The Soluble Adenylyl Cyclase Inhibitor LRE1 Prevents Hepatic Ischemia/Reperfusion Damage Through Improvement of Mitochondrial Function

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
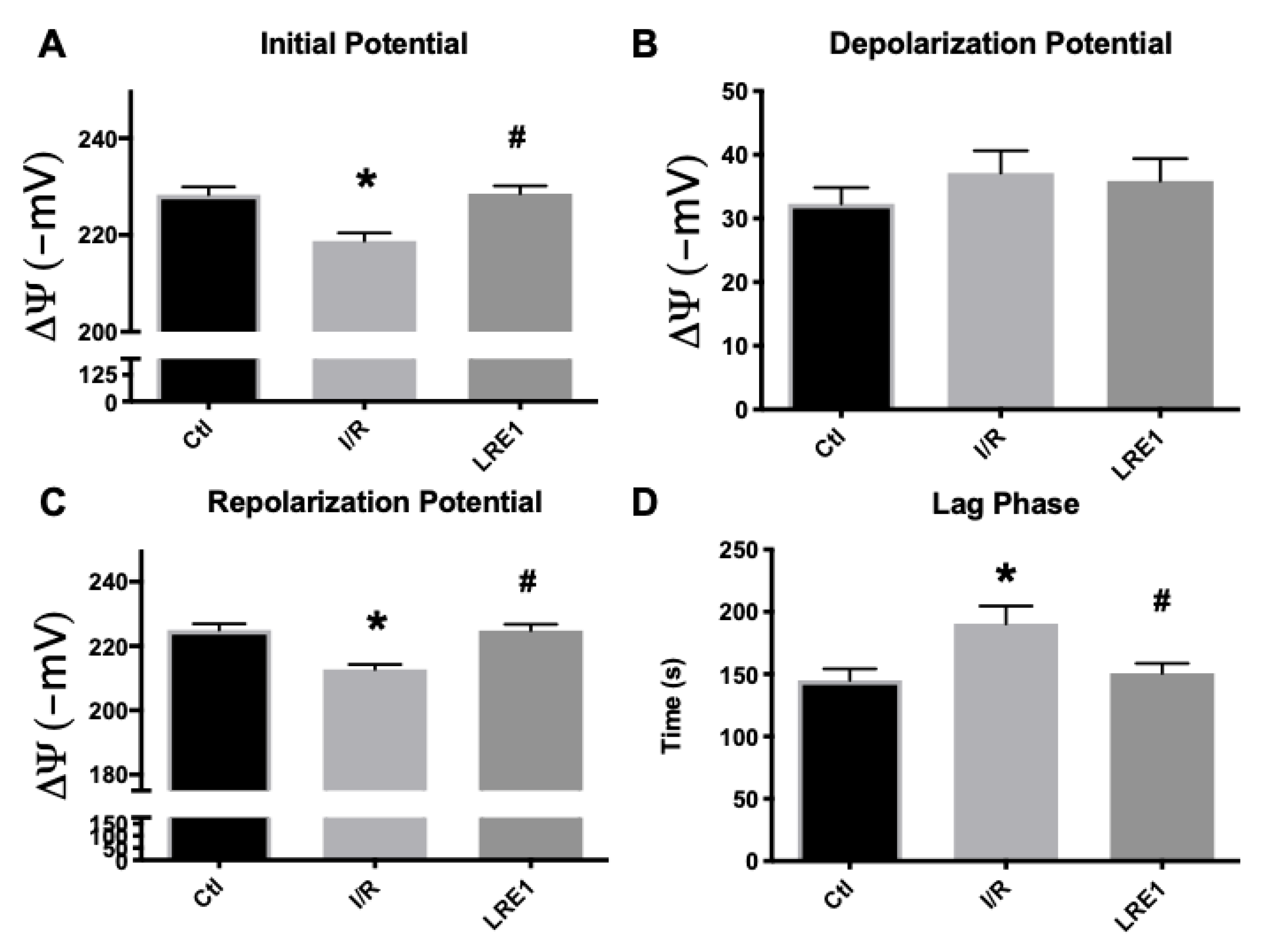
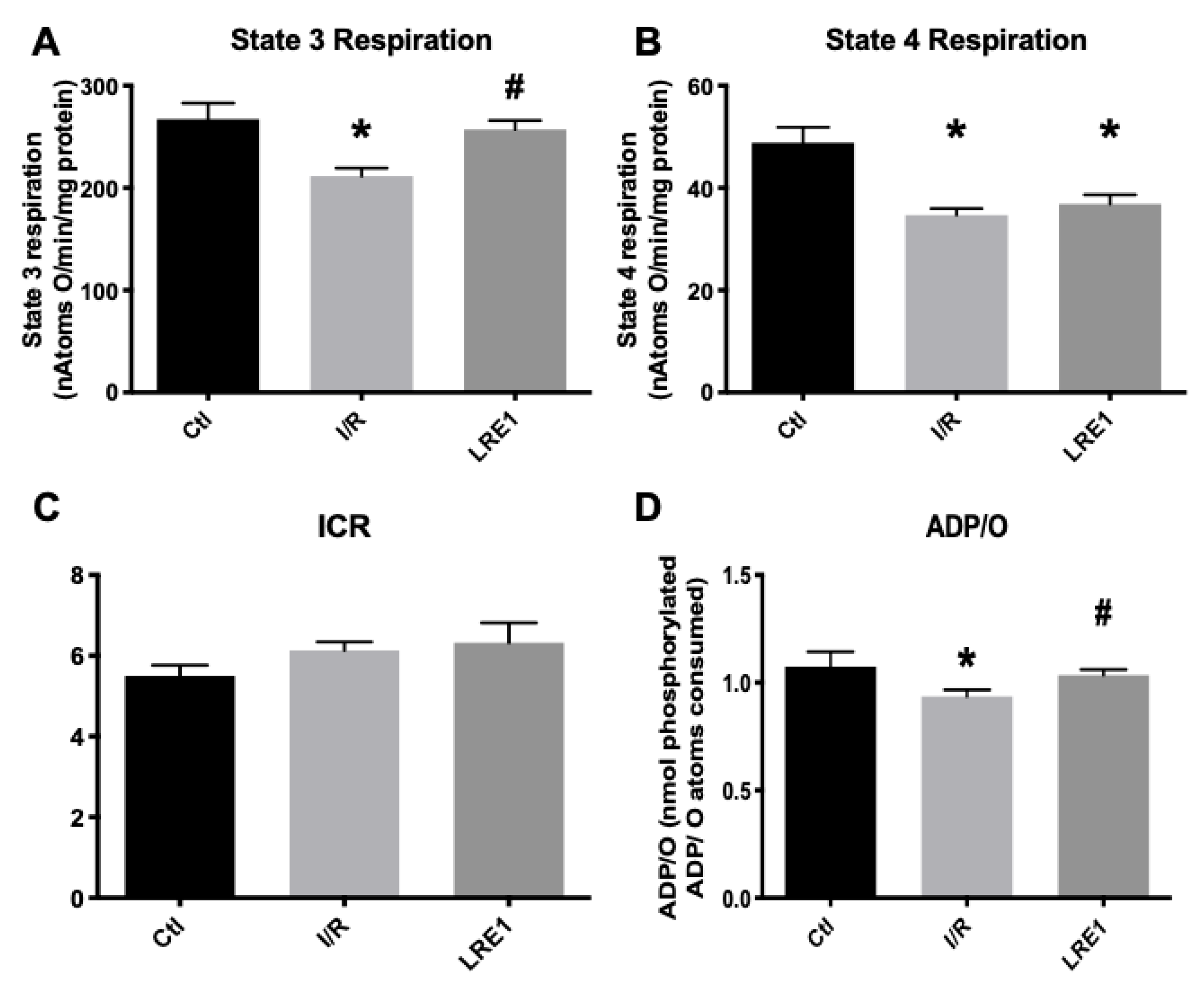
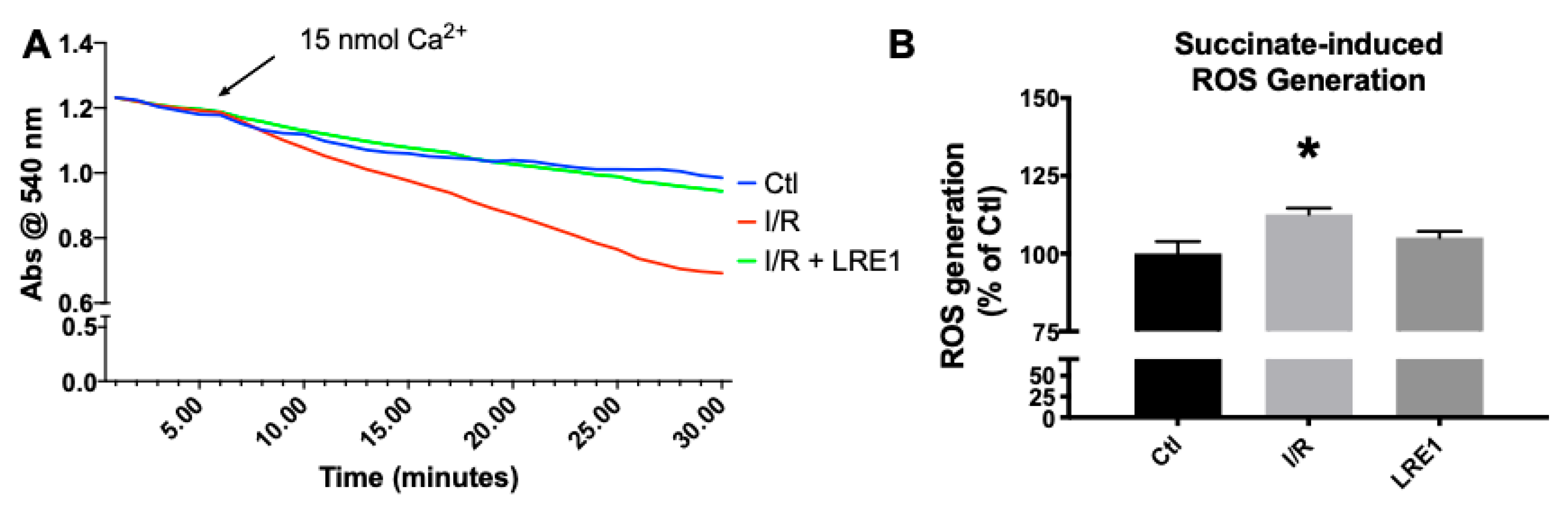
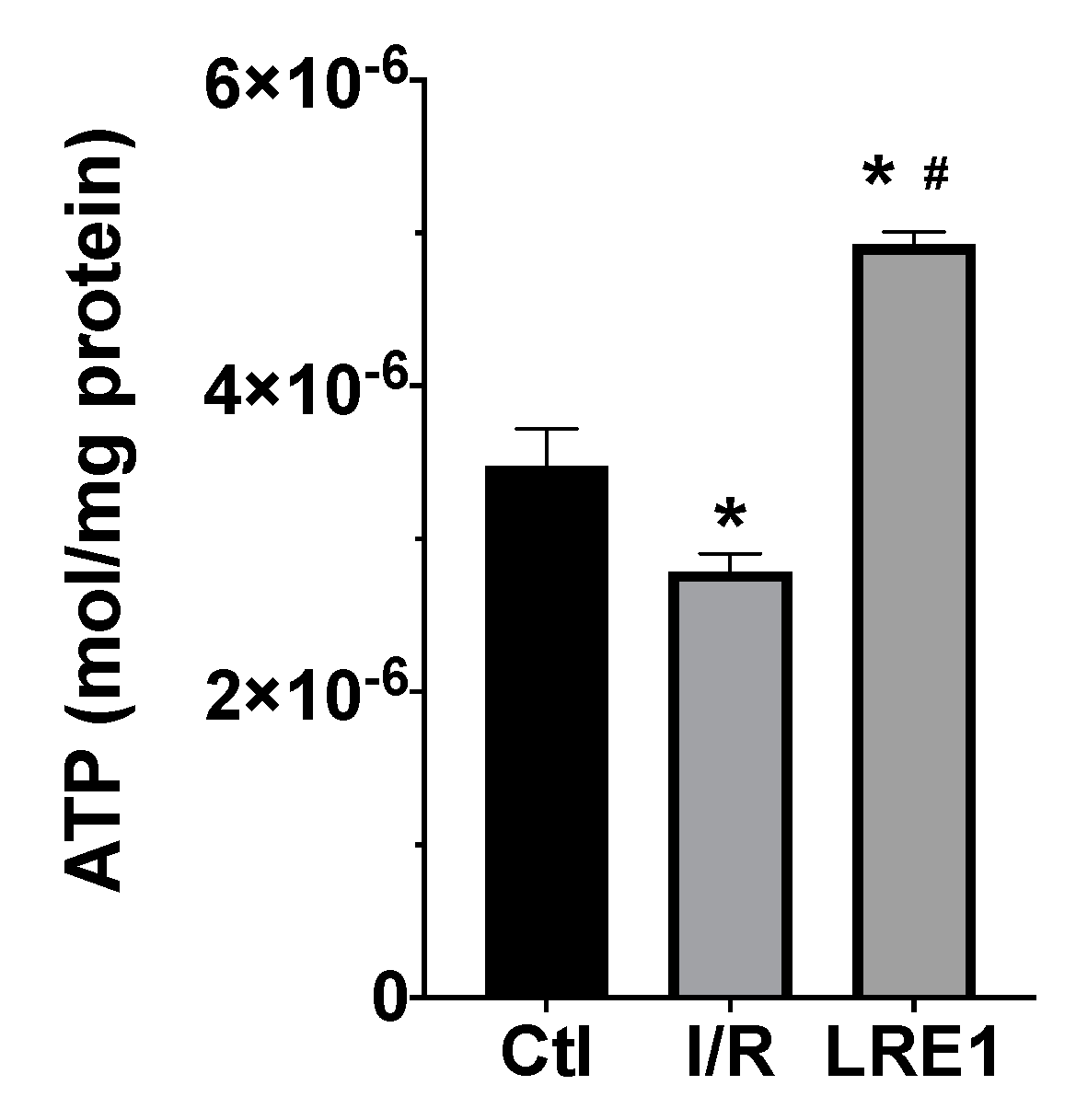
2.1. Hepatic Mitochondrial Bioenergetics
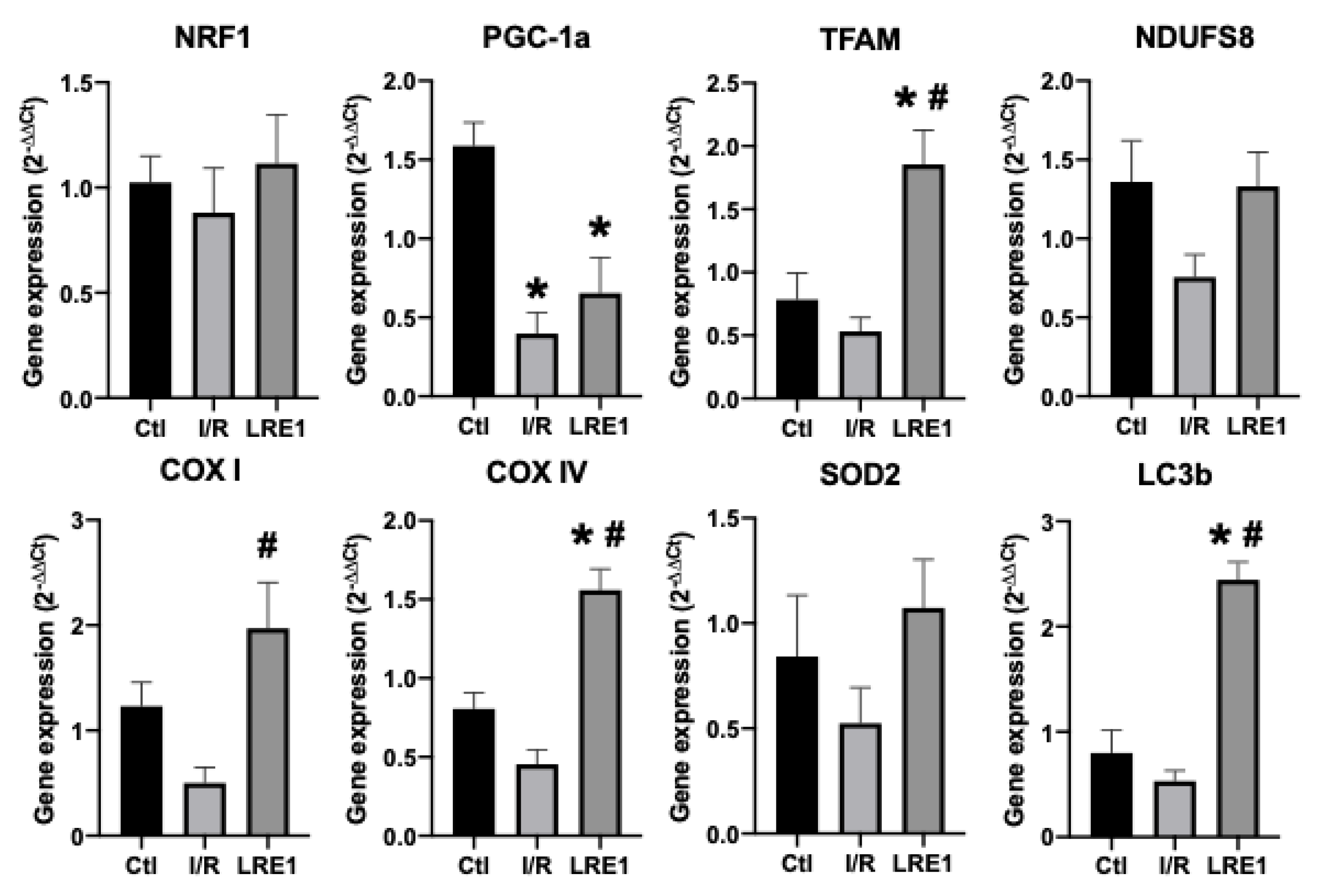
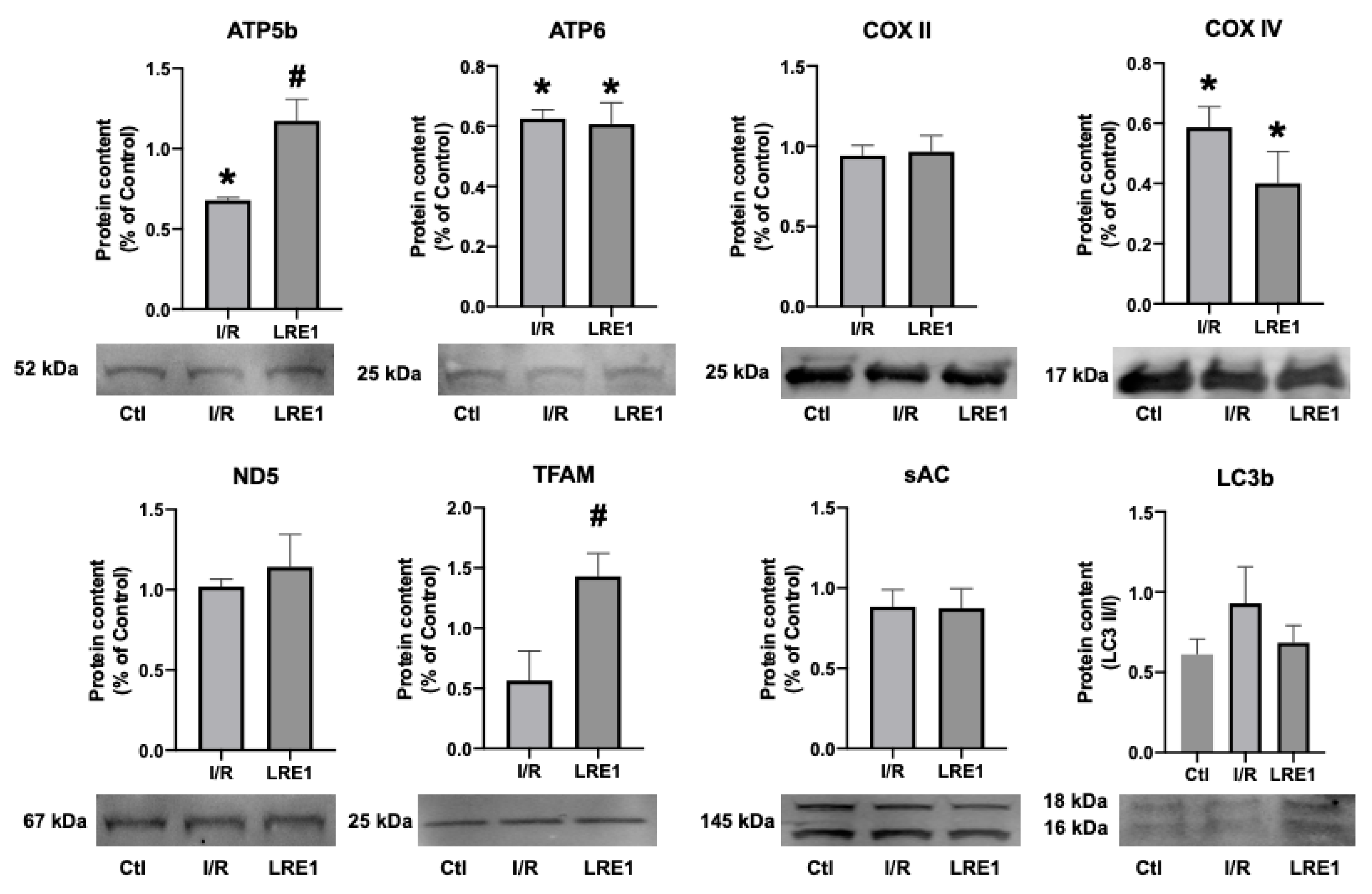
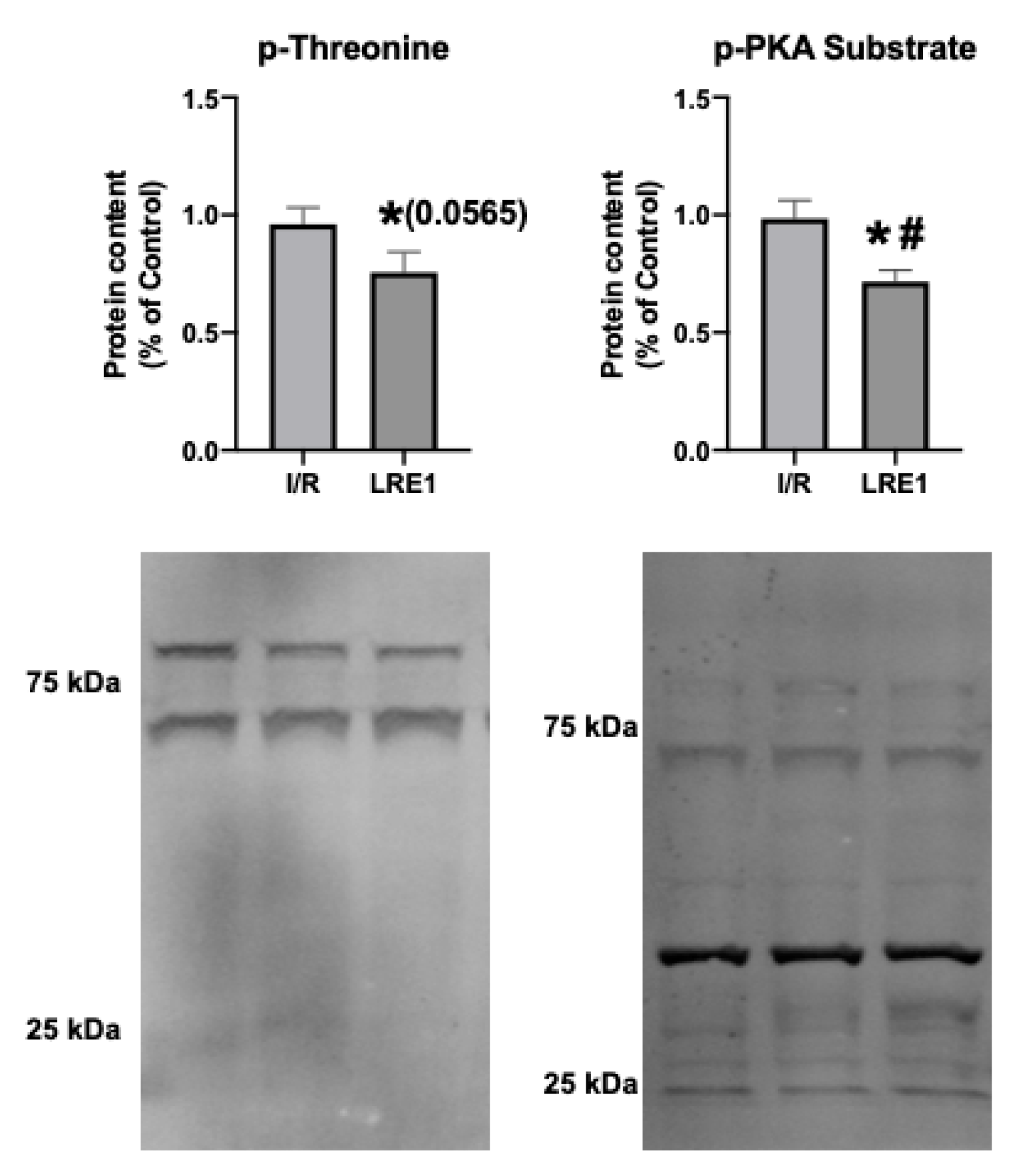
2.2. Gene Expression and Protein Content
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Surgical Induction of Ischemia/Reperfusion
4.4. Mitochondrial Isolation
4.5. Mitochondrial Oxygen Consumption Evaluation
4.6. Mitochondrial Membrane Potential (ΔΨ) Measurement
4.7. Mitochondrial Permeability Transition Recording
4.8. Mitochondrial Reactive Oxygen Species (ROS) Generation Quantification
4.9. Mitochondrial ATP Content Evaluation
4.10. qPCR
4.11. Western Blotting
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martins, R.M.; Pinto Rolo, A.; Soeiro Teodoro, J.; Furtado, E.; Caetano Oliveira, R.; Tralhão, J.G.; Marques Palmeira, C. Addition of Berberine to Preservation Solution in an Animal Model of Ex Vivo Liver Transplant Preserves Mitochondrial Function and Bioenergetics from the Damage Induced by Ischemia/Reperfusion. Int. J. Mol. Sci. 2011, 19, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.M.; Teodoro, J.S.; Furtado, E.; Rolo, A.P.; Palmeira, C.M.; Tralhão, J.G. Recent insights into mitochondrial targeting strategies in liver transplantation. Int. J. Med Sci. 2018, 15, 248. