Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
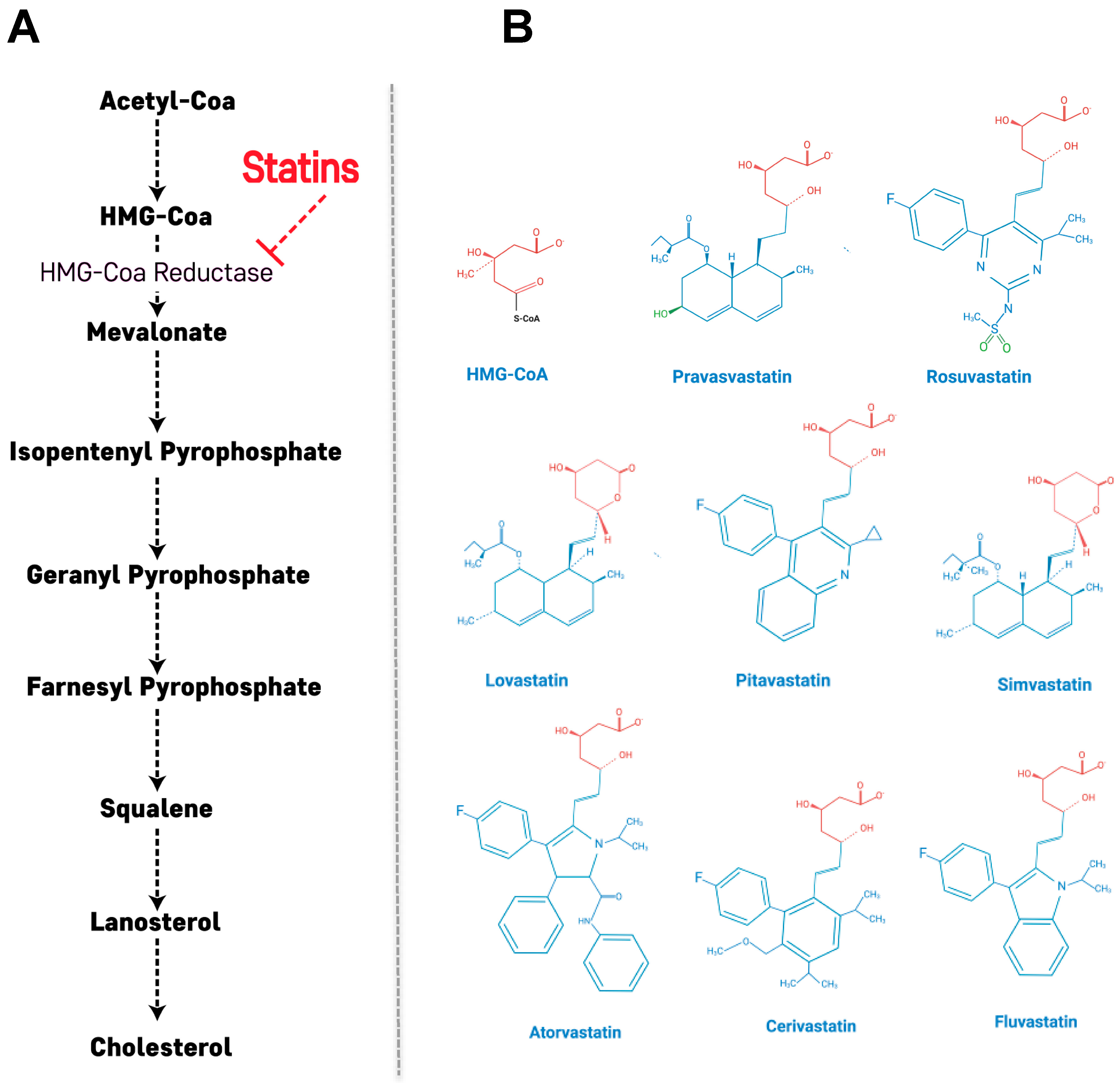
2. Primary Action of Statins: Cholesterol Biosynthetic Pathway
3. Beneficial Effects of Statins on Diabetic Complication and/or Inflammation in T2DM
4. Statin Therapy and Risk of Developing T2DM: Observational Studies, Clinical Trials and Meta-Analysis
5. Proposed Mechanisms for T2DM Development Induced by Statins
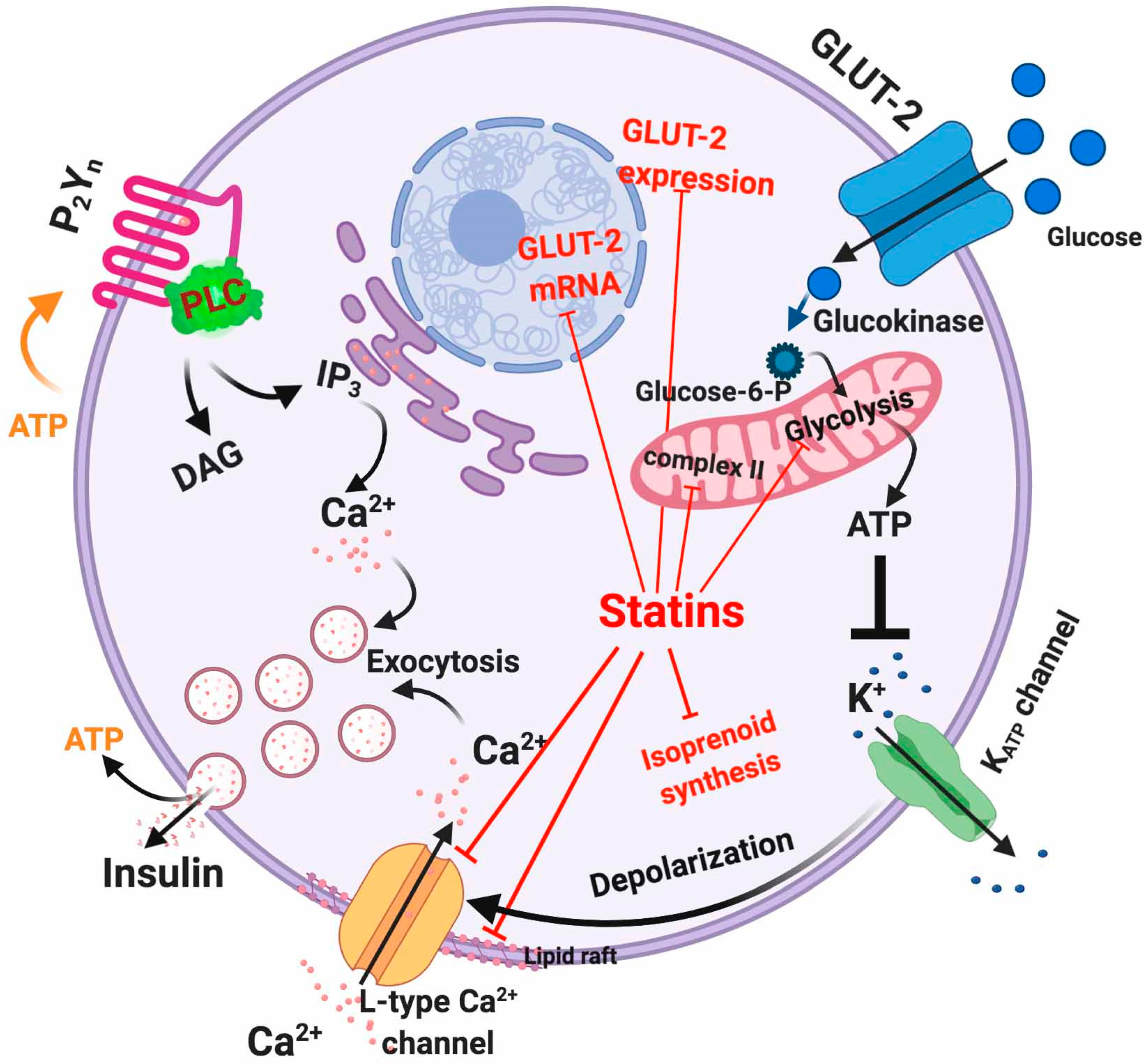
5.1. Dysfunctional Effects Caused by Statins in Pancreatic β-Cell
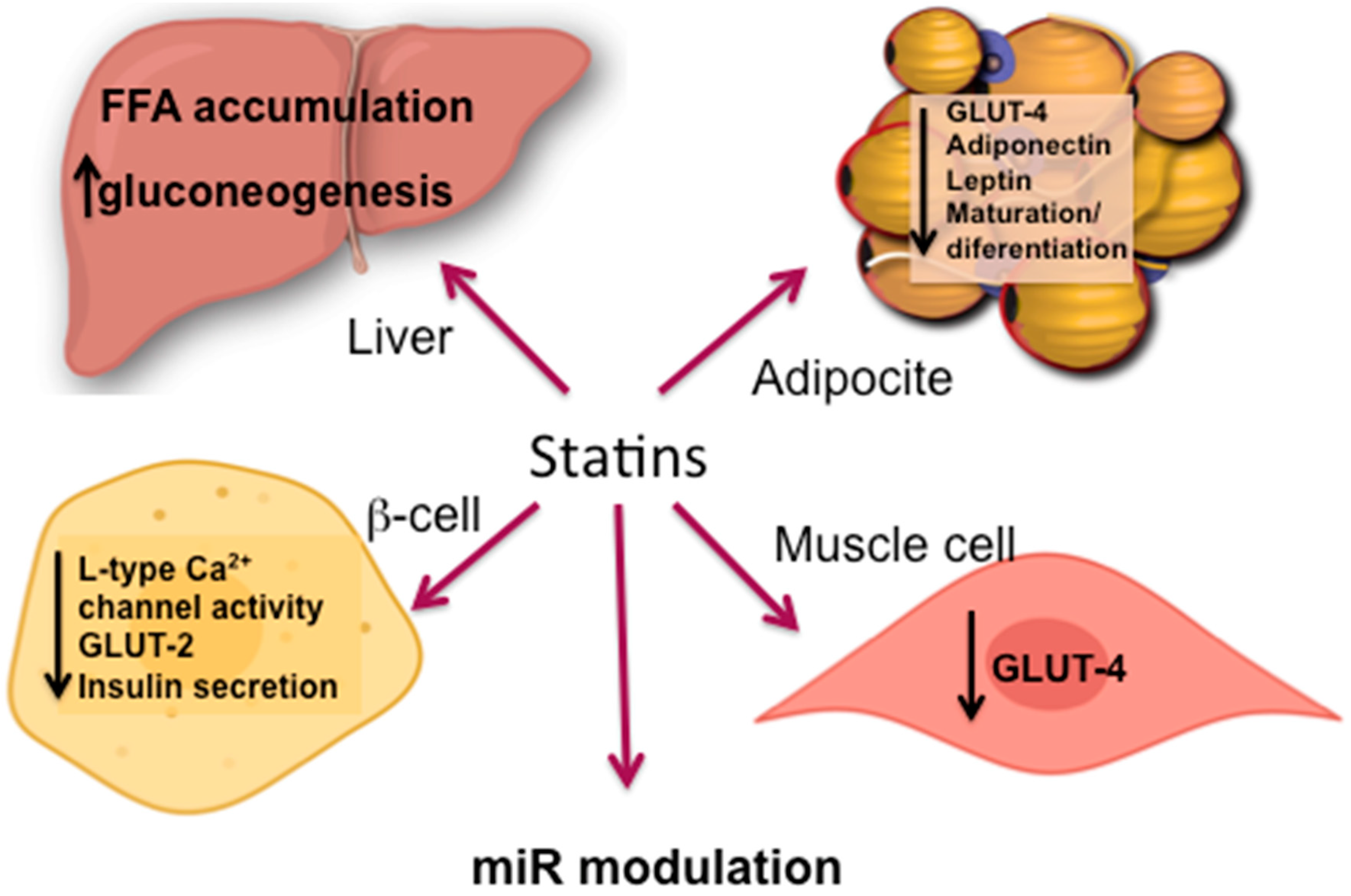
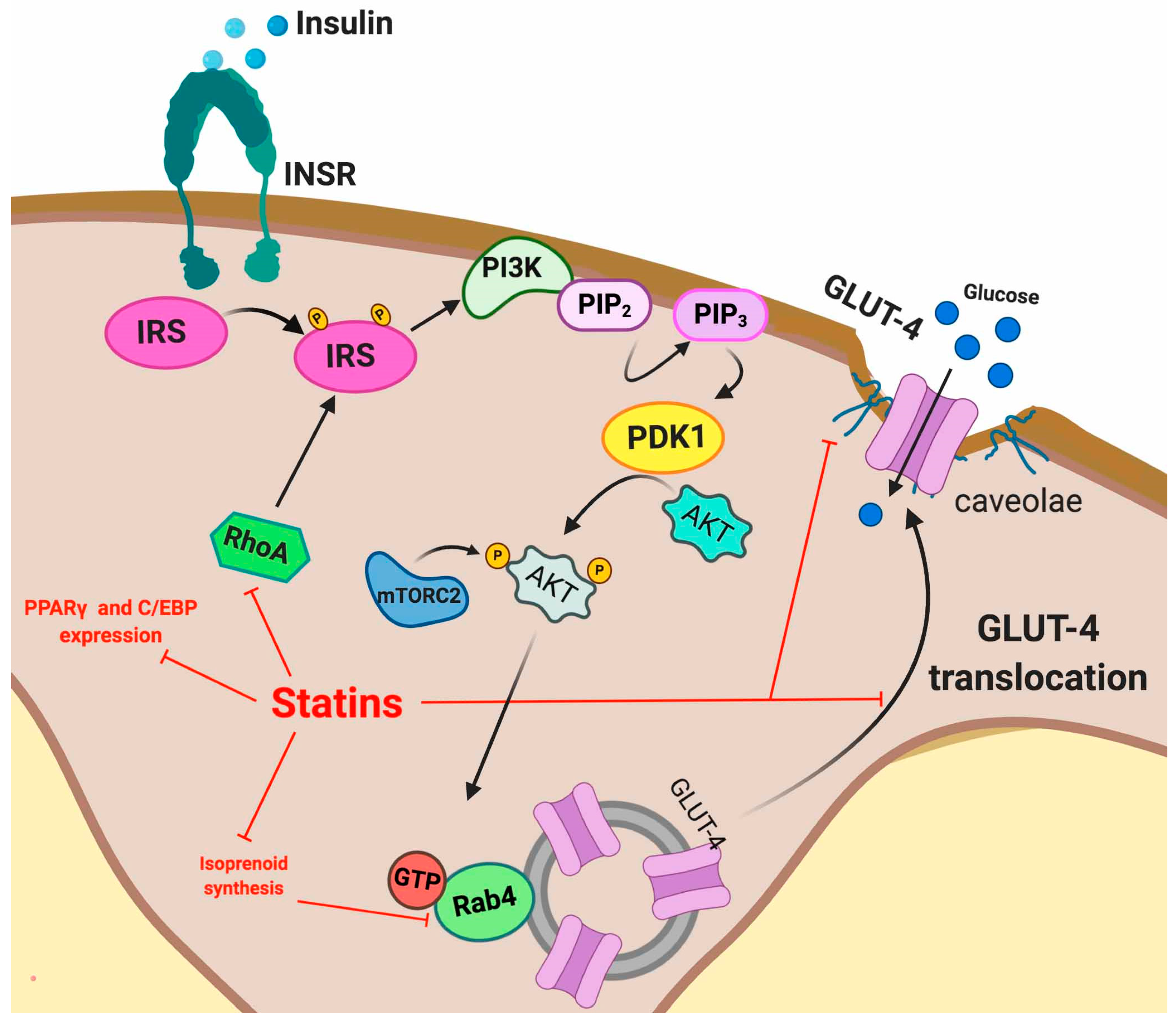
5.2. Statin Induced IR
5.2.1. Adipose Tissue
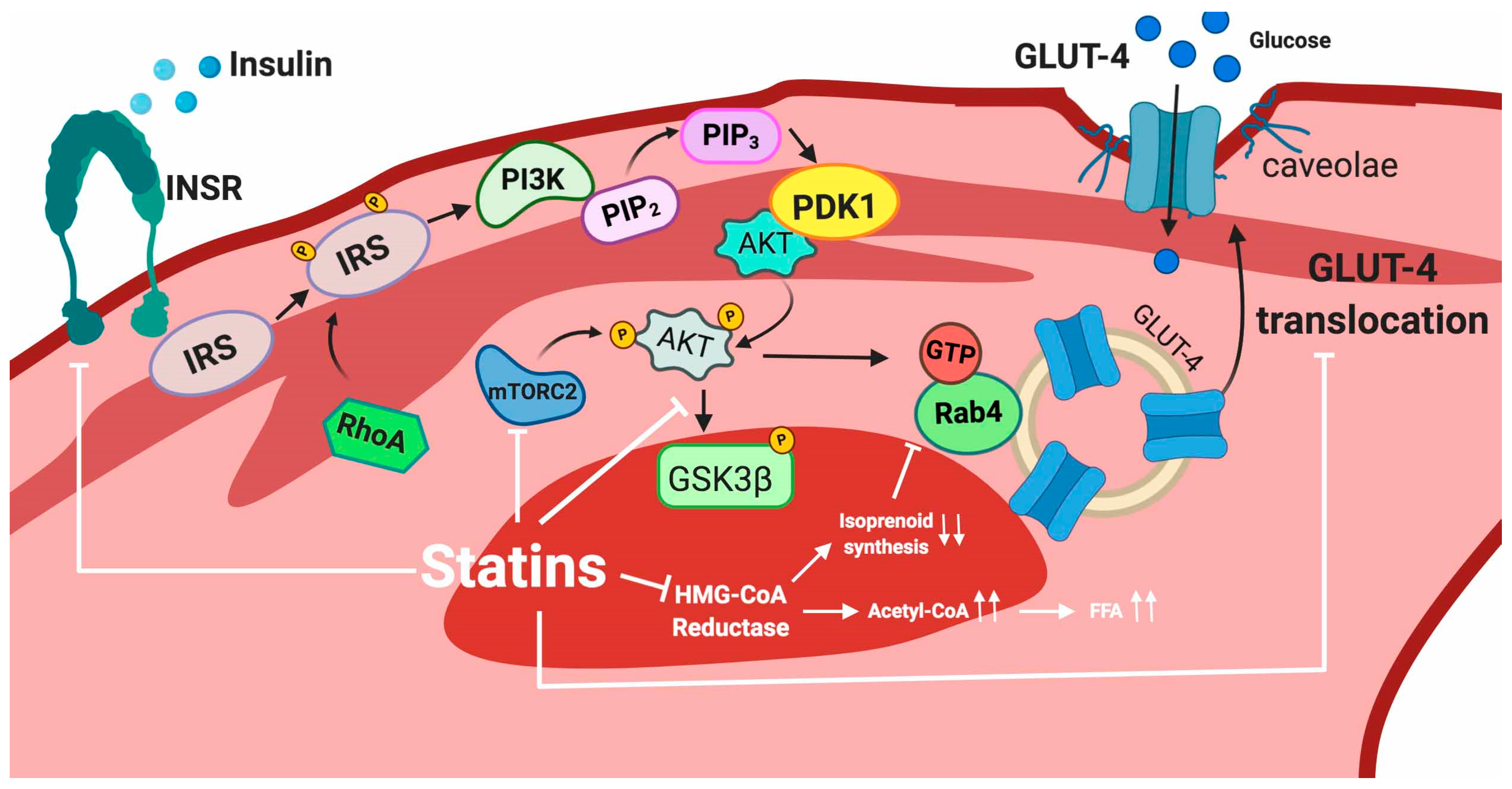
5.2.2. Skeletal Muscle
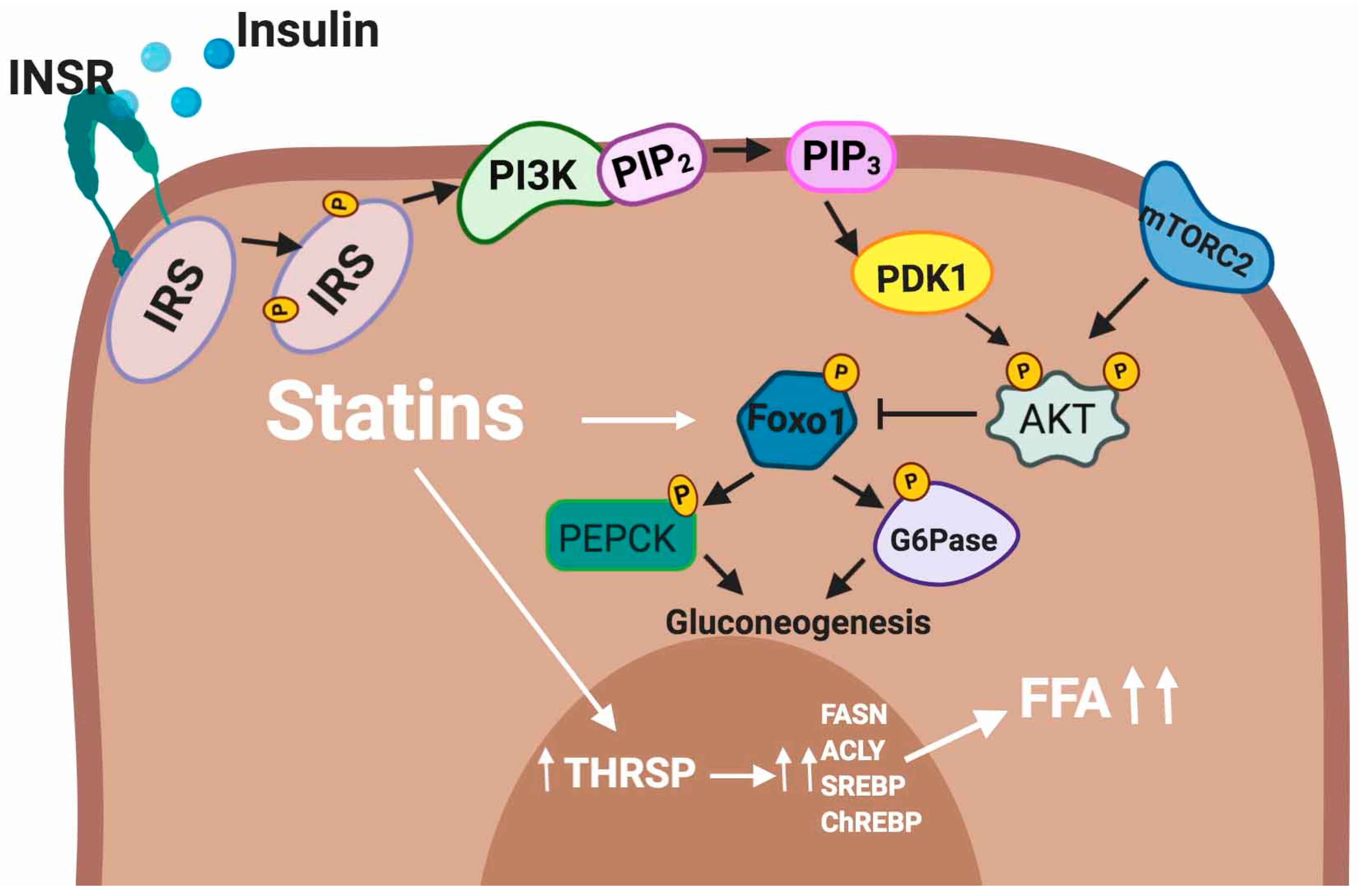
5.2.3. Liver
5.3. MicroRNAs and Impact of Statin Therapy on microRNA Expression Profile
5.3.1. miR Modulation of Cholesterol and Lipid Homeostasis
5.3.2. Modulation of Hepatic Glucose Production
5.3.3. Modulation of the Insulin Signaling Pathway
6. Differences in Diabetogenic Effects between Hydrophilic and Lipophilic Statins
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| Cav-1 | Caveolin 1 |
| CVD | Cardiovascular disease |
| FASN | Fatty-acid synthase |
| FFA | Free fatty acids |
| G-proteins | Small GTP-binding proteins |
| G6Pase | Glucose-6-phosphatase |
| GLUT-4 | Insulin-responsive glucose transporter 4 |
| GSIS | Stimulated insulin secretion |
| HMG-CoA | 3-hydroxy-3-methyl-glutaryl coenzyme-A |
| INSR | Insulin receptor |
| IRS | Insulin receptor substrates |
| LDL-C | LDL cholesterol |
| LDLR | LDL receptor |
| miR | MicroRNA |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PP2A | Protein phosphatase 2a |
| PP2CA | Protein phosphatase 2CA |
| PTPAses | Protein tyrosine phosphatases |
| PXR | Pregnane X receptor |
| RCT | Randomized control trials |
| SGK2 | Serum/glucocorticoid regulated kinase 2 |
| T2DM | Type 2 diabetes mellitus |
References
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.N.; Hitman, G.A.; Neil, H.A.W.; Livingstone, S.J.; Thomason, M.J.; Mackness, M.I.; Charlton-Menys, V.; Fuller, J.H. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): Multicentre randomised placebo-controlled trial. Lancet 2004, 364, 685–696. [Google Scholar] [CrossRef]
- Mihos, C.G.; Pineda, A.M.; Santana, O. Cardiovascular effects of statins, beyond lipid-lowering properties. Pharm. Res. 2014, 88, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Casula, M.; Mozzanica, F.; Scotti, L.; Tragni, E.; Pirillo, A.; Corrao, G.; Catapano, A.L. Statin use and risk of new-onset diabetes: A meta-analysis of observational studies. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 396–406. [Google Scholar] [CrossRef]
- Cederberg, H.; Stancakova, A.; Yaluri, N.; Modi, S.; Kuusisto, J.; Laakso, M. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: A 6 year follow-up study of the METSIM cohort. Diabetologia 2015, 58, 1109–1117. [Google Scholar] [CrossRef]
- Jones, M.; Tett, S.; Peeters, G.M.; Mishra, G.D.; Dobson, A. New-Onset Diabetes After Statin Exposure in Elderly Women: The Australian Longitudinal Study on Women’s Health. Drugs Aging 2017, 34, 203–209. [Google Scholar] [CrossRef]
- Lee, J.; Noh, Y.; Shin, S.; Lim, H.S.; Park, R.W.; Bae, S.K.; Oh, E.; Kim, G.J.; Kim, J.H.; Lee, S. Impact of statins on risk of new onset diabetes mellitus: A population-based cohort study using the Korean National Health Insurance claims database. Ther. Clin. Risk Manag. 2016, 12, 1533–1543. [Google Scholar] [CrossRef]
- Maki, K.C.; Diwadkar-Navsariwala, V.; Kramer, M.W. Statin use and risk for type 2 diabetes: What clinicians should know. Postgrad. Med. 2018, 130, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Crandall, J.P.; Mather, K.; Rajpathak, S.N.; Goldberg, R.B.; Watson, K.; Foo, S.; Ratner, R.; Barrett-Connor, E.; Temprosa, M. Statin use and risk of developing diabetes: Results from the Diabetes Prevention Program. BMJ Open Diabetes Res. Care 2017, 5, e000438. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Pradhan, A.; MacFadyen, J.G.; Libby, P.; Glynn, R.J. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: An analysis from the JUPITER trial. Lancet 2012, 380, 565–571. [Google Scholar] [CrossRef]
- Agarwala, A.; Kulkarni, S.; Maddox, T. The Association of Statin Therapy with Incident Diabetes: Evidence, Mechanisms, and Recommendations. Curr. Cardiol. Rep. 2018, 20, 50. [Google Scholar] [CrossRef] [PubMed]
- Alberton, M.; Wu, P.; Druyts, E.; Briel, M.; Mills, E.J. Adverse events associated with individual statin treatments for cardiovascular disease: An indirect comparison meta-analysis. QJM 2012, 105, 145–157. [Google Scholar] [CrossRef]
