The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome



Abstract
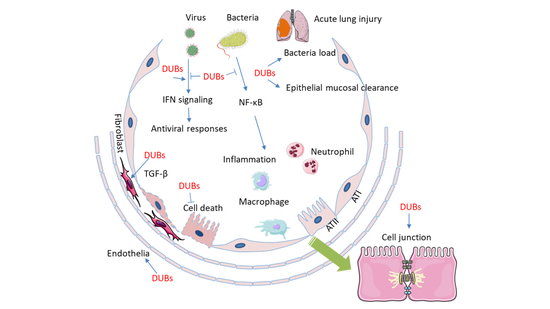
1. Introduction
2. Protein Ubiquitin Proteasomal Degradation and Deubiquitination
3. Molecular Mechanisms of DUBs in the Pathogenesis of ALI/ARDS
4. Deubiquitinating Enzymes Involved in ALI/ARDS
4.1. USPs
4.2. OTUs
4.3. JAMMs
4.4. OTHER DUBs
5. Potential Therapeutic Approaches Targeting DUBS in ALI/ARDS
6. Conclusions and Future Perspectives
Funding
Conflicts of Interest
Abbreviations
| ABRO1 | Abraxas Brother 1 |
| ALI | Acute lung injury |
| ALKBH3 | AlkB homologue 3 |
| AMSH | Anti-Müllerian hormone |
| ARDS | Acute respiratory distress syndrome |
| ASC | Apeck-like protein containing a CARD |
| ATXN3 | Ataxin3 |
| BRISC | BRCC36 isopeptidase complex |
| CBP | CREB-binding protein |
| cIAP-1 | Cellular inhibitor of apoptosis protein-1 |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CSN5 | COP9 signalosome 5 |
| COVID-19 | Coronavirus disease 2019 |
| CYLD | Cylindromatosis |
| DUBs | Deubiquitinating enzymes |
| E2F1 | E2F transcription factor 1 |
| EIF3 | Eukaryotic translation initiation factor 3 |
| HBO1 | Histone acetyltransferase binding to origin recognition complex 1 |
| HDAC2 | Histone deacetylase 2 |
| IFN | Interferon |
| IKKγ | IκB kinase γ |
| IL-1β | Interlukin-1β |
| IRF3 | Interferon regulatory factor 3 |
| JAMMs | Zn-JAB1/MPN/MOV34 domain metallopeptidase |
| LPA1 | Lysophosphatidic acid receptor 1 |
| LPS | Lipopolysaccharide |
| LUBAC | Linear ubiquitin chain assembly complex |
| MAVS | Mitochondria antiviral-signaling protein |
| MCL1 | Myeloid cell leukemia 1 |
| MINDYs | Motif interacting with ubiquitin - containing novel DUB family |
| MJDs | Machado-Josephin disease protein domain protease |
| NEMO | Nuclear factor (NF)-κB essential modulator |
| NALP7 | NACHT, LRR and PYD domains-containing protein 7 |
| NFAT | Nuclear factor of activated T cells |
| NICD1 | NOTCH1 intracellular domain |
| NLRP3 | NLR family pyrin domain containing 3 |
| MYSM1 | MPN domain containing (MPND, myb-like SWIRM and MPN domains 1 |
| OTUs | Ovarian tumor proteases |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PEG10 | Paternally expressed gene 10 |
| PLY | Pneumolysin |
| POH1 | Proteasome non-ATPase regulatory subunit 14 |
| PRPF8 | Pre-mRNA-processing-splicing factor 8 |
| PSMD7 | Proteasome non-ATPase regulatory subunit 7 |
| RIG-1 | Retinoic acid-inducible gene I |
| RIPK1 | Receptor-interacting protein kinase 1 |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| STAMBP | STAM-binding protein |
| STING | Stimulator of interferon |
| TAK1 | TGF-β-activated kinase 1 |
| TGFβ-1 | Transforming growth factor β-1 |
| TRIF | TIR domain-containing adaptor inducing interferon-β |
| TNF-α | Tumor necrosis factor-α |
| TRAF | Tumor necrosis factor receptor-associated factor |
| UBA | Ub-activating enzymes |
| UBC | Ub-conjugating enzymes |
| UCHs | Ubiquitin carboxy-terminal hydrolases |
| USPs | Ubiquitin-specific proteases |
References
- Han, S.; Mallampalli, R.K. The acute respiratory distress syndrome: From mechanism to translation. J. Immunol 2015, 194, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar]
- Matthay, M.A.; Song, Y.; Bai, C.; Jones, K.D. The acute respiratory distress syndrome in 2013. Transl. Respir. Med. 2013, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Confalonieri, M.; Salton, F.; Fabiano, F. Acute respiratory distress syndrome. Eur. Respir. Rev. 2017, 26, 160116. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Herold, S.; Gabrielli, N.M.; Vadasz, I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L665–L681. [Google Scholar] [CrossRef]
- Dada, L.A.; Sznajder, J.I. Mechanisms of pulmonary edema clearance during acute hypoxemic respiratory failure: Role of the Na,K-ATPase. Crit. Care Med. 2003, 31 (Suppl. 4), S248–S252. [Google Scholar] [CrossRef]
- Garcia, J.G.; Sznajder, J.I. Healthcare disparities in patients with acute respiratory distress syndrome. Toward Equity. Am. J. Respir Crit. Care Med. 2013, 188, 631–632. [Google Scholar] [CrossRef]
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early local immune defences in the respiratory tract. Nat. Rev. Immunol. 2017, 17, 7–20. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol 2011, 6, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Nuckton, T.J.; Alonso, J.A.; Kallet, R.H.; Daniel, B.M.; Pittet, J.F.; Eisner, M.D.; Matthay, M.A. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N. Engl. J. Med. 2002, 346, 1281–1286. [Google Scholar] [CrossRef]
