Kondratov, R.V. Deficiency of circadian clock protein BMAL1 in mice results in a low bone mass phenotype. Bone 2016, 84, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Z.; Man, C.W.; Guo, J.; Han, X.; Hu, Z.; Ng, T.B.; Zhao, Z.; Li, J.; Wang, W.; et al. The effect of exogenous melatonin on reducing scoliotic curvature and improving bone quality in melatonin-deficient C57BL/6J mice. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Li, S.; Liu, B.; Zhang, L.; Rong, L. Amyloid beta peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone 2014, 61, 164–175. [Google Scholar] [CrossRef]
- Zhu, P.; Zhang, Z.; Huang, X.; Liang, S.; Khandekar, N.; Song, Z.; Lin, S. RANKL reduces body weight and food intake via the modulation of hypothalamic NPY/CART expression. Int. J. Med. Sci. 2018, 15, 969–977. [Google Scholar] [CrossRef]
- Laksitorini, M.D.; Yathindranath, V.; Xiong, W.; Hombach-Klonisch, S.; Miller, D.W. Modulation of Wnt/β-catenin signaling promotes blood-brain barrier phenotype in cultured brain endothelial cells. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Eger, M.; Bader, M.; Bree, D.; Hadar, R.; Nemerovski, A.; Tam, J.; Levy, D.; Pick, C.G.; Gabet, Y. Bone Anabolic Response in the Calvaria Following Mild Traumatic Brain Injury is Mediated by the Cannabinoid-1 Receptor. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Seemann, R.; Graef, F.; Garbe, A.; Keller, J.; Huang, F.; Duda, G.; Schmidt-Bleek, K.; Schaser, K.D.; Tsitsilonis, S. Leptin-deficiency eradicates the positive effect of traumatic brain injury on bone healing: Histological analyses in a combined trauma mouse model. J. Musculoskelet. Neuronal Interact. 2018, 18, 32–41. [Google Scholar] [PubMed]
- Song, Y.; Han, G.X.; Chen, L.; Zhai, Y.Z.; Dong, J.; Chen, W.; Li, T.S.; Zhu, H.Y. The role of the hippocampus and the function of calcitonin gene-related peptide in the mechanism of traumatic brain injury accelerating fracture-healing. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1522–1531. [Google Scholar]
- Liu, X.; Zhou, C.; Li, Y.; Ji, Y.; Xu, G.; Wang, X.; Yan, J. SDF-1 Promotes Endochondral Bone Repair during Fracture Healing at the Traumatic Brain Injury Condition. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, H.W.; Fu, P.; Liu, B.L.; Jin, W.Z.; Duan, S.B.; Xue, J.; Liu, K.; Sun, Z.M.; Zeng, X.W. Leptin’s effect on accelerated fracture healing after traumatic brain injury. Neurol. Res. 2013, 35, 537–544. [Google Scholar] [CrossRef]
- Yang, S.; Ma, Y.; Liu, Y.; Que, H.; Zhu, C.; Liu, S. Arachidonic acid: A bridge between traumatic brain injury and fracture healing. J. Neurotrauma 2012, 29, 2696–2705. [Google Scholar] [CrossRef]