- Mills, E.J.; Wu, P.; Chong, G.; Ghement, I.; Singh, S.; Akl, E.A.; Eyawo, O.; Guyatt, G.; Berwanger, O.; Briel, M. Efficacy and safety of statin treatment for cardiovascular disease: A network meta-analysis of 170,255 patients from 76 randomized trials. QJM 2011, 104, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Navarese, E.P.; Buffon, A.; Andreotti, F.; Kozinski, M.; Welton, N.; Fabiszak, T.; Caputo, S.; Grzesk, G.; Kubica, A.; Swiatkiewicz, I.; et al. Meta-analysis of impact of different types and doses of statins on new-onset diabetes mellitus. Am. J. Cardiol. 2013, 111, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Rajpathak, S.N.; Kumbhani, D.J.; Crandall, J.; Barzilai, N.; Alderman, M.; Ridker, P.M. Statin therapy and risk of developing type 2 diabetes: A meta-analysis. Diabetes Care 2009, 32, 1924–1929. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; Preiss, D.; Murray, H.M.; Welsh, P.; Buckley, B.M.; de Craen, A.J.M.; Seshasai, S.R.K.; McMurray, J.J.; Freeman, D.J.; Jukema, J.W.; et al. Statins and risk of incident diabetes: A collaborative meta-analysis of randomised statin trials. Lancet 2010, 375, 735–742. [Google Scholar] [CrossRef]
- Betteridge, D.J.; Carmena, R. The diabetogenic action of statins-mechanisms and clinical implications. Nat. Rev. Endocrinol. 2016, 12, 99–110. [Google Scholar] [CrossRef]
- Shetty, G.K.; Economides, P.A.; Horton, E.S.; Mantzoros, C.S.; Veves, A. Circulating adiponectin and resistin levels in relation to metabolic factors, inflammatory markers, and vascular reactivity in diabetic patients and subjects at risk for diabetes. Diabetes Care 2004, 27, 2450–2457. [Google Scholar] [CrossRef]
- Kruit, J.K.; Brunham, L.R.; Verchere, C.B.; Hayden, M.R. HDL and LDL cholesterol significantly influence beta-cell function in type 2 diabetes mellitus. Curr. Opin. Lipidol. 2010, 21, 178–185. [Google Scholar] [CrossRef]
- Kruit, J.K.; Kremer, P.H.; Dai, L.; Tang, R.; Ruddle, P.; de Haan, W.; Brunham, L.R.; Verchere, C.B.; Hayden, M.R. Cholesterol efflux via ATP-binding cassette transporter A1 (ABCA1) and cholesterol uptake via the LDL receptor influences cholesterol-induced impairment of beta cell function in mice. Diabetologia 2010, 53, 1110–1119. [Google Scholar] [CrossRef]
- Chamberlain, L.H. Inhibition of isoprenoid biosynthesis causes insulin resistance in 3T3-L1 adipocytes. FEBS Lett. 2001, 507, 357–361. [Google Scholar] [CrossRef]
- Nakata, M.; Nagasaka, S.; Kusaka, I.; Matsuoka, H.; Ishibashi, S.; Yada, T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): Implications in glycaemic control. Diabetologia 2006, 49, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Paseban, M.; Butler, A.E.; Sahebkar, A. Mechanisms of statin-induced new-onset diabetes. J. Cell. Physiol. 2019, 234, 12551–12561. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. A gift from nature: The birth of the statins. Nat. Med. 2008, 14, 1050–1052. [Google Scholar] [CrossRef]
- Fong, C.W. Statins in therapy: Understanding their hydrophilicity, lipophilicity, binding to 3-hydroxy-3-methylglutaryl-CoA reductase, ability to cross the blood brain barrier and metabolic stability based on electrostatic molecular orbital studies. Eur. J. Med. Chem. 2014, 85, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B 2010, 86, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The low-density lipoprotein pathway and its relation to atherosclerosis. Annu. Rev. Biochem. 1977, 46, 897–930. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Walter, M.F.; Day, C.A.; Jacob, R.F. Intermolecular differences of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors contribute to distinct pharmacologic and pleiotropic actions. Am. J. Cardiol. 2005, 96, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharm. 2005, 19, 117–125. [Google Scholar] [CrossRef]
- Shitara, Y.; Sugiyama, Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: Drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol. Ther. 2006, 112, 71–105. [Google Scholar] [CrossRef]
- Davidson, M.H. Rosuvastatin: A highly efficacious statin for the treatment of dyslipidaemia. Expert Opin. Investig. Drugs 2002, 11, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Singh, G. Management of diabetic dyslipidemia: An update. World J. Diabetes 2019, 10, 280–290. [Google Scholar] [CrossRef]
- American Diabetes Association. 15. Diabetes Care in the Hospital: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42, S173–S181. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Chaudhuri, A. Targeting inflammation to reduce ASCVD in type 2 diabetes. J. Diabetes Complicat. 2019, 33, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Koksal, M.; Eren, M.A.; Turan, M.N.; Sabuncu, T. The effects of atorvastatin and rosuvastatin on oxidative stress in diabetic patients. Eur. J. Intern. Med. 2011, 22, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Berthold, H.K.; Gouni-Berthold, I.; Bohm, M.; Krone, W.; Bestehorn, K.P. Patterns and predictors of statin prescription in patients with type 2 diabetes. Cardiovasc. Diabetol. 2009, 8, 25. [Google Scholar] [CrossRef]
- Neil, H.A.; DeMicco, D.A.; Luo, D.; Betteridge, D.J.; Colhoun, H.M.; Durrington, P.N.; Livingstone, S.J.; Fuller, J.H.; Hitman, G.A.; Investigators, C.S. Analysis of efficacy and safety in patients aged 65–75 years at randomization: Collaborative Atorvastatin Diabetes Study (CARDS). Diabetes Care 2006, 29, 2378–2384. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists, C.; Kearney, P.M.; Blackwell, L.; Collins, R.; Keech, A.; Simes, J.; Peto, R.; Armitage, J.; Baigent, C. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: A meta-analysis. Lancet 2008, 371, 117–125. [Google Scholar] [CrossRef]
