Phosphorylation of the Chaperone-Like HspB5 Rescues Trafficking and Function of F508del-CFTR

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
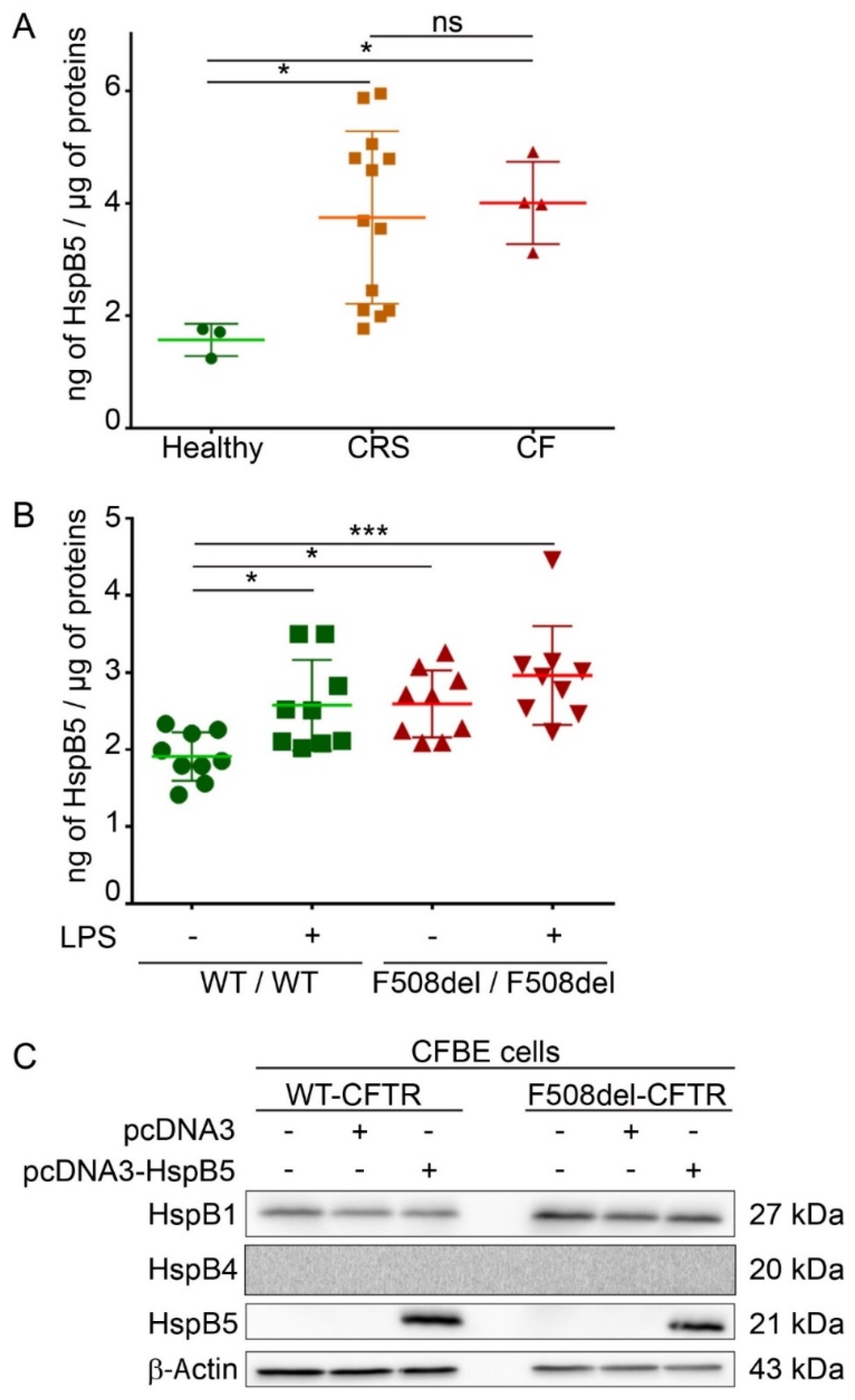
2.1. Endogenous Expression of HspB5 Protein in Different Cystic Fibrosis Models
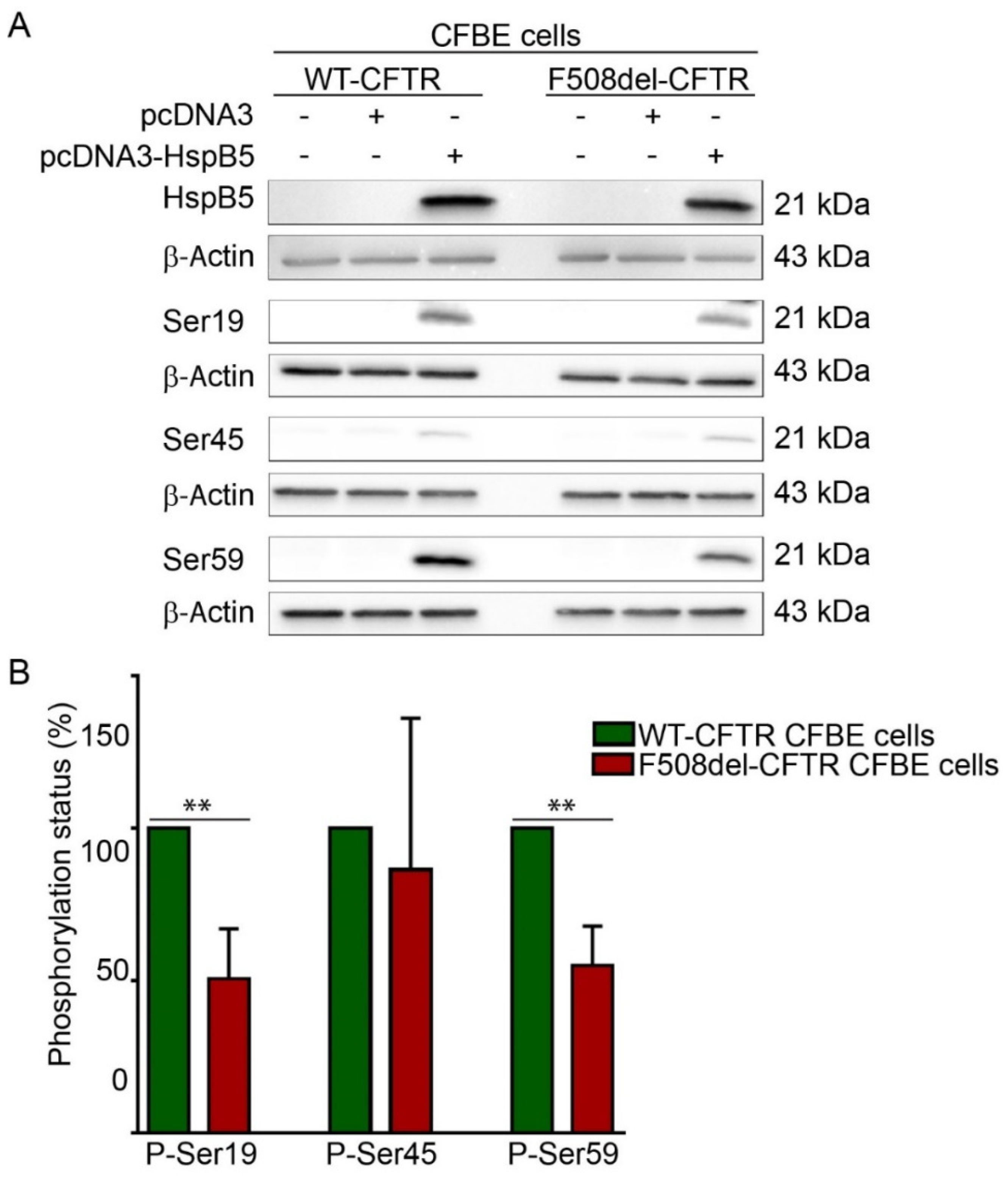
2.2. Phosphorylation Pattern of Overexpressed HspB5 is Altered in F508del-CFBE Cell Line Compared to WT-CFBE Cell Line
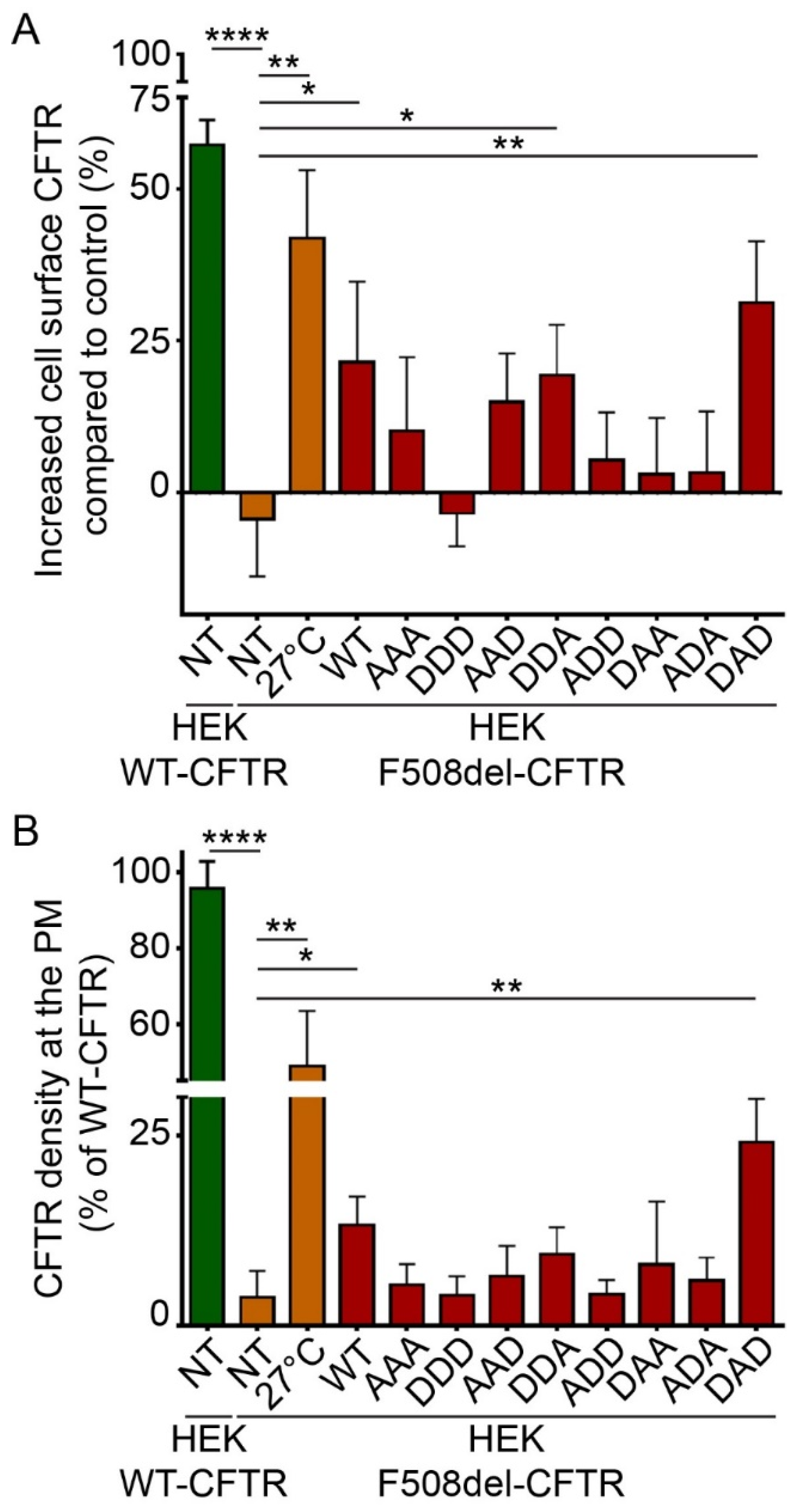
2.3. Phosphorylation-Dependent Ability of HspB5 to Rescue the Plasma Membrane Localization of F508del-CFTR
2.4. DAD-HspB5 Increases F508del-CFTR Stability
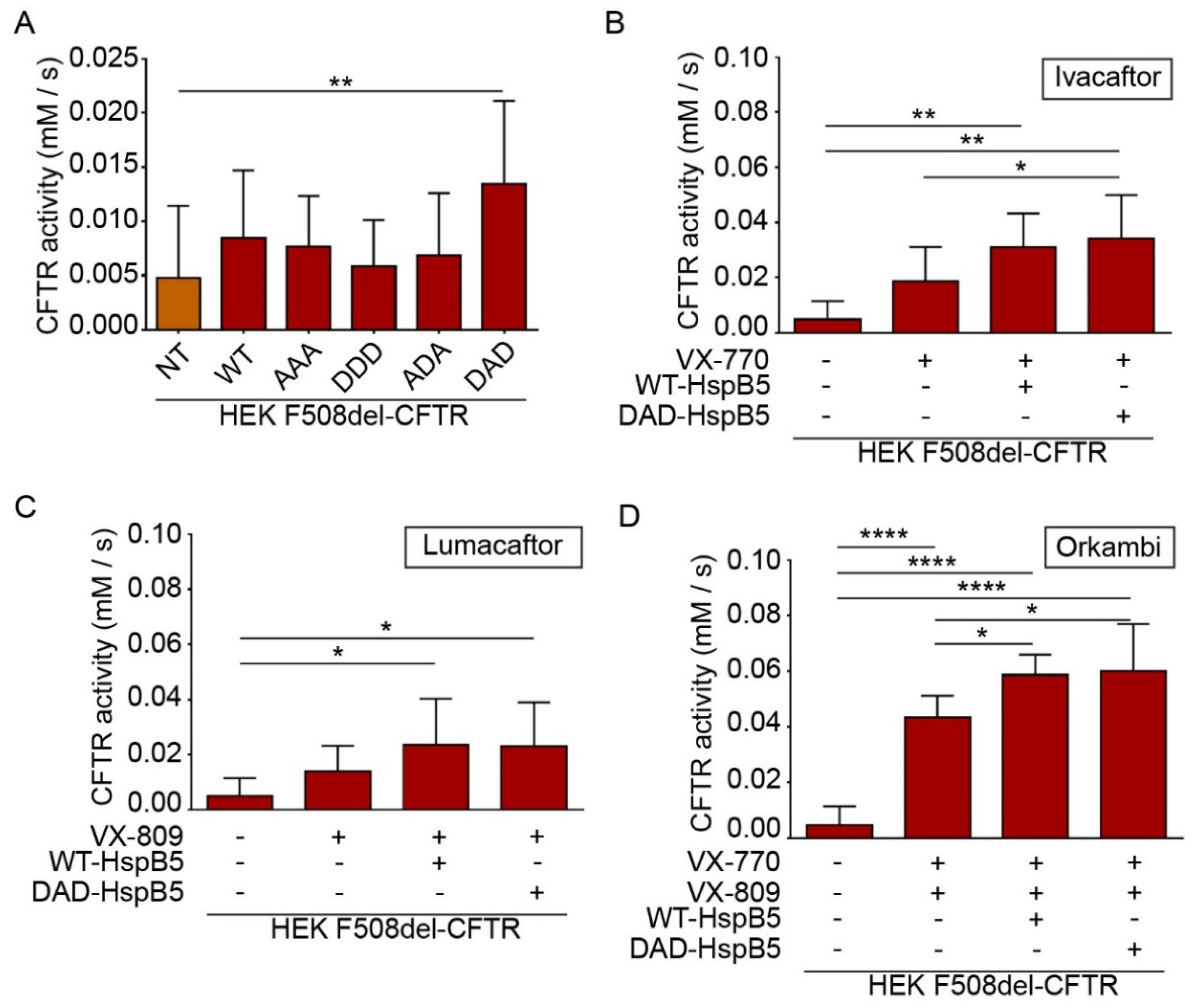
2.5. HspB5 and Its Phosphomimetic DAD Mutant Increase Halide Transport and Act Additively with Ivacaftor, Lumacaftor, and Orkambi Treatments
3. Discussion
4. Material and Methods
4.1. Cell Lines
4.2. Human Nasal Epithelial Cells
4.3. Mouse Lung Samples
4.4. Vector Constructs and Transfection
4.5. Immunofluorescence Confocal Microscopy
4.6. Total Cellular Protein Extraction and Western-Blotting
4.7. HspB5 ELISA Assay
4.8. ELISA-based Assay to Measure CFTR at the PM
4.9. Cell Surface Density Measurement of CFTR-3HA
4.10. YFP Halide-Exchange-Assay
4.11. Pulse (CHX)-Chase
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ellgaard, L.; Helenius, A. Quality Control in the Endoplasmic Reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef]
- Janovjak, H.; Struckmeier, J.; Hubain, M.; Kedrov, A.; Kessler, M.; Muller, D.J. Probing the Energy Landscape of the Membrane Protein Bacteriorhodopsin. Structure 2004, 12, 871–879. [Google Scholar] [CrossRef]
- Hung, M.C.; Link, W. Protein Localization in Disease and Therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef]
- Collins, F.S. Cystic Fibrosis: Molecular Biology and Therapeutic Implications. Science 1992, 256, 774–779. [Google Scholar] [CrossRef]