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G15–G26. [Google Scholar] [CrossRef]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Guan, L.-Y. Mechanisms of hepatic ischemia-reperfusion injury and protective effects of nitric oxide. World J. Gastrointest. Surg. 2014, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Alexandrino, H.; Varela, A.T.; Teodoro, J.S.; Martins, M.A.; Rolo, A.P.; Tralhão, J.G.; Palmeira, C.M.; Castro e Sousa, F. Mitochondrial bioenergetics and posthepatectomy liver dysfunction. Eur. J. Clin. Investig. 2016, 46, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Varela, A.T.; Simões, A.M.; Teodoro, J.S.; Duarte, F.V.; Gomes, A.P.; Palmeira, C.M.; Rolo, A.P. Indirubin-3′-oxime prevents hepatic I/R damage by inhibiting GSK-3 [beta] and mitochondrial permeability transition. Mitochondrion 2010, 10, 456–463. [Google Scholar] [CrossRef]
- Varela, A.T.; Rolo, A.P.; Palmeira, C.M. Fatty liver and ischemia/reperfusion: Are there drugs able to mitigate injury? Curr. Med. Chem. 2011, 18, 4987–5002. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Bienengraeber, M.; Stowe, D.F. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2011, 2, 13. [Google Scholar] [CrossRef]
- Jassem, W.; Heaton, N.D. The role of mitochondria in ischemia/reperfusion injury in organ transplantation. Kidney Int. 2004, 66, 514–517. [Google Scholar] [CrossRef]
- Jassem, W.; Fuggle, S.V.; Rela, M.; Koo, D.D.; Heaton, N.D. The role of mitochondria in ischemia/reperfusion injury. Transplantation 2002, 73, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Katwal, G.; Baral, D.; Fan, X.; Weiyang, H.; Zhang, X.; Ling, L.; Xiong, Y.; Ye, Q.; Wang, Y. SIRT3 a major player in attenuation of hepatic ischemia-reperfusion injury by reducing ROS via its downstream mediators: SOD2, CYP-D, and HIF-1a. Oxidative Med. Cell. Longev. 