- Collins, R.; Armitage, J.; Parish, S.; Sleigh, P.; Peto, R.; Heart Protection Study Collaborative, G. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: A randomised placebo-controlled trial. Lancet 2003, 361, 2005–2016. [Google Scholar] [CrossRef]
- Baker, W.L.; Talati, R.; White, C.M.; Coleman, C.I. Differing effect of statins on insulin sensitivity in non-diabetics: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2010, 87, 98–107. [Google Scholar] [CrossRef]
- Yada, T.; Nakata, M.; Shiraishi, T.; Kakei, M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br. J. Pharm. 1999, 126, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.A.; Gomes, T.; Camacho, X.; Juurlink, D.N.; Shah, B.R.; Mamdani, M.M. Risk of incident diabetes among patients treated with statins: Population based study. BMJ 2013, 346, f2610. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cai, R.; Yuan, Y.; Varghese, Z.; Moorhead, J.; Ruan, X.Z. Association between reductions in low-density lipoprotein cholesterol with statin therapy and the risk of new-onset diabetes: A meta-analysis. Sci. Rep. 2017, 7, 39982. [Google Scholar] [CrossRef] [PubMed]
- Zaharan, N.L.; Williams, D.; Bennett, K. Statins and risk of treated incident diabetes in a primary care population. Br. J. Clin. Pharm. 2013, 75, 1118–1124. [Google Scholar] [CrossRef]
- Laakso, M.; Kuusisto, J.; Stancakova, A.; Kuulasmaa, T.; Pajukanta, P.; Lusis, A.J.; Collins, F.S.; Mohlke, K.L.; Boehnke, M. The Metabolic Syndrome in Men study: A resource for studies of metabolic and cardiovascular diseases. J. Lipid Res. 2017, 58, 481–493. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef]
- Preiss, D.; Seshasai, S.R.; Welsh, P.; Murphy, S.A.; Ho, J.E.; Waters, D.D.; DeMicco, D.A.; Barter, P.; Cannon, C.P.; Sabatine, M.S.; et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: A meta-analysis. JAMA 2011, 305, 2556–2564. [Google Scholar] [CrossRef]
- Waters, D.D.; Ho, J.E.; DeMicco, D.A.; Breazna, A.; Arsenault, B.J.; Wun, C.C.; Kastelein, J.J.; Colhoun, H.; Barter, P. Predictors of new-onset diabetes in patients treated with atorvastatin: Results from 3 large randomized clinical trials. J. Am. Coll. Cardiol. 2011, 57, 1535–1545. [Google Scholar] [CrossRef]
- Livingstone, S.J.; Looker, H.C.; Akbar, T.; Betteridge, D.J.; Durrington, P.N.; Hitman, G.A.; Neil, H.A.; Fuller, J.H.; Colhoun, H.M. Effect of atorvastatin on glycaemia progression in patients with diabetes: An analysis from the Collaborative Atorvastatin in Diabetes Trial (CARDS). Diabetologia 2016, 59, 299–306. [Google Scholar] [CrossRef]
- Armitage, J.; Bowman, L.; Wallendszus, K.; Bulbulia, R.; Rahimi, K.; Haynes, R.; Parish, S.; Peto, R.; Collins, R. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: A double-blind randomised trial. Lancet 2010, 376, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Park, Z.H.; Juska, A.; Dyakov, D.; Patel, R.V. Statin-associated incident diabetes: A literature review. Consult. Pharm. 2014, 29, 317–334. [Google Scholar] [CrossRef]
- Cai, R.; Yuan, Y.; Zhou, Y.; Xia, W.; Wang, P.; Sun, H.; Yang, Y.; Huang, R.; Wang, S. Lower intensified target LDL-c level of statin therapy results in a higher risk of incident diabetes: A meta-analysis. PLoS ONE 2014, 9, e104922. [Google Scholar] [CrossRef]
- Thakker, D.; Nair, S.; Pagada, A.; Jamdade, V.; Malik, A. Statin use and the risk of developing diabetes: A network meta-analysis. Pharm. Drug Saf. 2016, 25, 1131–1149. [Google Scholar] [CrossRef] [PubMed]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E. Risk factors for type 2 diabetes mellitus: An exposure-wide umbrella review of meta-analyses. PLoS ONE 2018, 13, e0194127. [Google Scholar] [CrossRef]
- Joseph, J.; Svartberg, J.; Njolstad, I.; Schirmer, H. Incidence of and risk factors for type-2 diabetes in a general population: The Tromso Study. Scand. J. Public Health 2010, 38, 768–775. [Google Scholar] [CrossRef]
- Brault, M.; Ray, J.; Gomez, Y.H.; Mantzoros, C.S.; Daskalopoulou, S.S. Statin treatment and new-onset diabetes: A review of proposed mechanisms. Metabolism 2014, 63, 735–745. [Google Scholar] [CrossRef]
- Wang, H.J.; Park, J.Y.; Kwon, O.; Choe, E.Y.; Kim, C.H.; Hur, K.Y.; Lee, M.S.; Yun, M.; Cha, B.S.; Kim, Y.B.; et al. Chronic HMGCR/HMG-CoA reductase inhibitor treatment contributes to dysglycemia by upregulating hepatic gluconeogenesis through autophagy induction. Autophagy 2015, 11, 2089–2101. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhao, S.P. Different effects of statins on induction of diabetes mellitus: An experimental study. Drug Des. Dev. Ther. 2015, 9, 6211–6223. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Hamilton, M.P.; Mundy, D.I.; Chua, S.C.; Scherer, P.E. Impact of simvastatin on adipose tissue: Pleiotropic effects in vivo. Endocrinology 2009, 150, 5262–5272. [Google Scholar] [CrossRef] [PubMed]