- Riordan, J.R. Cftr Function and Prospects for Therapy. Annu. Rev. Biochem. 2008, 77, 701–726. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.D.; Canato, S.; Carvalho, A.S.; Botelho, H.M.; Aloria, K.; Amaral, M.D.; Matthiesen, R.; Falcao, A.O.; Farinha, C.M. Folding Status Is Determinant over Traffic-Competence in Defining Cftr Interactors in the Endoplasmic Reticulum. Cells 2019, 8, 353. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Okiyoneda, T. Peripheral Protein Quality Control as a Novel Drug Target for Cftr Stabilizer. Front. Pharmacol. 2018, 9, 1100. [Google Scholar] [CrossRef]
- Yang, Y.; Janich, S.; Cohn, J.A.; Wilson, J.M. The Common Variant of Cystic Fibrosis Transmembrane Conductance Regulator Is Recognized by Hsp70 and Degraded in a Pre-Golgi Nonlysosomal Compartment. Proc. Natl. Acad. Sci. USA 1993, 90, 9480–9484. [Google Scholar] [CrossRef]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 Co-Chaperone Chip Targets Immature Cftr for Proteasomal Degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Nogueira, P.; Mendes, F.; Penque, D.; Amaral, M.D. The Human Dnaj Homologue (Hdj)-1/Heat-Shock Protein (Hsp) 40 Co-Chaperone Is Required for the in Vivo Stabilization of the Cystic Fibrosis Transmembrane Conductance Regulator by Hsp70. Biochem. J. 2002, 366, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Bagdany, M.; Veit, G.; Fukuda, R.; Avramescu, R.G.; Okiyoneda, T.; Baaklini, I.; Singh, J.; Sovak, G.; Xu, H.; Apaja, P.M. Chaperones Rescue the Energetic Landscape of Mutant Cftr at Single Molecule and in Cell. Nat. Commun. 2017, 8, 398. [Google Scholar] [CrossRef]
- Ahner, A.; Nakatsukasa, K.; Zhang, H.; Frizzell, R.A.; Brodsky, J.L. Small Heat-Shock Proteins Select Deltaf508-Cftr for Endoplasmic Reticulum-Associated Degradation. Mol. Biol. Cell 2007, 18, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Liao, Y.; Ahner, A.; Larsen, M.B.; Wang, X.; Bertrand, C.A.; Frizzell, R.A. Different