2018, 2018, 2976957. [Google Scholar] [CrossRef] [PubMed]
- Lejay, A.; Meyer, A.; Schlagowski, A.I.; Charles, A.L.; Singh, F.; Bouitbir, J.; Pottecher, J.; Chakfé, N.; Zoll, J.; Geny, B. Mitochondria: Mitochondrial participation in ischemia-reperfusion injury in skeletal muscle. Int. J. Biochem. Cell Biol. 2014, 50, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP Produced inside Mitochondria Regulates Oxidative Phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Gerbino, A.; Lefkimmiatis, K. Shaping mitochondrial dynamics: The role of cAMP signaling. Biochem. Biophys. Res. Commun. 2018, 500, 65–74. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Russwurm, M.; Günnewig, K.; Gertz, M.; Zoidl, G.; Ramos, L.; Buck, J.; Levin, L.R.; Rassow, J.; Manfredi, G.; et al. A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J. Biol. Chem. 2011, 286, 30423–30432. [Google Scholar] [CrossRef]
- Zippin, J.H.; Chen, Y.; Nahirney, P.; Kamenetsky, M.; Wuttke, M.S.; Fischman, D.A.; Levin, L.R.; Buck, J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J. 2003, 17, 82–84. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Varin, A.; Nicolas, V.; Courilleau, D.; Mateo, P.; Caubère, C.; Rouet, P.; Gomez, A.M.; Vandecasteele, G.; et al. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis. 2016, 7, e2198. [Google Scholar] [CrossRef]