- Mancini, G.B.; Baker, S.; Bergeron, J.; Fitchett, D.; Frohlich, J.; Genest, J.; Gupta, M.; Hegele, R.A.; Ng, D.; Pearson, G.J.; et al. Diagnosis, Prevention, and Management of Statin Adverse Effects and Intolerance: Canadian Consensus Working Group Update (2016). Can. J. Cardiol. 2016, 32, S35–S65. [Google Scholar] [CrossRef]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Jahanshahi, P.; Wu, R.; Carter, J.D.; Nunemaker, C.S. Evidence of diminished glucose stimulation and endoplasmic reticulum function in nonoscillatory pancreatic islets. Endocrinology 2009, 150, 607–615. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rorsman, P.; Eliasson, L.; Renstrom, E.; Gromada, J.; Barg, S.; Gopel, S. The Cell Physiology of Biphasic Insulin Secretion. News Physiol. Sci. 2000, 15, 72–77. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, P.E.; Wheeler, M.B. Voltage-dependent K(+) channels in pancreatic beta cells: Role, regulation and potential as therapeutic targets. Diabetologia 2003, 46, 1046–1062. [Google Scholar] [CrossRef] [PubMed]
- Fridlyand, L.E.; Tamarina, N.; Philipson, L.H. Bursting and calcium oscillations in pancreatic beta-cells: Specific pacemakers for specific mechanisms. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E517–E532. [Google Scholar] [CrossRef]
- Gilon, P.; Shepherd, R.M.; Henquin, J.C. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J. Biol. Chem. 1993, 268, 22265–22268. [Google Scholar] [PubMed]
- Ashcroft, F.M.; Rorsman, P. K(ATP) channels and islet hormone secretion: New insights and controversies. Nat. Rev. Endocrinol. 2013, 9, 660–669. [Google Scholar] [CrossRef]
- Bertrand, G.; Chapal, J.; Loubatieres-Mariani, M.M. Potentiating synergism between adenosine diphosphate or triphosphate and acetylcholine on insulin secretion. Am. J. Physiol. 1986, 251, E416–E421. [Google Scholar] [CrossRef]
- Geisler, J.C.; Corbin, K.L.; Li, Q.; Feranchak, A.P.; Nunemaker, C.S.; Li, C. Vesicular nucleotide transporter-mediated ATP release regulates insulin secretion. Endocrinology 2013, 154, 675–684. [Google Scholar] [CrossRef]
- Jacques-Silva, M.C.; Correa-Medina, M.; Cabrera, O.; Rodriguez-Diaz, R.; Makeeva, N.; Fachado, A.; Diez, J.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; et al. ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proc. Natl. Acad. Sci. USA 2010, 107, 6465–6470. [Google Scholar] [CrossRef]
- Khan, S.; Yan-Do, R.; Duong, E.; Wu, X.; Bautista, A.; Cheley, S.; MacDonald, P.E.; Braun, M. Autocrine activation of P2Y1 receptors couples Ca (2+) influx to Ca (2+) release in human pancreatic beta cells. Diabetologia 2014, 57, 2535–2545. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, A.; Idevall-Hagren, O.; Tengholm, A. P2Y(1) receptor-dependent diacylglycerol signaling microdomains in beta cells promote insulin secretion. FASEB J. 2013, 27, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Xie, L.; Mihic, A.; Gao, X.; Chen, Y.; Gaisano, H.Y.; Tsushima, R.G. Inhibition of cholesterol biosynthesis impairs insulin secretion and voltage-gated calcium channel function in pancreatic beta-cells. Endocrinology 2008, 149, 5136–5145. [Google Scholar] [CrossRef]
- Sadighara, M.; Amirsheardost, Z.; Minaiyan, M.; Hajhashemi, V.; Naserzadeh, P.; Salimi, A.; Seydi, E.; Pourahmad, J. Toxicity of Atorvastatin on Pancreas Mitochondria: A Justification for Increased Risk of Diabetes Mellitus. Basic Clin. Pharm. Toxicol. 2017, 120, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Curry, L.; Almukhtar, H.; Alahmed, J.; Roberts, R.; Smith, P.A. Simvastatin Inhibits L-Type Ca2+-Channel Activity Through Impairment of Mitochondrial Function. Toxicol. Sci. 2019, 169, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Gould, G.W.; Holman, G.D. The glucose transporter family: Structure, function and tissue-specific expression. Biochem. J. 1993, 295 Pt 2, 329–341. [Google Scholar] [CrossRef]
- Thorens, B.; Sarkar, H.K.; Kaback, H.R.; Lodish, H.F. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 1988, 55, 281–290. [Google Scholar] [CrossRef]
- Baudry, A.; Leroux, L.; Jackerott, M.; Joshi, R.L. Genetic manipulation of insulin signaling, action and secretion in mice. Insights into glucose homeostasis and pathogenesis of type 2 diabetes. EMBO Rep. 2002, 3, 323–328. [Google Scholar] [CrossRef][Green Version]
- Zhou, J.; Li, W.; Xie, Q.; Hou, Y.; Zhan, S.; Yang, X.; Xu, X.; Cai, J.; Huang, Z. Effects of simvastatin on glucose metabolism in mouse MIN6 cells. J. Diabetes Res. 2014, 2014, 376570. [Google Scholar] [CrossRef]
- Roehrich, M.E.; Mooser, V.; Lenain, V.; Herz, J.; Nimpf, J.; Azhar, S.; Bideau, M.; Capponi, A.; Nicod, P.; Haefliger, J.A.; et al. Insulin-secreting beta-cell dysfunction induced by human lipoproteins. J. Biol. Chem. 2003, 278, 18368–18375. [Google Scholar] [CrossRef]
- Metz, S.A.; Rabaglia, M.E.; Stock, J.B.; Kowluru, A. Modulation of insulin secretion from normal rat islets by inhibitors of the post-translational modifications of GTP-binding proteins. Biochem. J. 1993, 295 Pt 1, 31–40. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Alessi, D.R. The insulin signalling pathway. Curr. Biol. 2002, 12, R236–R238. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Kiselyov, V.V.; Versteyhe, S.; Gauguin, L.; De Meyts, P. Harmonic oscillator model of the insulin and IGF1 receptors’ allosteric binding and activation. Mol. Syst. Biol. 2009, 5, 243. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.K.; Sriram, G.; Dipple, K.M. Insulin sensitivity predictions in individuals with obesity and type II diabetes mellitus using mathematical model of the insulin signal transduction pathway. Mol. Genet. Metab. 2016, 119, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef]
