A Timing Effect of 17-β Estradiol on Atherosclerotic Lesion Development in Female ApoE−/− Mice



Abstract
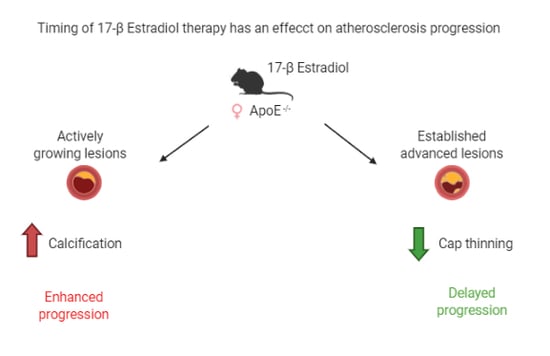
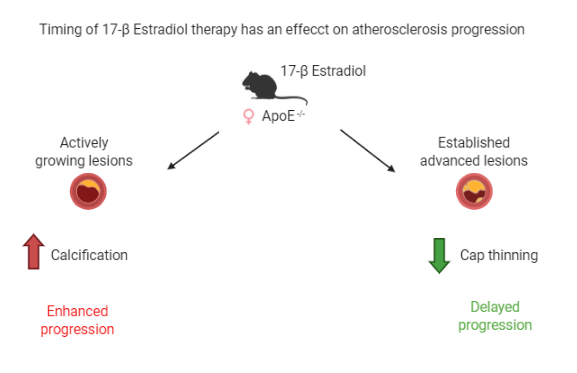
1. Introduction
2. Results
2.1. E2 Treatment Does Not Affect Physiological Parameters
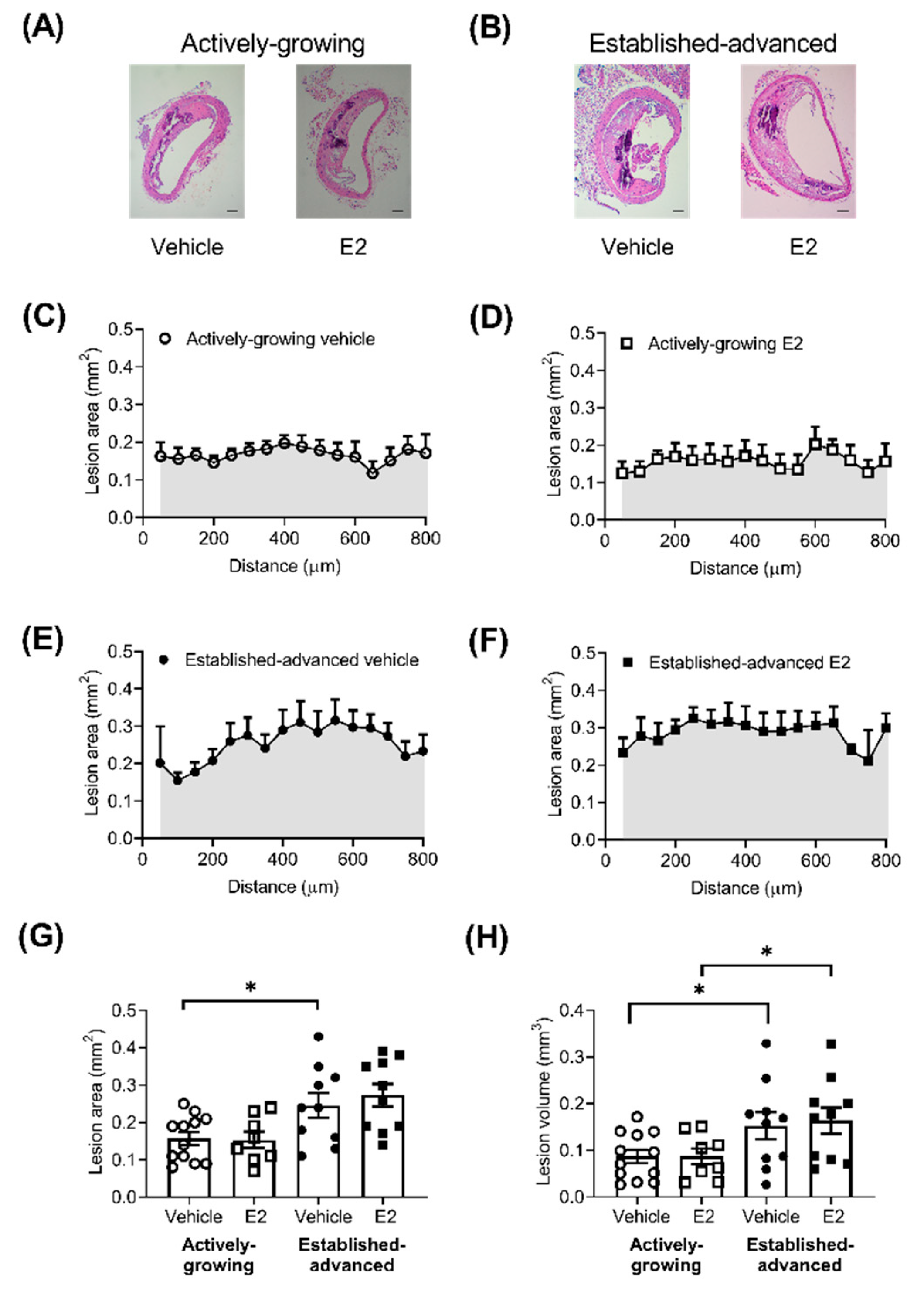
2.2. Atherosclerotic Lesion Burden Is Not Enhanced by E2
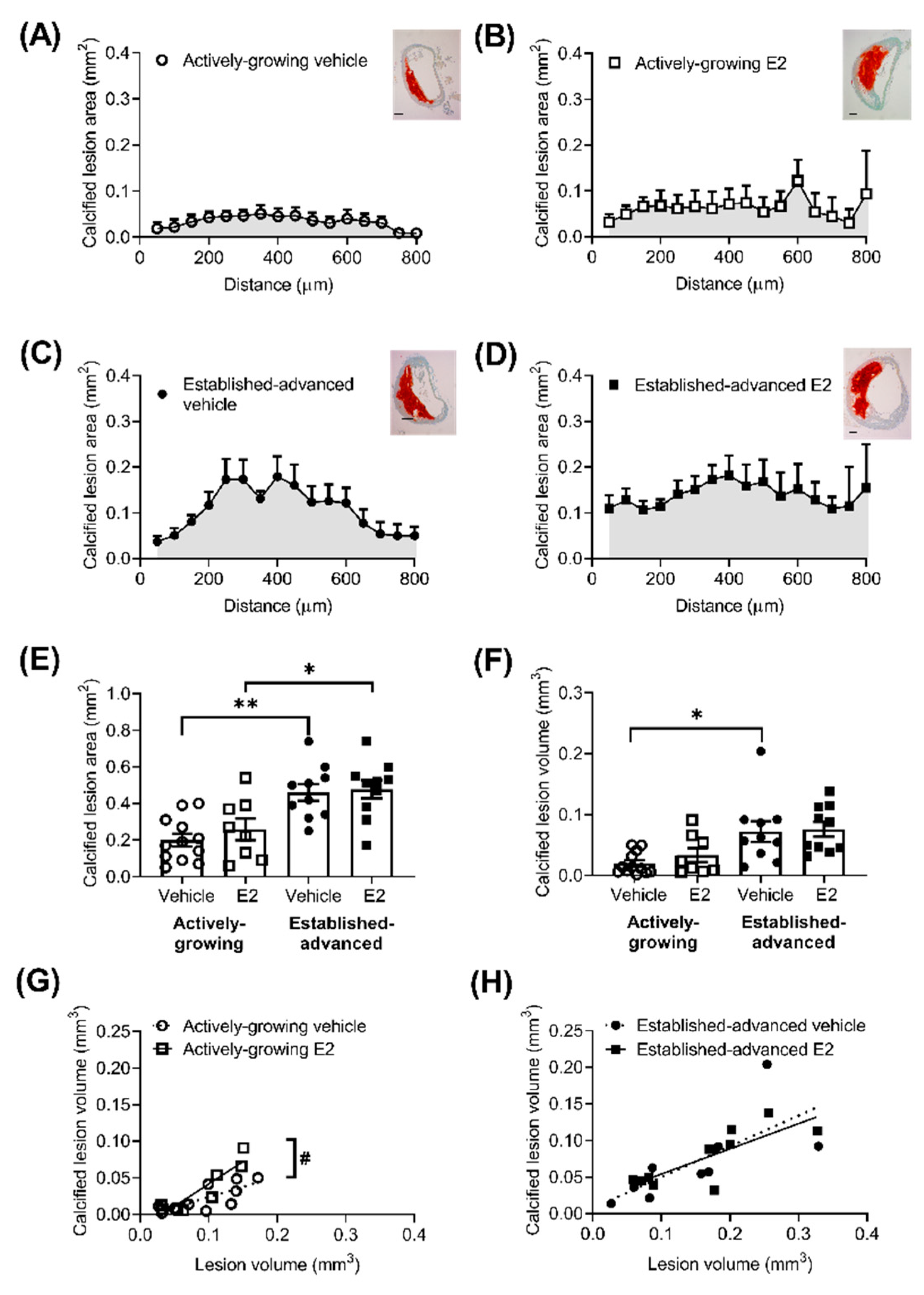
2.3. Composition of Growing Lesions is Affected by E2
2.4. Clinical Based Assessment of Atherosclerotic Lesions
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Histopathological Processing
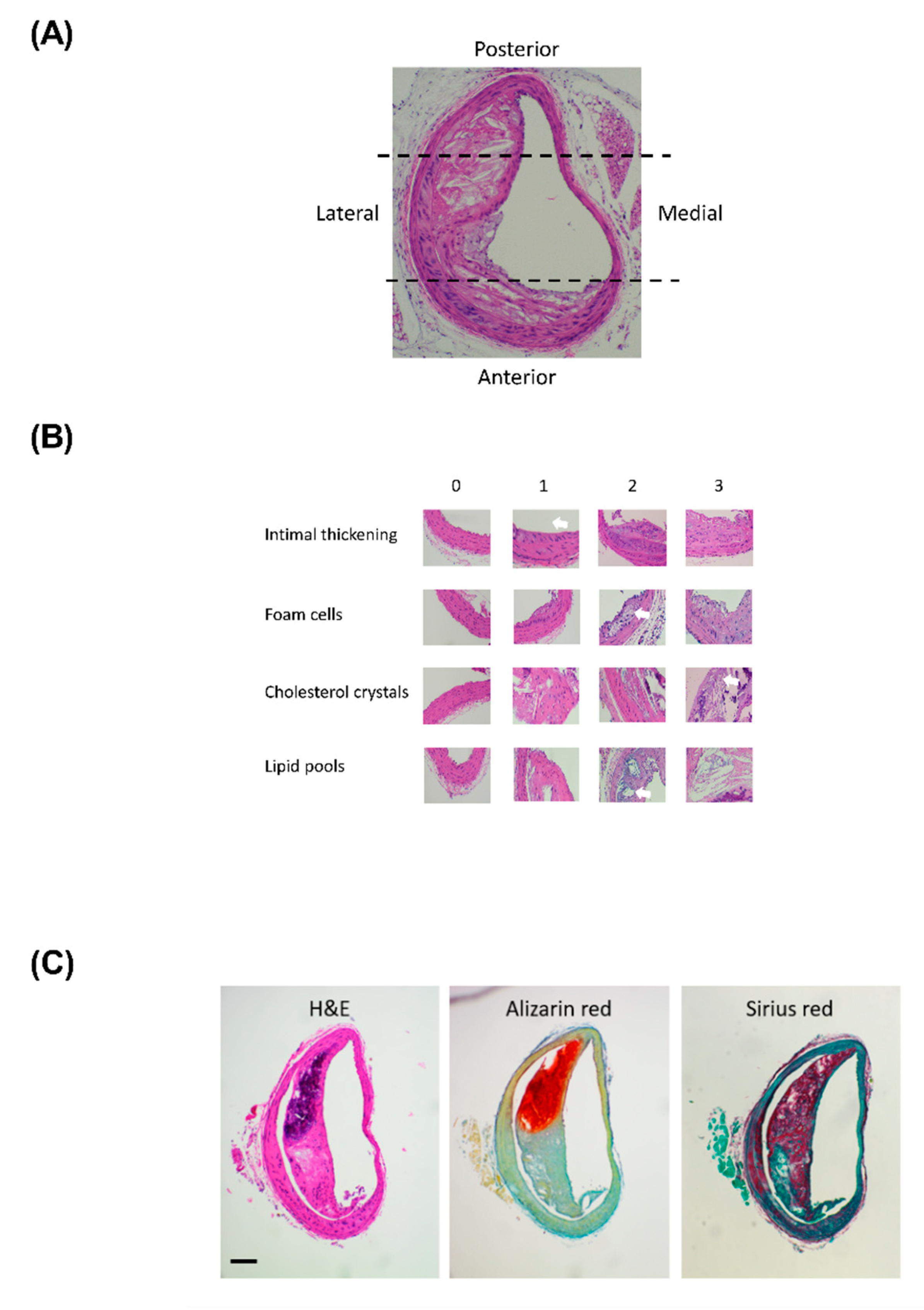
4.3. Lesion Assessment
4.3.1. Lesion Measurements
4.3.2. Lesion Cellular Composition
4.3.3. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AEC | Animal Ethics Committee |
| ANOVA | Analysis of variance |
| ApoE−/− | apolipoprotein E-null mouse |
| BCA | Brachiocephalic artery |
| CO2 | Carbon dioxide |
| E2 | 17β-estradiol |
| H&E | Hematoxylin and eosin |
| SEM | Standard error of mean |
| VSMC | Vascular smooth muscle cells |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Score | Lesion Stage | Lesion Type | Key Cellular Features |