- Signorile, A.; Santeramo, A.; Tamma, G.; Pellegrino, T.; D’Oria, S.; Lattanzio, P.; De Rasmo, D. Mitochondrial cAMP prevents apoptosis modulating Sirt3 protein level and OPA1 processing in cardiac myoblast cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 355–366. [Google Scholar] [CrossRef]
- Scher, M.B.; Vaquero, A.; Reinberg, D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; MacK, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under Caloric Restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Buler, M.; Aatsinki, S.M.; Izzi, V.; Hakkola, J. Metformin Reduces Hepatic Expression of SIRT3, the Mitochondrial Deacetylase Controlling Energy Metabolism. PLoS ONE 2012, 7, e49863. [Google Scholar] [CrossRef]
- Anderson, K.A.; Hirschey, M.D. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012, 52, 23–35. [Google Scholar]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Lefkimmiatis, K.; Leronni, D.; Hofer, A.M. The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J. Cell Biol. 2013, 202, 453–462. [Google Scholar] [CrossRef]
- Valsecchi, F.; Konrad, C.; Manfredi, G. Role of soluble adenylyl cyclase in mitochondria. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2555–2560. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Scalzotto, E.; Mongillo, M.; Pozzan, T. Mitochondrial Ca2+ uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab. 2013, 17, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, F.; Konrad, C.; D’Aurelio, M.; Ramos-Espiritu, L.S.; Stepanova, A.; Burstein, S.R.; Galkin, A.; Magranè, J.; Starkov, A.; Buck, J.; et al. Distinct intracellular sAC-cAMP domains regulate ER Ca2+ signaling and OXPHOS function. J. Cell Sci. 2017, 130, 3713–3727. [Google Scholar] [CrossRef]