- Magnani, N.D.; Dada, L.A.; Sznajder, J.I. Ubiquitin-proteasome signaling in lung injury. Transl. Res. 2018, 198, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, I.; Weiss, C.H.; Sznajder, J.I. Ubiquitination and proteolysis in acute lung injury. Chest 2012, 141, 763–771. [Google Scholar] [CrossRef][Green Version]
- Helenius, I.T.; Dada, L.A.; Sznajder, J.I. Role of ubiquitination in Na,K-ATPase regulation during lung injury. Proc. Am. Thorac. Soc. 2010, 7, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Xu, C.; Kim, N.G.; Gumbiner, B.M. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 2009, 8, 4032–4039. [Google Scholar] [CrossRef]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–690. [Google Scholar] [CrossRef]
- Weathington, N.M.; Sznajder, J.I.; Mallampalli, R.K. The emerging role of the ubiquitin proteasome in pulmonary biology and disease. Am. J. Respir Crit. Care Med. 2013, 188, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.D. Ubiquitination and deubiquitination: Targeting of proteins for degradation by the proteasome. Semin. Cell Dev. Biol. 2000, 11, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating enzymes in cellular signaling and disease regulation. IUBMB Life 2015, 67, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbe, S. Deubiquitylases from genes to organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. NY Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Mialki, R.K.; Zhao, J.; Wei, J.; Mallampalli, D.F.; Zhao, Y. Overexpression of USP14 protease reduces I-kappaB protein levels and increases cytokine release in lung epithelial cells. J. Biol. Chem. 2013, 288, 15437–15441. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Dong, S.; Bowser, R.K.; Khoo, A.; Zhang, L.; Jacko, A.M.; Zhao, Y.; Zhao, J. Regulation of the ubiquitylation and deubiquitylation of CREB-binding protein modulates histone acetylation and lung inflammation. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Guan, J.; Li, S.; Zhang, X.; Zheng, X. HSCARG downregulates NF-kappaB signaling by interacting with USP7 and inhibiting NEMO ubiquitination. Cell Death Dis. 2014, 5, e1229. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113, 3264–3275. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar]
- Zhao, J.; Wei, J.; Dong, S.; Bowser, R.K.; Zhang, L.; Jacko, A.M.; Zhao, Y. Destabilization of Lysophosphatidic Acid Receptor 1 Reduces Cytokine Release and Protects Against Lung Injury. EBioMedicine 2016, 10, 195–203. [Google Scholar] [CrossRef]
- Bomberger, J.M.; Barnaby, R.L.; Stanton, B.A. The deubiquitinating enzyme USP10 regulates the endocytic recycling of CFTR in airway epithelial cells. Channels (Austin) 2010, 4, 150–154. [Google Scholar] [CrossRef]
- Palazon-Riquelme, P.; Worboys, J.D.; Green, J.; Valera, A.; Martin-Sanchez, F.; Pellegrini, C.; Brough, D.; Lopez-Castejon, G. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Colleran, A.; Collins, P.E.; O’Carroll, C.; Ahmed, A.; Mao, X.; McManus, B.; Kiely, P.A.; Burstein, E.; Carmody, R.J. Deubiquitination of NF-kappaB by Ubiquitin-Specific Protease-7 promotes transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 618–623. [Google Scholar] [CrossRef]
- Li, L.; Wei, J.; Li, S.; Jacko, A.M.; Weathington, N.M.; Mallampalli, R.K.; Zhao, J.; Zhao, Y. The deubiquitinase USP13 stabilizes the anti-inflammatory receptor IL-1R8/Sigirr to suppress lung inflammation. EBioMedicine 2019, 45, 553–562. [Google Scholar] [CrossRef]
- Geng, J.; Huang, X.; Li, Y.; Xu, X.; Li, S.; Jiang, D.; Liang, J.; Jiang, D.; Wang, C.; Dai, H. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respir Res. 2015, 16, 124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, M.; Jing, Y.; Yin, X.; Ma, P.; Zhang, Z.; Wang, X.; Di, W.; Zhuang, G. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat. Commun 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Lim, J.H.; Jono, H.; Ha, U.H.; Xu, H.; Ishinaga, H.; Morino, S.; Xu, X.; Yan, C.; Kai, H.; et al. Tumor suppressor cylindromatosis acts as a negative regulator for Streptococcus pneumoniae-induced NFAT signaling. J. Biol. Chem. 2008, 283, 12546–12554. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, J.; Yu, J.; Zhang, W.; Huang, Y. Cylindromatosis (CYLD) inhibits Streptococcus pneumonia-induced plasminogen activator inhibitor-1 expression via interacting with TRAF-6. Biochem. Biophys. Res. Commun. 2015, 463, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Jono, H.; Kai, H.; Li, J.D. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J. Biol. Chem. 2005, 280, 41111–41121. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Jono, H.; Koga, T.; Woo, C.H.; Ishinaga, H.; Bourne, P.; Xu, H.; Ha, U.H.; Xu, H.; Li, J.D. Tumor suppressor CYLD acts as a negative regulator for non-typeable Haemophilus influenza-induced inflammation in the middle ear and lung of mice. PLoS ONE 2007, 2, e1032. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Jono, H.; Komatsu, K.; Woo, C.H.; Lee, J.; Miyata, M.; Matsuno, T.; Xu, X.; Huang, Y.; Zhang, W.; et al. CYLD negatively regulates transforming growth factor-beta-signalling via deubiquitinating Akt. Nat. Commun. 2012, 3, 771. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Stirling, B.; Derry, J.; Koga, T.; Jono, H.; Woo, C.H.; Xu, H.; Bourne, P.; Ha, U.H.; Ishinaga, H.; et al. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity 2007, 27, 349–360. [Google Scholar] [CrossRef]
- Vidal, S.; El Motiam, A.; Seoane, R.; Preitakaite, V.; Bouzaher, Y.H.; Gomez-Medina, S.; San Martin, C.; Rodriguez, D.; Rejas, M.T.; Baz-Martinez, M.; et al. Regulation of the Ebola Virus VP24 Protein by SUMO. J. Virol. 2019, 94. [Google Scholar] [CrossRef]
- Ali, A.; Raja, R.; Farooqui, S.R.; Ahmad, S.; Banerjea, A.C. USP7 deubiquitinase controls HIV-1 production by stabilizing Tat protein. Biochem. J. 2017, 474, 1653–1668. [Google Scholar] [CrossRef]
- Xiang, Q.; Ju, H.; Nicholas, J. USP7-Dependent Regulation of TRAF Activation and Signaling by a Viral Interferon Regulatory Factor Homologue. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Bomberger, J.M.; Ye, S.; Maceachran, D.P.; Koeppen, K.; Barnaby, R.L.; O’Toole, G.A.; Stanton, B.A. A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PLoS Pathog. 2011, 7, e1001325. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.; Sugino, T.; Nolte, H.; Andrade, J.; Zimmermann, B.; Shi, C.; Doddaballapur, A.; Ong, Y.T.; Wilhelm, K.; Fasse, J.W.D.; et al. Deubiquitinase USP10 regulates Notch signaling in the endothelium. Science 2019, 364, 188–193. [Google Scholar] [PubMed]
- Wang, D.; Zhao, J.; Li, S.; Wei, J.; Nan, L.; Mallampalli, R.K.; Weathington, N.M.; Ma, H.; Zhao, Y. Phosphorylated E2F1 is stabilized by nuclear USP11 to drive Peg10 gene expression and activate lung epithelial cells. J. Mol. Cell Biol. 2018, 10, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.M.; Yu, C.Y.; Yang, H.C.; Ko, S.H.; Liao, C.L.; Lin, Y.L. Ubiquitin-specific protease 13 regulates IFN signaling by stabilizing STAT1. J. Immunol. 2013, 191, 3328–3336. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Q.; Jing, Y.Y.; Zhang, M.; Wang, H.Y.; Cai, Z.; Liuyu, T.; Zhang, Z.D.; Xiong, T.C.; Wu, Y.; et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat. Commun. 2017, 8, 15534. [Google Scholar] [CrossRef]
- Lu, Y.; Qiu, Y.; Chen, P.; Chang, H.; Guo, L.; Zhang, F.; Ma, L.; Zhang, C.; Zheng, X.; Xiao, J.; et al. ER-localized Hrd1 ubiquitinates and inactivates Usp15 to promote TLR4-induced inflammation during bacterial infection. Nat. Microbiol. 2019, 4, 2331–2346. [Google Scholar] [CrossRef]
- Song, H.; Tao, L.; Chen, C.; Pan, L.; Hao, J.; Ni, Y.; Li, D.; Li, B.; Shi, G. USP17-mediated deubiquitination and stabilization of HDAC2 in cigarette smoke extract-induced inflammation. Int. J. Clin. Exp. Pathol. 2015, 8, 10707–10715. [Google Scholar]
- Lu, C.H.; Yeh, D.W.; Lai, C.Y.; Liu, Y.L.; Huang, L.R.; Lee, A.Y.; Jin, S.C.; Chuang, T.H. USP17 mediates macrophage-promoted inflammation and stemness in lung cancer cells by regulating TRAF2/TRAF3 complex formation. Oncogene 2018, 37, 6327–6340. [Google Scholar] [CrossRef]
- Lei, C.Q.; Wu, X.; Zhong, X.; Jiang, L.; Zhong, B.; Shu, H.B. USP19 Inhibits TNF-alpha- and IL-1beta-Triggered NF-kappaB Activation by Deubiquitinating TAK1. J. Immunol. 2019, 203, 259–268. [Google Scholar] [CrossRef]
- Wu, X.; Lei, C.; Xia, T.; Zhong, X.; Yang, Q.; Shu, H.B. Regulation of TRIF-mediated innate immune response by K27-linked polyubiquitination and deubiquitination. Nat. Commun. 2019, 10, 4115. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jin, S.; Wang, R.F. The BECN1-USP19 axis plays a role in the crosstalk between autophagy and antiviral immune responses. Autophagy 2016, 12, 1210–1211. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Liu, X.; Wang, X.; Liu, X.; Li, H.; Darnay, B.G.; Lin, X.; Sun, S.C.; Dong, C. Ubiquitin-specific protease 25 regulates TLR4-dependent innate immune responses through deubiquitination of the adaptor protein TRAF3. Sci Signal. 2013, 6, ra35. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Wang, D.; Fang, L.; Zhang, H.; Luo, R.; Shang, M.; Ouyang, C.; Ouyang, H.; Chen, H.; Xiao, S. Ubiquitin-specific proteases 25 negatively regulates virus-induced type I interferon signaling. PLoS ONE 2013, 8, e80976. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Zhang, M.; Zhang, M.X.; Ren, Y.; Jin, J.; Zhao, Q.; Pan, Z.; Wu, M.; Shu, H.B.; Dong, C.; et al. Induction of USP25 by viral infection promotes innate antiviral responses by mediating the stabilization of TRAF3 and TRAF6. Proc. Natl. Acad. Sci. USA 2015, 112, 11324–11329. [Google Scholar] [CrossRef]