- Song, Y.; Bi, L.; Zhang, Z.; Huang, Z.; Hou, W.; Lu, X.; Sun, P.; Han, Y. Increased levels of calcitonin gene-related peptide in serum accelerate fracture healing following traumatic brain injury. Mol. Med. Rep. 2012, 5, 432–438. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, P.; Wang, Y.; Han, N.; Tang, C.; Jiang, B. The influence of brain injury or peripheral nerve injury on calcitonin gene-related peptide concentration variation and fractures healing process. Artif. Cells Blood Substitutes Biotechnol. 2009, 37, 85–91. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, L.; Clark, J.C.M.; Dass, C.R.; Choong, P.F.M. Elevated leptin expression in a rat model of fracture and traumatic brain injury. J. Pharm. Pharmacol. 2008, 60, 1667–1672. [Google Scholar] [CrossRef]
- Boes, M.; Kain, M.; Kakar, S.; Nicholls, F.; Cullinane, D.; Gerstenfeld, L.; Einhorn, T.A.; Tornetta, P. Osteogenic effects of traumatic brain injury on experimental fracture-healing. J. Bone Jt. Surg. Ser. A 2006, 88, 738–743. [Google Scholar] [CrossRef]
- Khallaf, F.G.; Kehinde, E.O. The Effect of Serum from Acute Traumatic Brain or Spinal Cord Injury Patients on the Growth of Human Bone Marrow-Derived Mesenchymal Stem Cells. J. Trauma Treat. 2016, 5. [Google Scholar] [CrossRef]
- Dengler-Crish, C.M.; Ball, H.C.; Lin, L.; Novak, K.M.; Cooper, L.N. Evidence of Wnt/β-catenin alterations in brain and bone of a tauopathy mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 67, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Khrimian, L.; Obri, A.; Karsenty, G. Modulation of cognition and anxiety-like behavior by bone remodeling. Mol. Metab. 2017, 6, 1610–1615. [Google Scholar] [CrossRef] [PubMed]
- Inatani, M.; Irie, F.; Plump, A.S.; Tessier-Lavigne, M.; Yamaguchi, Y. Mammalian Brain Morphogenesis and Midline Axon Guidance Require Heparan Sulfate. Science 2003, 302, 1044–1046. [Google Scholar] [CrossRef]
- Irie, F.; Badie-Mahdavi, H.; Yamaguchi, Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc. Natl. Acad. Sci. USA 2012, 109, 5052–5056. [Google Scholar] [CrossRef] [PubMed]
- Suto, Y.; Nagata, K.; Ahmed, S.M.; Jacovides, C.L.; Browne, K.D.; Cognetti, J.; Johnson, V.E.; Leone, R.; Kaplan, L.J.; Smith, D.H.; et al. Cerebral Edema and Neurological Recovery after Traumatic Brain Injury Are Worsened if Accompanied by a Concomitant Long Bone Fracture. J. Neurotrauma 2019, 36, 609–618. [Google Scholar] [CrossRef] [PubMed]


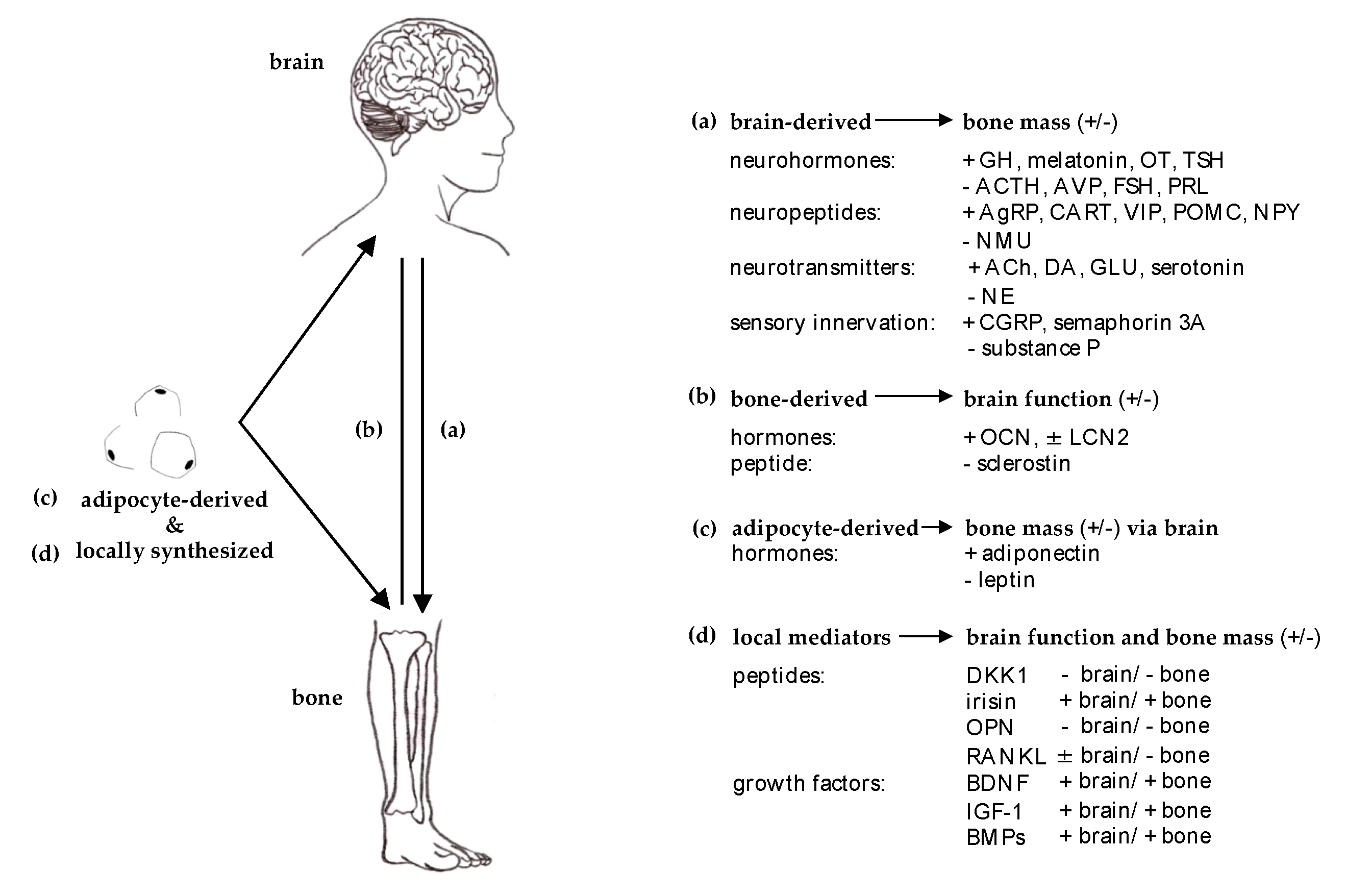{kind=link}
| Origin | Condition | Effect on Bone | Clinical Studies/Reviews | |
|---|---|---|---|---|
| brain | neuro-psychological causes | chronic stress and shift work |
| [13,14,25,41,42,44,45,157,158] reviewed by [159] |
| major depressive disorder (MDD) |
| [49,50,51,52,53,54,55,56,160] | ||
| stroke |
| [16,62,63,64,65,66,67,68,69,161,162] | ||
| dementia/Alzheimer’s disease (AD) |
| [8,73,74,75,76,77,78,86] reviewed by [163] | ||
| Parkinson’s disease (PD) |
| [23,87,88] | ||
| trauma | traumatic brain injury (TBI) |
| [19,116,121,128,129,130,131,132,133,134,139,140] [164,165] | |
| spinal cord injury (SCI) |
| [20,145,146,147,148,149] [164] reviewed by [166] | ||