Sumo Paralogues Determine the Fate of Wild-Type and Mutant Cftrs: Biogenesis Versus Degradation. Mol. Biol. Cell 2019, 30, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Combination of Correctors Rescue Deltaf508-Cftr by Reducing Its Association with Hsp40 and Hsp27. J. Biol. Chem. 2015, 290, 25636–25645. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Sabirzhanova, I.; Rapino, D.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Correctors Rescue Cftr Mutations in Nucleotide-Binding Domain 1 (Nbd1) by Modulating Proteostasis. Chembiochem 2016, 17, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.P.; Nagineni, C.N. Alpha B Subunit of Lens-Specific Protein Alpha-Crystallin Is Present in Other Ocular and Non-Ocular Tissues. Biochem. Biophys. Res. Commun. 1989, 158, 319–325. [Google Scholar] [CrossRef]
- Iwaki, T.; Kume-Iwaki, A.; Goldman, J.E. Cellular Distribution of Alpha B-Crystallin in Non-Lenticular Tissues. J. Histochem. Cytochem. 1990, 38, 31–39. [Google Scholar] [CrossRef]
- D’Agostino, M.; Lemma, V.; Chesi, G.; Stornaiuolo, M.; Serio, M.C.; D’Ambrosio, C.; Scaloni, A.; Polishchuk, R.; Bonatti, S. The Cytosolic Chaperone Alpha-Crystallin B Rescues Folding and Compartmentalization of Misfolded Multispan Transmembrane Proteins. J. Cell Sci. 2013, 126, 4160–4172. [Google Scholar] [CrossRef]
- Ciano, M.; Allocca, S.; Ciardulli, M.C.; della Volpe, L.; Bonatti, S.; D’Agostino, M. Differential Phosphorylation-Based Regulation of Alphab-Crystallin Chaperone Activity for Multipass Transmembrane Proteins. Biochem. Biophys. Res. Commun. 2016, 479, 325–330. [Google Scholar] [CrossRef]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Bel, N.S.; Grenet, D. Correction of Cftr Function in Nasal Epithelial Cells from Cystic Fibrosis Patients Predicts Improvement of Respiratory Function by Cftr Modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Fontaine, J.M.; Martin, J.L.; Sun, X.; Hoppe, A.D.; Welsh, M.J.; Benndorf, R.; Vicart, P. Myopathy-Associated Alphab-Crystallin Mutants: Abnormal Phosphorylation, Intracellular Location, and Interactions with Other Small Heat Shock Proteins. J. Biol. Chem. 2007, 282, 34276–34287. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.S.; Moyer, B.D.; Bannykh, S.; Yoo, H.M.; Riordan, J.R.; Balch, W.E. Non-Conventional Trafficking of the Cystic Fibrosis Transmembrane Conductance Regulator through the Early Secretory Pathway. J. Biol. Chem. 2002, 277, 11401–11409. [Google Scholar] [CrossRef]
- Peters, K.W.; Okiyoneda, T.; Balch, W.E.; Braakman, I.; Brodsky, J.L.; Guggino, W.B.; Penland, C.M.; Pollard, H.B.; Sorscher, E.J.; Skach, W.R. Cftr Folding Consortium: Methods Available for Studies of Cftr Folding and Correction. Methods Mol. Biol. 2011, 742, 335–353. [Google Scholar] [PubMed]
- Trzcinska-Daneluti, A.M.; Chen, A.; Nguyen, L.; Murchie, R.; Jiang, C.; Moffat, J.; Pelletier, L.; Rotin, D. Rna Interference Screen to Identify Kinases That Suppress Rescue of Deltaf508-Cftr. Mol. Cell. Proteomics 2015, 14, 1569–1583. [Google Scholar] [CrossRef]
- Ahner, A.; Gong, X.; Schmidt, B.Z.; Peters, K.W.; Rabeh, W.M.; Thibodeau, P.H.; Lukacs, G.L.; Frizzell, R.A. Small Heat Shock Proteins Target Mutant Cystic Fibrosis Transmembrane Conductance Regulator for Degradation Via a Small Ubiquitin-Like Modifier-Dependent Pathway. Mol. Biol. Cell 2013, 24, 74–84. [Google Scholar] [CrossRef]
- Galietta, L.V.; Jayaraman, S.; Verkman, A.S. Cell-Based Assay for High-Throughput Quantitative Screening of Cftr Chloride Transport Agonists. Am. J. Physiol. Cell Physiol. 2001, 281, C1734–C1742. [Google Scholar] [CrossRef]
- Pedemonte, N.; Zegarra-Moran, O.; Galietta, L.J. High-Throughput Screening of Libraries of Compounds to Identify Cftr Modulators. Methods Mol. Biol. 2011, 741, 13–21. [Google Scholar] [PubMed]