- Moraes-Vieira, P.M.; Saghatelian, A.; Kahn, B.B. GLUT4 Expression in Adipocytes Regulates De Novo Lipogenesis and Levels of a Novel Class of Lipids With Antidiabetic and Anti-inflammatory Effects. Diabetes 2016, 65, 1808–1815. [Google Scholar] [CrossRef]
- Koeppen, B.M.; Stanton, B.A. Berne & Levy Physiology; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Invest. 2016, 126, 12–22. [Google Scholar] [CrossRef]
- Li, R.; Chen, L.Z.; Zhao, S.P.; Huang, X.S. Inflammation Activation Contributes to Adipokine Imbalance in Patients with Acute Coronary Syndrome. PLoS ONE 2016, 11, e0151916. [Google Scholar] [CrossRef]
- Zhao, S.P.; Zhang, D.Q. Atorvastatin reduces interleukin-6 plasma concentration and adipocyte secretion of hypercholesterolemic rabbits. Clin. Chim. Acta 2003, 336, 103–108. [Google Scholar] [CrossRef]
- Takaguri, A.; Satoh, K.; Itagaki, M.; Tokumitsu, Y.; Ichihara, K. Effects of atorvastatin and pravastatin on signal transduction related to glucose uptake in 3T3L1 adipocytes. J. Pharm. Sci. 2008, 107, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, J.; Parpal, S.; Stralfors, P. Insulin-stimulated glucose uptake involves the transition of glucose transporters to a caveolae-rich fraction within the plasma membrane: Implications for type II diabetes. Mol. Med. 1996, 2, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, J.; Parpal, S.; Karlsson, M.; Ramsing, C.; Thorn, H.; Borg, M.; Lindroth, M.; Peterson, K.H.; Magnusson, K.E.; Stralfors, P. Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 1999, 13, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, J.; Kabayama, K.; Gohara, K.; Inokuchi, J. Dissociation of the insulin receptor from caveolae during TNFalpha-induced insulin resistance and its recovery by D-PDMP. FEBS Lett. 2012, 586, 191–195. [Google Scholar] [CrossRef]
- Cohen, A.W.; Razani, B.; Wang, X.B.; Combs, T.P.; Williams, T.M.; Scherer, P.E.; Lisanti, M.P. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am. J. Physiol. Cell Physiol. 2003, 285, C222–C235. [Google Scholar] [CrossRef]
- Breen, M.R.; Camps, M.; Carvalho-Simoes, F.; Zorzano, A.; Pilch, P.F. Cholesterol depletion in adipocytes causes caveolae collapse concomitant with proteosomal degradation of cavin-2 in a switch-like fashion. PLoS ONE 2012, 7, e34516. [Google Scholar] [CrossRef]
- Murata, M.; Peranen, J.; Schreiner, R.; Wieland, F.; Kurzchalia, T.V.; Simons, K. VIP21/caveolin is a cholesterol-binding protein. Proc. Natl. Acad. Sci. USA 1995, 92, 10339–10343. [Google Scholar] [CrossRef]
- Krautbauer, S.; Neumeier, M.; Eisinger, K.; Hader, Y.; Dada, A.; Schmitz, G.; Aslanidis, C.; Buechler, C. LDL but not HDL increases adiponectin release of primary human adipocytes. Exp. Mol. Pathol. 2013, 95, 325–329. [Google Scholar] [CrossRef]
- Carnagarin, R.; Dharmarajan, A.M.; Dass, C.R. Molecular aspects of glucose homeostasis in skeletal muscle--A focus on the molecular mechanisms of insulin resistance. Mol. Cell. Endocrinol. 2015, 417, 52–62. [Google Scholar] [CrossRef]
- Bradley, H.; Shaw, C.S.; Worthington, P.L.; Shepherd, S.O.; Cocks, M.; Wagenmakers, A.J. Quantitative immunofluorescence microscopy of subcellular GLUT4 distribution in human skeletal muscle: Effects of endurance and sprint interval training. Physiol. Rep. 2014, 2, e12085. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Somwar, R.; Bilan, P.J.; Liu, Z.; Jin, J.; Woodgett, J.R.; Klip, A. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol. Cell. Biol. 1999, 19, 4008–4018. [Google Scholar] [CrossRef] [PubMed]
- Funaki, M.; Randhawa, P.; Janmey, P.A. Separation of insulin signaling into distinct GLUT4 translocation and activation steps. Mol. Cell. Biol. 2004, 24, 7567–7577. [Google Scholar] [CrossRef] [PubMed]
- Govers, R.; Coster, A.C.; James, D.E. Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol. Cell. Biol. 2004, 24, 6456–6466. [Google Scholar] [CrossRef]
- Sadler, J.B.; Bryant, N.J.; Gould, G.W.; Welburn, C.R. Posttranslational modifications of GLUT4 affect its subcellular localization and translocation. Int. J. Mol. Sci. 2013, 14, 9963–9978. [Google Scholar] [CrossRef]
- Yaluri, N.; Modi, S.; Kokkola, T. Simvastatin induces insulin resistance in L6 skeletal muscle myotubes by suppressing insulin signaling, GLUT4 expression and GSK-3beta phosphorylation. Biochem. Biophys. Res. Commun. 2016, 480, 194–200. [Google Scholar] [CrossRef]
- Sun, B.; Zhong, Z.; Wang, F.; Xu, J.; Xu, F.; Kong, W.; Ling, Z.; Shu, N.; Li, Y.; Wu, T.; et al. Atorvastatin impaired glucose metabolism in C2C12 cells partly via inhibiting cholesterol-dependent glucose transporter 4 translocation. Biochem. Pharm. 2018, 150, 108–119. [Google Scholar] [CrossRef]
- Sanvee, G.M.; Panajatovic, M.V.; Bouitbir, J.; Krahenbuhl, S. Mechanisms of insulin resistance by simvastatin in C2C12 myotubes and in mouse skeletal muscle. Biochem. Pharm. 2019, 164, 23–33. [Google Scholar] [CrossRef]