- Ramos-Espiritu, L.; Kleinboelting, S.; Navarrete, F.A.; Alvau, A.; Visconti, P.E.; Valsecchi, F.; Starkov, A.; Manfredi, G.; Buck, H.; Adura, C.; et al. Discovery of LRE1 as a specific and allosteric inhibitor of soluble adenylyl cyclase. Nat. Chem. Biol. 2016, 12, 838–844. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Isobe, K.; Jung, H.J.; Yang, C.R.; Claxton, J.N.; Sandoval, P.; Burg, M.B.; Raghuram, V.; Knepper, M.A. Systems-level identification of PKA-dependent signaling in epithelial cells. Proc. Natl. Acad. Sci. USA 2017, 114, E8875–E8884. [Google Scholar] [CrossRef]
- Turnham, R.E.; Scott, J.D. Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Gene 2016, 577, 101–108. [Google Scholar] [CrossRef]
- Sun, W.; Liu, C.; Chen, Q.; Liu, N.; Yan, Y.; Liu, B. SIRT3: A new regulator of cardiovascular diseases. Oxidative Med. Cell. Longev. 2018, 2018, 7293861. [Google Scholar] [CrossRef]
- Rossetti, A.; Togliatto, G.; Rolo, A.P.; Teodoro, J.S.; Granata, R.; Ghigo, E.; Columbano, A.; Palmeira, C.M.; Brizzi, M.F. Unacylated ghrelin prevents mitochondrial dysfunction in a model of ischemia/reperfusion liver injury. Cell Death Discov. 2017, 3, 1–11. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Varela, A.T.; Duarte, F.V.; Gomes, A.P.; Palmeira, C.M.; Rolo, A.P. Indirubin and NAD+prevent mitochondrial ischaemia/reperfusion damage in fatty livers. Eur. J. Clin. Investig. 2018, 48, e12932. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.S.; Palmeira, C.M.; Rolo, A.P. Mitochondrial membrane potential (ΔΨ) fluctuations associated with the metabolic states of mitochondria. Methods Mol. Biol. 2018, 1782, 109–119. [Google Scholar]