- Rhoden, K.J.; Cianchetta, S.; Stivani, V.; Portulano, C.; Galietta, L.J.; Romeo, G. Cell-Based Imaging of Sodium Iodide Symporter Activity with the Yellow Fluorescent Protein Variant Yfp-H148q/I152l. Am. J. Physiol. Cell Physio.l 2007, 292, C814–C823. [Google Scholar] [CrossRef] [PubMed]
- Eckford, P.D.W.; McCormack, J.; Munsie, L.; He, G.; Stanojevic, S.; Pereira, S.L.; Ho, K.; Avolio, J.; Bartlett, C.; Yang, J.Y. The Cf Canada-Sick Kids Program in Individual Cf Therapy: A Resource for the Advancement of Personalized Medicine in Cf. J. Cyst. Fibros. 2019, 18, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Laskowska, E.; Matuszewska, E.; Kuczynska-Wisnik, D. Small Heat Shock Proteins and Protein-Misfolding Diseases. Curr. Pharm. Biotechnol. 2010, 11, 146–157. [Google Scholar] [CrossRef]
- Zhu, Z.; Reiser, G. The Small Heat Shock Proteins, Especially Hspb4 and Hspb5 Are Promising Protectants in Neurodegenerative Diseases. Neurochem. Int. 2018, 115, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Wu, J.; Li, W.; Chen, Z.; Yang, L. Progression of the Role of Cryab in Signaling Pathways and Cancers. Onco Targets Ther. 2019, 12, 4129. [Google Scholar] [CrossRef]
- Arrigo, A.P.; Gibert, B. Hspb1, Hspb5 and Hspb4 in Human Cancers: Potent Oncogenic Role of Some of Their Client Proteins. Cancers (Basel) 2014, 6, 333–365. [Google Scholar] [CrossRef]
- Simon, S.; Arrigo, A.P. Beneficial and Deleterious, the Dual Role of Shsps in Human Diseases: Implications for Therapeutic Strategies. In Small Stress Proteins and Human Diseases; Simon, S., Arrigo, A.P., Eds.; Nova Science Publishers Inc.: Hauppauge Smithtown, NY, USA, 2011. [Google Scholar]
- Arrigo, A.P.; Simon, S.; Gibert, B.; Kretz-Remy, C.; Nivon, M.; Czekalla, A.; Guillet, D.; Moulin, M.; Diaz-Latoud, C.; Vicart, P. Hsp27 (Hspb1) and Alphab-Crystallin (Hspb5) as Therapeutic Targets. FEBS Lett. 2007, 581, 3665–3674. [Google Scholar] [CrossRef] [PubMed]