- Zhong, B.; Liu, X.; Wang, X.; Chang, S.H.; Liu, X.; Wang, A.; Reynolds, J.M.; Dong, C. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat. Immunol. 2012, 13, 1110–1117. [Google Scholar] [CrossRef]
- Long, C.; Lai, Y.; Li, J.; Huang, J.; Zou, C. LPS promotes HBO1 stability via USP25 to modulate inflammatory gene transcription in THP-1 cells. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 773–782. [Google Scholar] [CrossRef]
- Long, C.; Lai, Y.; Li, T.; Nyunoya, T.; Zou, C. Cigarette smoke extract modulates Pseudomonas aeruginosa bacterial load via USP25/HDAC11 axis in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L252–L263. [Google Scholar] [CrossRef]
- Li, S.; Wang, D.; Zhao, J.; Weathington, N.M.; Shang, D.; Zhao, Y. The deubiquitinating enzyme USP48 stabilizes TRAF2 and reduces E-cadherin-mediated adherens junctions. FASEB J. 2018, 32, 230–242. [Google Scholar] [CrossRef]
- Nan, L.; Jacko, A.M.; Tan, J.; Wang, D.; Zhao, J.; Kass, D.J.; Ma, H.; Zhao, Y. Ubiquitin carboxyl-terminal hydrolase-L5 promotes TGFbeta-1 signaling by de-ubiquitinating and stabilizing Smad2/Smad3 in pulmonary fibrosis. Sci. Rep. 2016, 6, 33116. [Google Scholar] [CrossRef]
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol. Cell 2013, 50, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.J.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215.e20–1230.e20. [Google Scholar] [CrossRef]
- Zuo, Y.; Feng, Q.; Jin, L.; Huang, F.; Miao, Y.; Liu, J.; Xu, Y.; Chen, X.; Zhang, H.; Guo, T.; et al. Regulation of the linear ubiquitination of STAT1 controls antiviral interferon signaling. Nat. Commun. 2020, 11, 1146. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zheng, H.; Mao, A.P.; Zhong, B.; Li, Y.; Liu, Y.; Gao, Y.; Ran, Y.; Tien, P.; Shu, H.B. Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J. Biol. Chem. 2010, 285, 4291–4297. [Google Scholar] [CrossRef] [PubMed]
- Mulas, F.; Wang, X.; Song, S.; Nishanth, G.; Yi, W.; Brunn, A.; Larsen, P.K.; Isermann, B.; Kalinke, U.; Barragan, A.; et al. The deubiquitinase OTUB1 augments NF-kappaB-dependent immune responses in dendritic cells in infection and inflammation by stabilizing UBC13. Cell Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, J.; Cheng, X.; Zhao, B.; Manyam, G.C.; Zhang, L.; Schluns, K.; Li, P.; Wang, J.; Sun, S.C. The deubiquitinase Otub1 controls the activation of CD8(+) T cells and NK cells by regulating IL-15-mediated priming. Nat. Immunol. 2019, 20, 879–889. [Google Scholar] [CrossRef]
- Edelmann, M.J.; Kramer, H.B.; Altun, M.; Kessler, B.M. Post-translational modification of the deubiquitinating enzyme otubain 1 modulates active RhoA levels and susceptibility to Yersinia invasion. FEBS J. 2010, 277, 2515–2530. [Google Scholar] [CrossRef]
- Jahan, A.S.; Biquand, E.; Munoz-Moreno, R.; Le Quang, A.; Mok, C.K.; Wong, H.H.; Teo, Q.W.; Valkenburg, S.A.; Chin, A.W.H.; Man Poon, L.L.; et al. OTUB1 Is a Key Regulator of RIG-I-Dependent Immune Signaling and Is Targeted for Proteasomal Degradation by Influenza A NS1. Cell Rep. 2020, 30, 1570.e6–1584.e6. [Google Scholar] [CrossRef]
- Herhaus, L.; Al-Salihi, M.; Macartney, T.; Weidlich, S.; Sapkota, G.P. OTUB1 enhances TGFbeta signalling by inhibiting the ubiquitylation and degradation of active SMAD2/3. Nat. Commun. 2013, 4, 2519. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, J.; Qian, L.; Feng, Q.; Wang, X.; Yuan, Y.; Zuo, Y.; Cheng, Q.; Miao, Y.; Guo, T.; et al. Induction of OTUD1 by RNA viruses potently inhibits innate immune responses by promoting degradation of the MAVS/TRAF3/TRAF6 signalosome. PLoS Pathog. 2018, 14, e1007067. [Google Scholar] [CrossRef]