| Effect on Brain | ||||
| bone | chronic disorders | osteoporosis |
| see each condition/disorder for reference; reviewed by [15] |
| genetic | cleidocranial dysplasia (CCD) |
| reviewed by [90] [91,92] | |
| hereditary multiple exostoses (HME) |
| [101,102,103] | ||
| sclerosteosis and van Buchem disease |
| [99,100] [167] | ||
| trauma | fracture (Fx) |
| [23] [168,169] reviewed by [22] | |
| Effect on Brain and Bone | ||||
| brain and bone | genetic | Coffin-Lowry syndrome (CLS) |
| [94] |
| trauma | complex regional pain syndrome (CRPS) |
| reviewed by [21] [104,105,106,107] |
| Origin | Condition | Effect on Bone | Model | Pre-Clinical Studies/Reviews | |
|---|---|---|---|---|---|
| brain | neuro-psychological causes | chronic stress and shift work |
| in vitro mouse mouse mouse/rat mouse | [43] [46,47] [48] [37,289,408] [409] |
| depression |
| rat | [57] | ||
| stroke |
| mouse | [71] | ||
| dementia/Alzheimer’s disease (AD) |
| in vitro in vitro | [81,410] [411,412] reviewed by [163] | ||
| Parkinson’s disease (PD) |
| mouse | [89] | ||
| trauma | traumatic brain injury (TBI) |
| mouse/rat rat mouse mouse mouse mouse rat mouse rabbit rat rat rat rat rat | [117,118] [126] [141,142] [144] [413] [414] [415] [416] [417] [418] [419] [420] [421] [422] | |
| spinal cord injury (SCI) |
| in vitro | [423] | ||
| Effect on Brain | |||||
| bone | chronic disorders | osteoporosis |
| mouse | [424] |
| genetic | cleidocranial dysplasia (CCD) |
| mouse | [425] | |
| hereditary multiple exostoses (HME) |
| mouse mouse | [426] [427] | ||
| trauma | fracture (Fx) |
| mouse | [155,156,428] | |
| Effect on Brain and Bone | |||||
| brain and bone | genetic | Coffin-Lowry syndrome (CLS) |
| mouse mouse | [95] reviewed by [94] [97] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otto, E.; Knapstein, P.-R.; Jahn, D.; Appelt, J.; Frosch, K.-H.; Tsitsilonis, S.; Keller, J. Crosstalk of Brain and Bone—Clinical Observations and Their Molecular Bases. Int. J. Mol. Sci. 2020, 21, 4946. https://doi.org/10.3390/ijms21144946
Otto E, Knapstein P-R, Jahn D, Appelt J, Frosch K-H, Tsitsilonis S, Keller J. Crosstalk of Brain and Bone—Clinical Observations and Their Molecular Bases. International Journal of Molecular Sciences. 2020; 21(14):4946. https://doi.org/10.3390/ijms21144946
Chicago/Turabian StyleOtto, Ellen, Paul-Richard Knapstein, Denise Jahn, Jessika Appelt, Karl-Heinz Frosch, Serafeim Tsitsilonis, and Johannes Keller. 2020. "Crosstalk of Brain and Bone—Clinical Observations and Their Molecular Bases" International Journal of Molecular Sciences 21, no. 14: 4946. https://doi.org/10.3390/ijms21144946
APA StyleOtto, E., Knapstein, P.-R., Jahn, D., Appelt, J., Frosch, K.-H., Tsitsilonis, S., & Keller, J. (2020). Crosstalk of Brain and Bone—Clinical Observations and Their Molecular Bases. International Journal of Molecular Sciences, 21(14), 4946. https://doi.org/10.3390/ijms21144946