- Bakthisaran, R.; Tangirala, R.; Ch, M.R. Small Heat Shock Proteins: Role in Cellular Functions and Pathology. Biochim. Biophys. Acta 2015, 1854, 291–319. [Google Scholar] [CrossRef]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in Cystic Fibrosis: An Update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Guo, Y.; Huang, Z.; Zhao, H.; Zhou, M.; Huang, Y.; Wen, D.; Song, J.; Zhu, Z.; Sun, M. Small Heat Shock Protein Cryab Inhibits Intestinal Mucosal Inflammatory Responses and Protects Barrier Integrity through Suppressing Ikkbeta Activity. Mucosal Immunol. 2019, 12, 1291–1303. [Google Scholar] [CrossRef]
- Van Noort, J.M.; Bsibsi, M.; Nacken, P.J.; Gerritsen, W.H.; Amor, S.; Holtman, I.R.; Boddeke, E.; van Ark, I.; Leusink-Muis, T.; Folkerts, G. Activation of an Immune-Regulatory Macrophage Response and Inhibition of Lung Inflammation in a Mouse Model of Copd Using Heat-Shock Protein Alpha B-Crystallin-Loaded Plga Microparticles. Biomaterials 2013, 34, 831–840. [Google Scholar] [CrossRef]
- Van Noort, J.M.; Bsibsi, M.; Nacken, P.J.; Verbeek, R.; Venneker, E.H. Therapeutic Intervention in Multiple Sclerosis with Alpha B-Crystallin: A Randomized Controlled Phase Iia Trial. PLoS ONE 2015, 10, e0143366. [Google Scholar] [CrossRef]
- Fung, G.; Wong, J.; Berhe, F.; Mohamud, Y.; Xue, Y.C.; Luo, H. Phosphorylation and Degradation of Alphab-Crystallin During Enterovirus Infection Facilitates Viral Replication and Induces Viral Pathogenesis. Oncotarget 2017, 8, 74767–74780. [Google Scholar] [CrossRef] [PubMed]
- Svedin, E.; Utorova, R.; Huhn, M.H.; Larsson, P.G.; Stone, V.M.; Garimella, M.; Lind, K.; Hagglof, T.; Pincikova, T.; Laitinen, O.H. A Link between a Common Mutation in Cftr and Impaired Innate and Adaptive Viral Defense. J. Infect. Dis. 2017, 216, 1308–1317. [Google Scholar] [CrossRef]
- Gruenert, D.C.; Willems, M.; Cassiman, J.J.; Frizzell, R.A. Established Cell Lines Used in Cystic Fibrosis Research. J. Cyst. Fibros. 2004, 3 (Suppl. 2), 191–196. [Google Scholar] [CrossRef]