- Bonifacio, A.; Sanvee, G.M.; Brecht, K.; Kratschmar, D.V.; Odermatt, A.; Bouitbir, J.; Krahenbuhl, S. IGF-1 prevents simvastatin-induced myotoxicity in C2C12 myotubes. Arch. Toxicol. 2017, 91, 2223–2234. [Google Scholar] [CrossRef]
- Li, W.; Liang, X.; Zeng, Z.; Yu, K.; Zhan, S.; Su, Q.; Yan, Y.; Mansai, H.; Qiao, W.; Yang, Q.; et al. Simvastatin inhibits glucose uptake activity and GLUT4 translocation through suppression of the IR/IRS-1/Akt signaling in C2C12 myotubes. Biomed. Pharm. 2016, 83, 194–200. [Google Scholar] [CrossRef]
- Kain, V.; Kapadia, B.; Misra, P.; Saxena, U. Simvastatin may induce insulin resistance through a novel fatty acid mediated cholesterol independent mechanism. Sci. Rep. 2015, 5, 13823. [Google Scholar] [CrossRef] [PubMed]
- Roden, M. How free fatty acids inhibit glucose utilization in human skeletal muscle. News Physiol. Sci. 2004, 19, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Shu, N.; Xu, P.; Wang, F.; Zhong, Z.; Sun, B.; Li, F.; Zhang, M.; Zhao, K.; Tang, X.; et al. Involvement of pregnane X receptor in the impaired glucose utilization induced by atorvastatin in hepatocytes. Biochem. Pharm. 2016, 100, 98–111. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corra, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, S.; Negishi, M. Statin-activated nuclear receptor PXR promotes SGK2 dephosphorylation by scaffolding PP2C to induce hepatic gluconeogenesis. Sci. Rep. 2015, 5, 14076. [Google Scholar] [CrossRef]
- Brookheart, R.T.; Michel, C.I.; Schaffer, J.E. As a matter of fat. Cell Metab. 2009, 10, 9–12. [Google Scholar] [CrossRef]
- Wakil, S.J.; Abu-Elheiga, L.A. Fatty acid metabolism: Target for metabolic syndrome. J. Lipid Res. 2009, 50, S138–S143. [Google Scholar] [CrossRef]
- LaFave, L.T.; Augustin, L.B.; Mariash, C.N. S14: Insights from knockout mice. Endocrinology 2006, 147, 4044–4047. [Google Scholar] [CrossRef]
- Zhu, Q.; Anderson, G.W.; Mucha, G.T.; Parks, E.J.; Metkowski, J.K.; Mariash, C.N. The Spot 14 protein is required for de novo lipid synthesis in the lactating mammary gland. Endocrinology 2005, 146, 3343–3350. [Google Scholar] [CrossRef]
- Colbert, C.L.; Kim, C.W.; Moon, Y.A.; Henry, L.; Palnitkar, M.; McKean, W.B.; Fitzgerald, K.; Deisenhofer, J.; Horton, J.D.; Kwon, H.J. Crystal structure of Spot 14, a modulator of fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18820–18825. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Williams, M.D.; Mitchell, G.M. MicroRNAs in insulin resistance and obesity. Exp. Diabetes Res. 2012, 2012, 484696. [Google Scholar] [CrossRef]
- Ambros, V. MicroRNA pathways in flies and worms: Growth, death, fat, stress, and timing. Cell 2003, 113, 673–676. [Google Scholar] [CrossRef]
- Fernandez-Hernando, C.; Ramirez, C.M.; Goedeke, L.; Suarez, Y. MicroRNAs in metabolic disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 178–185. [Google Scholar] [CrossRef]
- Davalos, A.; Goedeke, L.; Smibert, P.; Ramirez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef]
- Marquart, T.J.; Allen, R.M.; Ory, D.S.; Baldan, A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. USA 2010, 107, 12228–12232. [Google Scholar] [CrossRef]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Naar, A.M. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef]
- Rayner, K.J.; Suarez, Y.; Davalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernandez-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Clerbaux, L.A.; Haumont, O.; Lanthier, N.; Das, A.K.; Burant, C.F.; Leclercq, I.A.; MacDougald, O.A.; Bommer, G.T. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J. Biol. Chem. 2010, 285, 33652–33661. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.M.; Marquart, T.J.; Albert, C.J.; Suchy, F.J.; Wang, D.Q.; Ananthanarayanan, M.; Ford, D.A.; Baldan, A. miR-33 controls the expression of biliary transporters, and mediates statin- and diet-induced hepatotoxicity. EMBO Mol. Med. 2012, 4, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Zhang, L.H.; Kang, M.H.; Abraham, T.; Bhattacharjee, A.; Warnock, G.L.; Verchere, C.B.; Hayden, M.R. miR-33a modulates ABCA1 expression, cholesterol accumulation, and insulin secretion in pancreatic islets. Diabetes 2012, 61, 653–658. [Google Scholar] [CrossRef]
- Kruit, J.K.; Wijesekara, N.; Fox, J.E.; Dai, X.Q.; Brunham, L.R.; Searle, G.J.; Morgan, G.P.; Costin, A.J.; Tang, R.; Bhattacharjee, A.; et al. Islet cholesterol accumulation due to loss of ABCA1 leads to impaired exocytosis of insulin granules. Diabetes 2011, 60, 3186–3196. [Google Scholar] [CrossRef]
- Lee, A.K.; Yeung-Yam-Wah, V.; Tse, F.W.; Tse, A. Cholesterol elevation impairs glucose-stimulated Ca(2+) signaling in mouse pancreatic beta-cells. Endocrinology 2011, 152, 3351–3361. [Google Scholar] [CrossRef]
- Takwi, A.A.; Li, Y.; Becker Buscaglia, L.E.; Zhang, J.; Choudhury, S.; Park, A.K.; Liu, M.; Young, K.H.; Park, W.Y.; Martin, R.C.; et al. A statin-regulated microRNA represses human c-Myc expression and function. EMBO Mol. Med. 2012, 4, 896–909. [Google Scholar] [CrossRef]