|---|---|---|---|
| 0 | No lesion | Normal | No abnormal features |
| 1 | Very early | Initial lesion | Minimal foam cells and intimal thickening |
| 2 | Early | Fatty streak | Increased intimal thickening and foam cells |
| 3 | Developing | Intermediate lesion | Extensive foam cell collection |
| 4 | Late-developing | Atheroma lesion | Lipid core and cholesterol crystals |
| 5 | Actively-growing | Fibroatheroma | Fibrosis and calcification |
| 6 | Established-advanced | Complicated lesion | Excessive calcification |

References
- Dubey, R.K.; Imthurn, B.; Barton, M.; Jackson, E.K. Vascular consequences of menopause and hormone therapy: Importance of timing of treatment and type of estrogen. Cardiovasc. Res. 2005, 66, 295–306. [Google Scholar] [CrossRef]
- Speth, R.C.; D’Ambra, M.; Ji, H.; Sandberg, K. A heartfelt message, estrogen replacement therapy: Use it or lose it. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1765–H1778. [Google Scholar] [CrossRef]
- Zhao, D.; Guallar, E.; Ouyanh, P.; Subramanya, V.; Vaidya, D.; Ndumele, C.E.; Lima, J.A.; Allison, M.A.; Shah, S.J.; Bertoni, A.G.; et al. Endogenous Sex Hormones and Incident Cardiovascular Disease in Post-Menopausal Women. J. Am. Coll. Cardiol. 2018, 71, 2555–2566. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J.; Lobo, R.A.; Shoupe, D.; Sevanian, A.; Mahrer, P.R.; Slezer, R.H.; Liu, C.R.; Liu, C.H.; Azen, S.P. Estrogen in the prevention of atherosclerosis. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2001, 135, 939–953. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J.; Henderson, V.W.; Shoupe, D.; Budoff, M.J.; Hwang-Levine, J.; Li, Y.; Feng, M.; Dunstin, L.; Kono, N.; et al. Vascular Effects of Early versus Late Postmenopausal Treatment with Estradiol. N. Engl. J. Med. 2016, 374, 1221–1231. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J.; Azen, S.P.; Lobo, R.A.; Shoupe, D.; Mahrer, M.M.; Faxon, D.P.; Cashin-Hemphill, L.; Sanmarco, M.E.; French, W.J.; et al. Hormone therapy and the progression of coronary-artery atherosclerosis in postmenopausal women. N. Engl. J. Med. 2003, 349, 535–545. [Google Scholar] [CrossRef]