- Illek, B.; Maurisse, R.; Wahler, L.; Kunzelmann, K.; Fischer, H.; Gruenert, D.C. Cl Transport in Complemented Cf Bronchial Epithelial Cells Correlates with Cftr Mrna Expression Levels. Cell. Physiol. Biochem. 2008, 22, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Schwiebert, E.M.; Zeitlin, P.L.; Kuo, W.L.; Stanton, B.A.; Gruenert, D.C. An Immortalized Cystic Fibrosis Tracheal Epithelial Cell Line Homozygous for the Delta F508 Cftr Mutation. Am. J. Respir. Cell Mol. Biol. 1993, 8, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Rocca, J.; Manin, S.; Hulin, A.; Aissat, A.; Verbecq-Morlot, W.; Pruliere-Escabasse, V.; Wohlhuter-Haddad, A.; Epaud, R.; Fanen, P.; Tarze, A. New Use for an Old Drug: Cox-Independent Anti-Inflammatory Effects of Sulindac in Models of Cystic Fibrosis. Br. J. Pharmacol. 2016, 173, 1728–1741. [Google Scholar] [CrossRef]
- Van Doorninck, J.H.; French, P.J.; Verbeek, E.; Peters, R.H.; Morreau, H.; Bijman, J.; Scholte, B.J. A Mouse Model for the Cystic Fibrosis Delta F508 Mutation. EMBO J. 1995, 14, 4403–4411. [Google Scholar] [CrossRef] [PubMed]
- Rogalla, T.; Ehrnsperger, M.; Preville, X.; Kotlyarov, A.; Lutsch, G.; Ducasse, C.; Paul, C.; Wieske, M.; Arrigo, A.P.; Buchner, J. Regulation of Hsp27 Oligomerization, Chaperone Function, and Protective Activity against Oxidative Stress/Tumor Necrosis Factor Alpha by Phosphorylation. J. Biol. Chem. 1999, 274, 18947–18956. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.J.; Zijlstra, M.P.; Kanon, B.; van Waarde-Verhagen, M.A.; Brunt, E.R.; Oosterveld-Hut, H.M.; Carra, S.; Sibon, O.C.; Kampinga, H.H. Hspb7 Is the Most Potent Polyq Aggregation Suppressor within the Hspb Family of Molecular Chaperones. Hum. Mol. Genet. 2010, 19, 4677–4693. [Google Scholar] [CrossRef]