- Palmeira, C.M.; Moreno, A.J.; Madeira, V.M.C. Interactions of herbicides 2,4-D and dinoseb with liver mitochondrial bioenergetics. Toxicol. Appl. Pharmacol. 1994, 127, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Gornall, A.G.; Bardawill, C.J.; David, M.M. Determination of serum proteins by means of the biuret reaction. J. Biol. Chem. 1949, 177, 751–766. [Google Scholar] [PubMed]
- Rolo, A.P.; Oliveira, P.J.; Moreno, A.J.; Palmeira, C.M. Bile acids affect liver mitochondrial bioenergetics: Possible relevance for cholestasis therapy. Toxicol. Sci. Off. J. Soc. Toxicol. 2000, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G.R. The respiratory chain and oxidative phosphorylation. Adv. Enzymol. Relat. Subj. Biochem. 2006, 17, 65–134. [Google Scholar]
- Kamo, N.; Muratsugu, M.; Hongoh, R.; Kobatake, Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J. Membr. Biol. 1979, 49, 105–121. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Wallace, K.B. Benzoquinone inhibits the voltage-dependent induction of the mitochondrial permeability transition caused by redox-cycling naphthoquinones. Toxicol. Appl. Pharmacol. 1997, 143, 338–347. [Google Scholar] [CrossRef]
- Zhou, S.; Palmeira, C.M.; Wallace, K.B. Doxorubicin-induced persistent oxidative stress to cardiac myocytes. Toxicol. Lett. 2001, 121, 151–157. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI Reagent). Cold Spring Harb. Protoc. 2010, 5, pdb-prot5439. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence | NCBI Nucleotide’s Accession Number |
|---|---|---|
| COX I Forward | CCA GTA TTA GCA GCA GGT ATC | KY_754542.1 |