- Lu, D.; Song, J.; Sun, Y.; Qi, F.; Liu, L.; Jin, Y.; McNutt, M.A.; Yin, Y. Mutations of deubiquitinase OTUD1 are associated with autoimmune disorders. J. Autoimmun. 2018, 94, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, D.; Wang, P.; Zhao, Y.; You, F. OTUD1 Negatively Regulates Type I IFN Induction by Disrupting Noncanonical Ubiquitination of IRF3. J. Immunol. 2020, 204, 1904–1918. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mudge, M.C.; Soll, J.M.; Rodrigues, R.B.; Byrum, A.K.; Schwarzkopf, E.A.; Bradstreet, T.R.; Gygi, S.P.; Edelson, B.T.; Mosammaparast, N. OTUD4 Is a Phospho-Activated K63 Deubiquitinase that Regulates MyD88-Dependent Signaling. Mol. Cell. 2018, 69, 505.e5–516.e5. [Google Scholar] [CrossRef]
- Liuyu, T.; Yu, K.; Ye, L.; Zhang, Z.; Zhang, M.; Ren, Y.; Cai, Z.; Zhu, Q.; Lin, D.; Zhong, B. Induction of OTUD4 by viral infection promotes antiviral responses through deubiquitinating and stabilizing MAVS. Cell Res. 2019, 29, 67–79. [Google Scholar] [CrossRef]
- Zhao, Y.; Majid, M.C.; Soll, J.M.; Brickner, J.R.; Dango, S.; Mosammaparast, N. Noncanonical regulation of alkylation damage resistance by the OTUD4 deubiquitinase. EMBO J. 2015, 34, 1687–1703. [Google Scholar] [CrossRef]
- Boone, D.L.; Turer, E.E.; Lee, E.G.; Ahmad, R.C.; Wheeler, M.T.; Tsui, C.; Hurley, P.; Chien, M.; Chai, S.; Hitotsumatsu, O.; et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004, 5, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Ma, A.; Harhaj, E.W. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 2010, 327, 1135–1139. [Google Scholar] [CrossRef]
- Li, Z.; Jia, Y.; Han, S.; Wang, X.; Han, F.; Zhang, J.; Zhang, W.; Guan, H.; Hu, D. Klf4 Alleviates Lipopolysaccharide-Induced Inflammation by Inducing Expression of MCP-1 Induced Protein 1 to Deubiquitinate TRAF6. Cell Physiol. Biochem. 2018, 47, 2278–2290. [Google Scholar] [CrossRef]
- Feng, Q.; Miao, Y.; Ge, J.; Yuan, Y.; Zuo, Y.; Qian, L.; Liu, J.; Cheng, Q.; Guo, T.; Zhang, L.; et al. ATXN3 Positively Regulates Type I IFN Antiviral Response by Deubiquitinating and Stabilizing HDAC3. J. Immunol. 2018, 201, 675–687. [Google Scholar] [CrossRef]
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. JOSD1 inhibits mitochondrial apoptotic signalling to drive acquired chemoresistance in gynaecological cancer by stabilizing MCL1. Cell Death Differ. 2020, 27, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Zhang, Y.; Zhao, P.; Qian, L.; Yuan, Y.; Liu, J.; Cheng, Q.; Xu, W.; Zuo, Y.; et al. JOSD1 Negatively Regulates Type-I Interferon Antiviral Activity by Deubiquitinating and Stabilizing SOCS1. Viral Immunol. 2017, 30, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Wang, B.; Xu, G.; Yang, Z.; Tang, M.; Ma, A.; Jing, T.; Xu, X.; Zhang, X.; et al. POH1 deubiquitinates pro-interleukin-1beta and restricts inflammasome activity. Nat. Commun. 2018, 9, 4225. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell. 2013, 49, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Bednash, J.S.; Weathington, N.; Londino, J.; Rojas, M.; Gulick, D.L.; Fort, R.; Han, S.; McKelvey, A.C.; Chen, B.B.; Mallampalli, R.K. Targeting the deubiquitinase STAMBP inhibits NALP7 inflammasome activity. Nat. Commun. 2017, 8, 15203. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L709–L725. [Google Scholar] [CrossRef]
- Duan, M.; Hibbs, M.L.; Chen, W. The contributions of lung macrophage and monocyte heterogeneity to influenza pathogenesis. Immunol. Cell Biol. 2017, 95, 225–235. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185.e6–197.e6. [Google Scholar] [CrossRef]
- Brummelkamp, T.R.; Nijman, S.M.; Dirac, A.M.; Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003, 424, 797–801. [Google Scholar] [CrossRef]
- Cai, J.; Culley, M.K.; Zhao, Y.; Zhao, J. The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell 2018, 9, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Tian, S.; Hu, W.; Niu, L.; Liu, H.; Xu, H.; Xiao, S.Y. Pulmonary Pathology of Early-Phase 2019 Novel Coronavirus (COVID-19) Pneumonia in Two Patients with Lung Cancer. J. Thorac. Oncol. 2020, 15, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, P1517–P1520. [Google Scholar] [CrossRef]
- Bekes, M.; Rut, W.; Kasperkiewicz, P.; Mulder, M.P.; Ovaa, H.; Drag, M.; Lima, C.D.; Huang, T.T. SARS hCoV papain-like protease is a unique Lys48 linkage-specific di-distributive deubiquitinating enzyme. Biochem. J. 2015, 468, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Garcia, G.; Arumugaswami, V.; Svendsen, C.N. Human iPSC-Derived Cardiomyocytes are Susceptible to SARS-CoV-2 Infection. bioRxiv 2020. [Google Scholar]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016.e19–1035.e19. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020. [Google Scholar]