- Fanen, P.; Clain, J.; Labarthe, R.; Hulin, P.; Girodon, E.; Pagesy, P.; Goossens, M.; Edelman, A. Structure-Function Analysis of a Double-Mutant Cystic Fibrosis Transmembrane Conductance Regulator Protein Occurring in Disorders Related to Cystic Fibrosis. FEBS Lett. 1999, 452, 371–374. [Google Scholar] [CrossRef]
- Caci, E.; Caputo, A.; Hinzpeter, A.; Arous, N.; Fanen, P.; Sonawane, N.; Verkman, A.S.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. Evidence for Direct Cftr Inhibition by Cftr(Inh)-172 Based on Arg347 Mutagenesis. Biochem. J. 2008, 413, 135–142. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degrugillier, F.; Aissat, A.; Prulière-Escabasse, V.; Bizard, L.; Simonneau, B.; Decrouy, X.; Jiang, C.; Rotin, D.; Fanen, P.; Simon, S. Phosphorylation of the Chaperone-Like HspB5 Rescues Trafficking and Function of F508del-CFTR. Int. J. Mol. Sci. 2020, 21, 4844. https://doi.org/10.3390/ijms21144844
Degrugillier F, Aissat A, Prulière-Escabasse V, Bizard L, Simonneau B, Decrouy X, Jiang C, Rotin D, Fanen P, Simon S. Phosphorylation of the Chaperone-Like HspB5 Rescues Trafficking and Function of F508del-CFTR. International Journal of Molecular Sciences. 2020; 21(14):4844. https://doi.org/10.3390/ijms21144844
Chicago/Turabian StyleDegrugillier, Fanny, Abdel Aissat, Virginie Prulière-Escabasse, Lucie Bizard, Benjamin Simonneau, Xavier Decrouy, Chong Jiang, Daniela Rotin, Pascale Fanen, and Stéphanie Simon. 2020. "Phosphorylation of the Chaperone-Like HspB5 Rescues Trafficking and Function of F508del-CFTR" International Journal of Molecular Sciences 21, no. 14: 4844. https://doi.org/10.3390/ijms21144844
APA StyleDegrugillier, F., Aissat, A., Prulière-Escabasse, V., Bizard, L., Simonneau, B., Decrouy, X., Jiang, C., Rotin, D., Fanen, P., & Simon, S. (2020). Phosphorylation of the Chaperone-Like HspB5 Rescues Trafficking and Function of F508del-CFTR. International Journal of Molecular Sciences, 21(14), 4844. https://doi.org/10.3390/ijms21144844