| COX I Reverse | CCG AAG AAT CAG AAT AGG TGT T | |
| COX IV Forward | GGC AGA ATG TTG GCT ACC | NM_017202.1 |
| COX IV Reverse | GCA TAG TCT TCA CTC TTC ACA A | |
| LC3b Forward | CTT CAG GTG TGC AAT GCT GG | NM_022867.2 |
| LC3b Reverse | TGG CTC TCT TCC TGT TGC TG | |
| NDUFS8 Forward | AGT GTA TCT ACT GTG GTT | NM_001106322.2 |
| NDUFS8 Reverse | TAG CTT CTC CTT GTT GTA | |
| NRF1 Forward | GGA TTC ATT ATG GCG GAA GTA A | NM_001100708.1 |
| NRF1 Reverse | AGT TGC TGT GGC GAG TTA | |
| PGC-1α Forward | CTG CTC TTG AGA ATG GAT ATA CTT | NM_031347.1 |
| PGC-1α Reverse | CATACT TGC TCT TGG TGG AA | |
| SOD2 Forward | CAC TGT GGC TGA GCT GTT GT | NM_017051.2 |
| SOD2 Reverse | TCC AAG CAA TTC AAG CCT CT | |
| TFAM Forward | AAA TGG CTG AAG TTG GGC GAA GTG | BC062022.1 |
| TFAM Reverse | AGC TTC TTG TGC CCA ATC CCA ATG | |
| 18S Forward | GTA ACC CGT TGA ACC CCA TT | NR_046239.1 |
| 18S Reverse | CCA TCC AAT CGG TAG TAG CG |
| Antibody Name | Source Host | Supplier | Reference Number# |
|---|---|---|---|
| Acetylated lysines | Rabbit | Cell Signaling Technologies | 9441 |
| ADCY10/sAC | Rabbit | Abcam | Ab82854 |
| ATP5B | Rabbit | Aviva Systems Biology | ARP48185 |
| ATP6 | Rabbit | LifeSpan BioSciences | LS-C352532 |
| COX II | Rabbit | LifeSpan BioSciences | LS-C330986 |
| COX IV | Rabbit | Thermo-Fisher | PA5-19471 |
| ND5 | Rabbit | LifeSpan BioSciences | LS-C368770 |
| LC3B | Rabbit | Sigma-Aldrich | L7543 |
| p-PKA substrates | Rabbit | Cell Signaling Technologies | 9624 |
| SIRT3 | Rabbit | Cell Signaling Technologies | 2627 |
| TFAM | Rabbit | Aviva Systems Biology | ARP36993 |
| p-threonine | Mouse | Qiagen | W10132 |