- Renatus, M.; Parrado, S.G.; D’Arcy, A.; Eidhoff, U.; Gerhartz, B.; Hassiepen, U.; Pierrat, B.; Riedl, R.; Vinzenz, D.; Worpenberg, S.; et al. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure 2006, 14, 1293–1302. [Google Scholar] [CrossRef]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Finerty, P.J., Jr.; Mackenzie, F.; Newman, E.M.; Dhe-Paganon, S. Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8). J. Biol. Chem. 2006, 281, 38061–38070. [Google Scholar] [CrossRef]
- Hu, M.; Li, P.; Song, L.; Jeffrey, P.D.; Chenova, T.A.; Wilkinson, K.D.; Cohen, R.E.; Shi, Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005, 24, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.R.; et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 2000, 25, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Massoumi, R. CYLD: A deubiquitination enzyme with multiple roles in cancer. Future Oncol. 2011, 7, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israel, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef]
- Jono, H.; Lim, J.H.; Chen, L.F.; Xu, H.; Trompouki, E.; Pan, Z.K.; Mosialos, G.; Li, J.D. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: Evidence for a novel inducible autoregulatory feedback pathway. J. Biol. Chem. 2004, 279, 36171–36174. [Google Scholar] [CrossRef]
- Lim, J.H.; Ha, U.H.; Woo, C.H.; Xu, H.; Li, J.D. CYLD is a crucial negative regulator of innate immune response in Escherichia coli pneumonia. Cell Microbiol. 2008, 10, 2247–2256. [Google Scholar] [CrossRef]
- Daviet, L.; Colland, F. Targeting ubiquitin specific proteases for drug discovery. Biochimie 2008, 90, 270–283. [Google Scholar] [CrossRef]
- Ritorto, M.S.; Ewan, R.; Perez-Oliva, A.B.; Knebel, A.; Buhrlage, S.J.; Wightman, M.; Kelly, S.M.; Wood, N.T.; Virdee, S.; Gray, N.S.; et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun 2014, 5, 4763. [Google Scholar] [CrossRef]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Hacker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Pietras, E.M.; He, J.Q.; Kang, J.R.; Liu, S.Y.; Oganesyan, G.; Shahangian, A.; Zarnegar, B.; Shiba, T.L.; Wang, Y.; et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006, 25, 3257–3263. [Google Scholar] [CrossRef]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A deubiquitinase that regulates type I interferon production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Li, F.; Long, Y.; Zheng, J. Chloroquine attenuates lipopolysaccharide-induced inflammatory responses through upregulation of USP25. Can. J. Physiol. Pharmacol. 2017, 95, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.; Hospenthal, M.K.; Geurink, P.P.; Elliott, P.R.; Akutsu, M.; Arnaudo, N.; Ekkebus, R.; Kulathu, Y.; Wauer, T.; El Oualid, F.; et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 2013, 154, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F. Linear ubiquitination signals in adaptive immune responses. Immunol. Rev. 2015, 266, 222–236. [Google Scholar] [CrossRef]
- Fiil, B.K.; Gyrd-Hansen, M. OTULIN deficiency causes auto-inflammatory syndrome. Cell Res. 2016, 26, 1176–1177. [Google Scholar] [CrossRef][Green Version]
- Juang, Y.C.; Landry, M.C.; Sanches, M.; Vittal, V.; Leung, C.C.; Ceccarelli, D.F.; Mateo, A.R.; Pruneda, J.N.; Mao, D.Y.; Szilard, R.K.; et al. OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Mol. Cell 2012, 45, 384–397. [Google Scholar] [CrossRef]
- Nakada, S.; Tai, I.; Panier, S.; Al-Hakim, A.; Iemura, S.; Juang, Y.C.; O’Donnell, L.; Kumakubo, A.; Munro, M.; Sicheri, F.; et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 2010, 466, 941–946. [Google Scholar] [CrossRef]
- Wang, T.; Yin, L.; Cooper, E.M.; Lai, M.Y.; Dickey, S.; Pickart, C.M.; Fushman, D.; Wilkinson, K.D.; Cohen, R.E.; Wolberger, C. Evidence for bidentate substrate binding as the basis for the K48 linkage specificity of otubain 1. J. Mol. Biol. 2009, 386, 1011–1023. [Google Scholar] [CrossRef]
- Wiener, R.; Zhang, X.; Wang, T.; Wolberger, C. The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 2012, 483, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Matmati, M.; Jacques, P.; Maelfait, J.; Verheugen, E.; Kool, M.; Sze, M.; Geboes, L.; Louagie, E.; Mc Guire, C.; Vereecke, L.; et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat. Genet. 2011, 43, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Liu, J.; Wu, X.; Liu, S.; Li, G.; Han, C.; Song, L.; Li, Z.; Wang, Q.; Wang, J.; et al. Histone methyltransferase Ash1l suppresses interleukin-6 production and inflammatory autoimmune diseases by inducing the ubiquitin-editing enzyme A20. Immunity 2013, 39, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Hinojosa, C.A.; Akula Suresh Babu, R.; Rahman, M.M.; Fernandes, G.; Boyd, A.R.; Orihuela, C.J. Elevated A20 contributes to age-dependent macrophage dysfunction in the lungs. Exp. Gerontol. 2014, 54, 58–66. [Google Scholar] [CrossRef]