- Zhang, H.; Lamon, B.D.; Moran, G.; Sun, T.; Gotto, A.M., Jr.; Hajjar, D.P. Pitavastatin Differentially Modulates MicroRNA-Associated Cholesterol Transport Proteins in Macrophages. PLoS ONE 2016, 11, e0159130. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, J.F.; Chen, W.J.; Tang, S.L.; Mo, Z.C.; Tang, Y.Y.; Li, Y.; Wang, J.L.; Liu, X.Y.; Peng, J.; et al. MicroRNA-27a/b regulates cellular cholesterol efflux, influx and esterification/hydrolysis in THP-1 macrophages. Atherosclerosis 2014, 234, 54–64. [Google Scholar] [CrossRef]
- Chen, W.J.; Yin, K.; Zhao, G.J.; Fu, Y.C.; Tang, C.K. The magic and mystery of microRNA-27 in atherosclerosis. Atherosclerosis 2012, 222, 314–323. [Google Scholar] [CrossRef]
- Vickers, K.C.; Shoucri, B.M.; Levin, M.G.; Wu, H.; Pearson, D.S.; Osei-Hwedieh, D.; Collins, F.S.; Remaley, A.T.; Sethupathy, P. MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology 2013, 57, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.L.; Khosroheidari, M.; Eddy, E.; Done, S.C. MicroRNA-27a decreases the level and efficiency of the LDL receptor and contributes to the dysregulation of cholesterol homeostasis. Atherosclerosis 2015, 242, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Michaely, P.; Li, W.P.; Anderson, R.G.; Cohen, J.C.; Hobbs, H.H. The modular adaptor protein ARH is required for low density lipoprotein (LDL) binding and internalization but not for LDL receptor clustering in coated pits. J. Biol. Chem. 2004, 279, 34023–34031. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.J.; Go, G.W.; Singh, R.; Liu, W.; Keramati, A.R.; Mani, A. LRP6 protein regulates low density lipoprotein (LDL) receptor-mediated LDL uptake. J. Biol. Chem. 2012, 287, 1335–1344. [Google Scholar] [CrossRef]
- He, G.; Gupta, S.; Yi, M.; Michaely, P.; Hobbs, H.H.; Cohen, J.C. ARH is a modular adaptor protein that interacts with the LDL receptor, clathrin, and AP-2. J. Biol. Chem. 2002, 277, 44044–44049. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.M.; Lockstone, H.E.; Taylor, J.M.; Ria, M.; Barrett, A.; Collins, S.; Kaisaki, P.; Argoud, K.; Fernandez, C.; Travers, M.E.; et al. Global microRNA expression profiles in insulin target tissues in a spontaneous rat model of type 2 diabetes. Diabetologia 2010, 53, 1099–1109. [Google Scholar] [CrossRef]
- Marquart, T.J.; Allen, R.M.; Chen, M.R.; Dorn, G.W., II; Matkovich, S.J.; Baldan, A. Statins Stimulate Hepatic Glucose Production via the miR-183/96/182 Cluster. BioRxiv 2019. [Google Scholar] [CrossRef]
- Hakkola, J.; Rysa, J.; Hukkanen, J. Regulation of hepatic energy metabolism by the nuclear receptor PXR. Biochim. Biophys. Acta 2016, 1859, 1072–1082. [Google Scholar] [CrossRef]
- Oh, K.J.; Park, J.; Kim, S.S.; Oh, H.; Choi, C.S.; Koo, S.H. TCF7L2 modulates glucose homeostasis by regulating CREB- and FoxO1-dependent transcriptional pathway in the liver. PLoS Genet 2012, 8, e1002986. [Google Scholar] [CrossRef]
- Neve, B.; Le Bacquer, O.; Caron, S.; Huyvaert, M.; Leloire, A.; Poulain-Godefroy, O.; Lecoeur, C.; Pattou, F.; Staels, B.; Froguel, P. Alternative human liver transcripts of TCF7L2 bind to the gluconeogenesis regulator HNF4alpha at the protein level. Diabetologia 2014, 57, 785–796. [Google Scholar] [CrossRef]
- Jin, T. Current Understanding on Role of the Wnt Signaling Pathway Effector TCF7L2 in Glucose Homeostasis. Endocr. Rev. 2016, 37, 254–277. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.; Shao, W.; Chiang, Y.T.; Jin, T. The Wnt signaling pathway effector TCF7L2 is upregulated by insulin and represses hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1166–E1176. [Google Scholar] [CrossRef] [PubMed]
- Norton, L.; Fourcaudot, M.; Abdul-Ghani, M.A.; Winnier, D.; Mehta, F.F.; Jenkinson, C.P.; Defronzo, R.A. Chromatin occupancy of transcription factor 7-like 2 (TCF7L2) and its role in hepatic glucose metabolism. Diabetologia 2011, 54, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Karolina, D.S.; Armugam, A.; Tavintharan, S.; Wong, M.T.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE 2011, 6, e22839. [Google Scholar] [CrossRef]
- Alipoor, B.; Ghaedi, H.; Meshkani, R.; Torkamandi, S.; Saffari, S.; Iranpour, M.; Omrani, M.D. Association of MiR-146a Expression and Type 2 Diabetes Mellitus: A Meta-Analysis. Int. J. Mol. Cell. Med. 2017, 6, 156–163. [Google Scholar] [CrossRef]
- Yang, Y.M.; Seo, S.Y.; Kim, T.H.; Kim, S.G. Decrease of microRNA-122 causes hepatic insulin resistance by inducing protein tyrosine phosphatase 1B, which is reversed by licorice flavonoid. Hepatology 2012, 56, 2209–2220. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, C.; Cheng, J.; Chen, B.; Ke, Q.; Lv, Z.; Wu, J.; Zhou, Y. MicroRNA-145 suppresses hepatocellular carcinoma by targeting IRS1 and its downstream Akt signaling. Biochem. Biophys. Res. Commun. 2014, 446, 1255–1260. [Google Scholar] [CrossRef]
- Wen, F.; Yang, Y.; Jin, D.; Sun, J.; Yu, X.; Yang, Z. MiRNA-145 is involved in the development of resistin-induced insulin resistance in HepG2 cells. Biochem. Biophys. Res. Commun. 2014, 445, 517–523. [Google Scholar] [CrossRef]
- Docrat, T.F.; Nagiah, S.; Krishnan, A.; Naidoo, D.B.; Chuturgoon, A.A. Atorvastatin induces MicroRNA-145 expression in HEPG2 cells via regulation of the PI3K/AKT signalling pathway. Chem. Biol. Interact. 2018, 287, 32–40. [Google Scholar] [CrossRef]