| Biotin-XX, antimouse IgG | Goat | Thermo Fisher | W10132 |
| Biotin-XX, antirabbit IgG | Goat | Thermo Fisher | W10142 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teodoro, J.S.; Amorim, J.A.; Machado, I.F.; Castela, A.C.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P.; Palmeira, C.M. The Soluble Adenylyl Cyclase Inhibitor LRE1 Prevents Hepatic Ischemia/Reperfusion Damage Through Improvement of Mitochondrial Function. Int. J. Mol. Sci. 2020, 21, 4896. https://doi.org/10.3390/ijms21144896
Teodoro JS, Amorim JA, Machado IF, Castela AC, Steegborn C, Sinclair DA, Rolo AP, Palmeira CM. The Soluble Adenylyl Cyclase Inhibitor LRE1 Prevents Hepatic Ischemia/Reperfusion Damage Through Improvement of Mitochondrial Function. International Journal of Molecular Sciences. 2020; 21(14):4896. https://doi.org/10.3390/ijms21144896
Chicago/Turabian StyleTeodoro, João S., João A. Amorim, Ivo F. Machado, Ana C. Castela, Clemens Steegborn, David A. Sinclair, Anabela P. Rolo, and Carlos M. Palmeira. 2020. "The Soluble Adenylyl Cyclase Inhibitor LRE1 Prevents Hepatic Ischemia/Reperfusion Damage Through Improvement of Mitochondrial Function" International Journal of Molecular Sciences 21, no. 14: 4896. https://doi.org/10.3390/ijms21144896
APA StyleTeodoro, J. S., Amorim, J. A., Machado, I. F., Castela, A. C., Steegborn, C., Sinclair, D. A., Rolo, A. P., & Palmeira, C. M. (2020). The Soluble Adenylyl Cyclase Inhibitor LRE1 Prevents Hepatic Ischemia/Reperfusion Damage Through Improvement of Mitochondrial Function. International Journal of Molecular Sciences, 21(14), 4896. https://doi.org/10.3390/ijms21144896