- Wang, B.; Ma, A.; Zhang, L.; Jin, W.L.; Qian, Y.; Xu, G.; Qiu, B.; Yang, Z.; Liu, Y.; Xia, Q.; et al. POH1 deubiquitylates and stabilizes E2F1 to promote tumour formation. Nat. Commun. 2015, 6, 8704. [Google Scholar] [CrossRef]
- Cooper, E.M.; Cutcliffe, C.; Kristiansen, T.Z.; Pandey, A.; Pickart, C.M.; Cohen, R.E. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009, 28, 621–631. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshikawa, A.; Yamagata, A.; Mimura, H.; Yamashita, M.; Ookata, K.; Nureki, O.; Iwai, K.; Komada, M.; Fukai, S. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature 2008, 455, 358–362. [Google Scholar] [CrossRef]
- Fletcher, A.J.; Mallery, D.L.; Watkinson, R.E.; Dickson, C.F.; James, L.C. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21. Proc. Natl. Acad. Sci. USA 2015, 112, 10014–10019. [Google Scholar] [CrossRef]
- McCullough, J.; Row, P.E.; Lorenzo, O.; Doherty, M.; Beynon, R.; Clague, M.J.; Urbe, S. Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Curr. Biol. 2006, 16, 160–165. [Google Scholar] [CrossRef]
- Feng, L.; Wang, J.; Chen, J. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J. Biol. Chem. 2010, 285, 30982–30988. [Google Scholar] [CrossRef] [PubMed]
- Carolan, B.J.; Heguy, A.; Harvey, B.G.; Leopold, P.L.; Ferris, B.; Crystal, R.G. Up-regulation of expression of the ubiquitin carboxyl-terminal hydrolase L1 gene in human airway epithelium of cigarette smokers. Cancer Res. 2006, 66, 10729–10740. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Shen, X. Ubiquitin carboxyl-terminal hydrolases: Involvement in cancer progression and clinical implications. Cancer Metastasis Rev. 2017, 36, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Niu, X.; Li, Z.; Yu, Y.; Ye, X.; Lu, S.; Chen, Z. Effect of ubiquitin carboxy-terminal hydrolase 37 on apoptotic in A549 cells. Cell Biochem. Funct. 2011, 29, 142–148. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef]
- Niu, J.; Azfer, A.; Zhelyabovska, O.; Fatma, S.; Kolattukudy, P.E. Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP). J. Biol. Chem. 2008, 283, 14542–14551. [Google Scholar] [CrossRef]
- Skalniak, L.; Mizgalska, D.; Zarebski, A.; Wyrzykowska, P.; Koj, A.; Jura, J. Regulatory feedback loop between NF-kappaB and MCP-1-induced protein 1 RNase. FEBS J. 2009, 276, 5892–5905. [Google Scholar] [CrossRef]
- Huang, S.; Miao, R.; Zhou, Z.; Wang, T.; Liu, J.; Liu, G.; Chen, Y.E.; Xin, H.B.; Zhang, J.; Fu, M. MCPIP1 negatively regulates toll-like receptor 4 signaling and protects mice from LPS-induced septic shock. Cell Signal. 2013, 25, 1228–1234. [Google Scholar] [CrossRef]
- Liang, J.; Wang, J.; Azfer, A.; Song, W.; Tromp, G.; Kolattukudy, P.E.; Fu, M. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J. Biol. Chem. 2008, 283, 6337–6346. [Google Scholar] [CrossRef]
- Liang, J.; Saad, Y.; Lei, T.; Wang, J.; Qi, D.; Yang, Q.; Kolattukudy, P.E.; Fu, M. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J. Exp. Med. 2010, 207, 2959–2973. [Google Scholar] [CrossRef]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Chien, H.L.; Lin, S.Y.; Chang, B.L.; Yu, H.P.; Tang, W.C.; Lin, Y.L. MCPIP1 ribonuclease exhibits broad-spectrum antiviral effects through viral RNA binding and degradation. Nucleic Acids Res. 2013, 41, 3314–3326. [Google Scholar] [CrossRef] [PubMed]
- Pruneda, J.N.; Durkin, C.H.; Geurink, P.P.; Ovaa, H.; Santhanam, B.; Holden, D.W.; Komander, D. The Molecular Basis for Ubiquitin and Ubiquitin-like Specificities in Bacterial Effector Proteases. Mol. Cell 2016, 63, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Elkin, B.A.; Knaap, R.C.; Johnson, G.G.; Dalebout, T.J.; Ninaber, D.K.; van Kasteren, P.B.; Bredenbeek, P.J.; Snijder, E.J.; Kikkert, M.; Mark, B.L. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014, 289, 34667–34682. [Google Scholar] [CrossRef]
- Lei, J.; Mesters, J.R.; Drosten, C.; Anemuller, S.; Ma, Q.; Hilgenfeld, R. Crystal structure of the papain-like protease of MERS coronavirus reveals unusual, potentially druggable active-site features. Antiviral Res. 2014, 109, 72–82. [Google Scholar] [CrossRef]
- Frias-Staheli, N.; Giannakopoulos, N.V.; Kikkert, M.; Taylor, S.L.; Bridgen, A.; Paragas, J.; Richt, J.A.; Rowland, R.R.; Schmaljohn, C.S.; Lenschow, D.J.; et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2007, 2, 404–416. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| DUBs | Target Genes | Function |