- Tang, C.Y.; Man, X.F.; Guo, Y.; Tang, H.N.; Tang, J.; Zhou, C.L.; Tan, S.W.; Wang, M.; Zhou, H.D. IRS-2 Partially Compensates for the Insulin Signal Defects in IRS-1(-/-) Mice Mediated by miR-33. Mol. Cells 2017, 40, 123–132. [Google Scholar] [CrossRef]
- Ugi, S.; Imamura, T.; Maegawa, H.; Egawa, K.; Yoshizaki, T.; Shi, K.; Obata, T.; Ebina, Y.; Kashiwagi, A.; Olefsky, J.M. Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 2004, 24, 8778–8789. [Google Scholar] [CrossRef] [PubMed]
- Nigi, L.; Grieco, G.E.; Ventriglia, G.; Brusco, N.; Mancarella, F.; Formichi, C.; Dotta, F.; Sebastiani, G. MicroRNAs as Regulators of Insulin Signaling: Research Updates and Potential Therapeutic Perspectives in Type 2 Diabetes. Int. J. Mol. Sci. 2018, 19, 3705. [Google Scholar] [CrossRef] [PubMed]
- Lashine, Y.A.; Salah, S.; Aboelenein, H.R.; Abdelaziz, A.I. Correcting the expression of miRNA-155 represses PP2Ac and enhances the release of IL-2 in PBMCs of juvenile SLE patients. Lupus 2015, 24, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Li, P.; Wang, Z.; Chen, J.; Lin, Z.; Liang, X.; Mo, Y. Rosuvastatin may reduce the incidence of cardiovascular events in patients with acute coronary syndromes receiving percutaneous coronary intervention by suppressing miR-155/SHIP-1 signaling pathway. Cardiovasc. Ther. 2014, 32, 276–282. [Google Scholar] [CrossRef]
- Ishikawa, M.; Okajima, F.; Inoue, N.; Motomura, K.; Kato, T.; Takahashi, A.; Oikawa, S.; Yamada, N.; Shimano, H. Distinct effects of pravastatin, atorvastatin, and simvastatin on insulin secretion from a beta-cell line, MIN6 cells. J. Atheroscler. Thromb. 2006, 13, 329–335. [Google Scholar] [CrossRef]
- Mita, T.; Watada, H.; Nakayama, S.; Abe, M.; Ogihara, T.; Shimizu, T.; Uchino, H.; Hirose, T.; Kawamori, R. Preferable effect of pravastatin compared to atorvastatin on beta cell function in Japanese early-state type 2 diabetes with hypercholesterolemia. Endocr. J. 2007, 54, 441–447. [Google Scholar] [CrossRef]
- Kostapanos, M.S.; Liamis, G.L.; Milionis, H.J.; Elisaf, M.S. Do statins beneficially or adversely affect glucose homeostasis? Curr. Vasc. Pharm. 2010, 8, 612–631. [Google Scholar] [CrossRef]
- Urbano, F.; Bugliani, M.; Filippello, A.; Scamporrino, A.; Di Mauro, S.; Di Pino, A.; Scicali, R.; Noto, D.; Rabuazzo, A.M.; Averna, M.; et al. Atorvastatin but Not Pravastatin Impairs Mitochondrial Function in Human Pancreatic Islets and Rat beta-Cells. Direct Effect of Oxidative Stress. Sci. Rep. 2017, 7, 11863. [Google Scholar] [CrossRef]
- Elmendorf, J.S.; Pessin, J.E. Insulin signaling regulating the trafficking and plasma membrane fusion of GLUT4-containing intracellular vesicles. Exp. Cell Res. 1999, 253, 55–62. [Google Scholar] [CrossRef]
- Khan, A.H.; Pessin, J.E. Insulin regulation of glucose uptake: A complex interplay of intracellular signalling pathways. Diabetologia 2002, 45, 1475–1483. [Google Scholar] [CrossRef]
- Bliznakov, E.G. Diabetes and the role of isoprenoid biosynthesis. FEBS Lett. 2002, 525, 169–170. [Google Scholar] [CrossRef]
- Authors/Task Force Members; Ryden, L.; Grant, P.J.; Anker, S.D.; Berne, C.; Cosentino, F.; Danchin, N.; Deaton, C.; Escaned, J.; Hammes, H.P.; et al. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: The Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD). Eur. Heart J. 2013, 34, 3035–3087. [Google Scholar] [CrossRef]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, S1–S45. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Jakicic, J.M.; Ard, J.D.; de Jesus, J.M.; Houston Miller, N.; Hubbard, V.S.; Lee, I.M.; Lichtenstein, A.H.; Loria, C.M.; Millen, B.E.; et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, S76–S99. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 7. Approaches to Glycemic Treatment. Diabetes Care 2016, 39 (Suppl. S1), S52–S59. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of hyperglycemia in type 2 diabetes, 2015: A patient-centered approach: Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015, 38, 140–149. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galicia-Garcia, U.; Jebari, S.; Larrea-Sebal, A.; Uribe, K.B.; Siddiqi, H.; Ostolaza, H.; Benito-Vicente, A.; Martín, C. Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights. Int. J. Mol. Sci. 2020, 21, 4725. https://doi.org/10.3390/ijms21134725
Galicia-Garcia U, Jebari S, Larrea-Sebal A, Uribe KB, Siddiqi H, Ostolaza H, Benito-Vicente A, Martín C. Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights. International Journal of Molecular Sciences. 2020; 21(13):4725. https://doi.org/10.3390/ijms21134725
Chicago/Turabian StyleGalicia-Garcia, Unai, Shifa Jebari, Asier Larrea-Sebal, Kepa B. Uribe, Haziq Siddiqi, Helena Ostolaza, Asier Benito-Vicente, and César Martín. 2020. "Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights" International Journal of Molecular Sciences 21, no. 13: 4725. https://doi.org/10.3390/ijms21134725
APA StyleGalicia-Garcia, U., Jebari, S., Larrea-Sebal, A., Uribe, K. B., Siddiqi, H., Ostolaza, H., Benito-Vicente, A., & Martín, C. (2020). Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights. International Journal of Molecular Sciences, 21(13), 4725. https://doi.org/10.3390/ijms21134725