|---|---|---|
| CYLD | TAK1 [43] | Negatively regulates S.p. induced NFAT signaling [43] |
| TRAF6 [44] | Inhibits S.p. induced PAI-1 expression [44] | |
| TRAF6/TRAF7 [45,46] | Regulates TLR4 signaling [45] Inhibits inflammation [46] | |
| AKT [47] | Regulates TGF- β signaling [47] | |
| PAI-1 [48] | Regulates acute lung injury [48] | |
| USP-7 | NLRP3 [38] | Regulates NLRP3 inflammasome activation [38] |
| NF-κB [39], NEMO [33] | Regulates NF-κB signaling [33,39] | |
| VP24 [49] | Involves in virus replication [49] | |
| Tat [50] | Involves in virus production [50] | |
| TRAF3/TRAF6 [51] | Modulates antiviral signaling [51] | |
| TRAF6/IKKγ [34] | Regulates TLR signaling [34] | |
| USP-10 | CFTR [37,52] | Epithelial mucosal clearance [37,52] |
| NICD1 [53] | Regulates Notch signaling [53] | |
| USP-11 | E2F1 [54] | Regulates lung epithelia proliferation and wound healing [54] |
| LPA1 [36] | Enhances inflammation [36] | |
| USP-13 | IL-1R8/Sigirr [40] | Suppresses lung inflammation [40] |
| PTEN [41] | Regulates cell apoptosis [41] | |
| MCL1 [42] | Regulates transformation of fibroblasts [42] | |
| STAT1 [55] | Regulates IFN Signaling [55] | |
| STING [56] | Negatively regulates antiviral responses [56] | |
| USP-14 | I-kB [31] | Increases cytokine release [31] |
| CBP [32] | Lung inflammation [32] | |
| USP-15 | IκBα [57] | NF-κB activation [57] |
| USP-17 | HDAC2 [58] | Reverses glucocorticoid resistance [58] |
| TRAF2/TRAF3 [59] | Lung inflammation [59] | |
| USP-19 | TAK1 [60] | Inhibits NF-κB activation [60] |
| TRIF [61] | Inactivates TLR3/4-mediated innate immune responses [61] | |
| BECN1 [62] | Promotes formation of autophagosomes and inhibits DDX58/RIG-I-mediated type I interferon signaling [62] | |
| USP-25 | TRAF3 [63] | Regulates TLR4-dependent Innate Immune Responses [63] |
| RIG-I/TRAF2/TRAF6 [64] | Negatively regulates virus-induced type I interferon signaling [64] | |
| TRAF3/TRAF6 [65] | Promotes Innate Antiviral Responses [65] | |
| TRAF5 and TRAF6 [66] | Regulates IL-17 signaling [66] | |
| HBO1 [67] | Modulates macrophage inflammation [67] | |
| HDAC11 [68] | Modulates bacteria load [68] | |
| USP-48 | TRAF2 [69] | Reduces E-cadherin-mediated adherence junctions [69] |
| UCHL5(UCH37) | Smad2/Smad3 [70] | Promotes TGFβ-1 signaling [70] |
| OTULIN | Met-1 [71,72,73] | Prevents inflammation [71,72,73] |
| STAT1 [74] | Controls antiviral signaling [74] | |
| OTUB1 | TRAF3/TRAF6 [75] | Negatively regulates virus-triggered type I IFN induction [75] |
| UBC13 [76] | Augments NF-κB-dependent Immune Responses [76] | |
| AKT [77] | Controls the activation of CD8 + T Cells and NK Cells [77] | |
| RhoA [78] | Increases bacteria uptake [78] | |
| RIG-1 [79] | Activates RIG-I signaling cascade and antiviral responses [79] | |
| Smad2/3 [80] | Enhances TGFβ signaling [80] | |
| OTUD1 | MAVS/TRAF3/TRAF6 [81] | Inhibits Innate Immune Responses [81] |
| IRF3 [82,83] | Maintains immune homeostasis [82] Negatively regulates Type I IFN induction [83] | |
| OTUD4 | MyD88 [84] | Suppresses TLR signaling [84] |
| MAVS [85] | Regulates innate antiviral responses [85] | |
| ALKBH3 [86] | Regulates DNA damage [86] | |
| A20 | TRAF6 [87] | Restricts TLR signals [87] |
| TRAF2/TRAF6/Ubc13/UbcH5c [88] | Inhibits NF-kappa B Signaling [88] | |
| MCPIP1 | TRAF6 [89] | Impedes NF-κB and inflammatory signaling [89] |
| ATXN3 | HDAC3 [90] | Positively regulates type I IFN antiviral response [90] |
| JOSD1 | MCL [91] | Inhibits mitochondrial apoptotic signaling [91] |
| SOCS1 [92] | Inhibits type I IFN signaling and antiviral response [92] | |
| POH1 | pro-IL-1β [93] | Negatively regulates the immune response [93] |
| BRCC3 | NLRP3 [94] | Promotes the inflammasome activation [94] |
| STAMBP | NALP7 [95] | Reduces pro-inflammatory stress [95] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Zou, C. The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 4842. https://doi.org/10.3390/ijms21144842
Li T, Zou C. The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences. 2020; 21(14):4842. https://doi.org/10.3390/ijms21144842
Chicago/Turabian StyleLi, Tiao, and Chunbin Zou. 2020. "The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome" International Journal of Molecular Sciences 21, no. 14: 4842. https://doi.org/10.3390/ijms21144842
APA StyleLi, T., & Zou, C. (2020). The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences, 21(14), 4842. https://doi.org/10.3390/ijms21144842