- Miller, V.M.; Naftolin, F.; Asthana, S.; Black, D.M.; Brintom, E.A.; Budoff, M.J.; Cedars, M.I.; Dowling, N.M.; Gleason, C.E.; Hodis, H.D.; et al. The Kronos Early Estrogen Prevention Study (KEEPS): What have we learned? Menopause 2019, 26, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Glisic, M.; Mujaj, B.; Rueda-Ochoa, O.L.; Asllanaj, E.; Laven, J.S.E.; Kavousi, M.; Ikram, M.K.; Vernooij, M.K.; Ikram, S.A.; Franco, O.H.; et al. Associations of Endogenous Estradiol and Testosterone Levels With Plaque Composition and Risk of Stroke in Subjects With Carotid Atherosclerosis. Circ. Res. 2018, 122, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Rennenberg, R.J.; Kessels, A.G.H.; Schurgers, L.J.; van Engelshovem, J.M.A.; de Leeuw, P.W.; Kroon, A.A. Vascular calcifications as a marker of increased cardiovascular risk: A meta-analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Christian, R.C.; Harrington, S.; Edwards, W.D.; Oberg, A.L.; Fitzpatrick, L.A. Estrogen status correlates with the calcium content of coronary atherosclerotic plaques in women. J. Clin. Endocrinol. Metab. 2002, 87, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Inoue, F.; Yamaguchi, S.; Ueshima, K.; Fujimoto, T.; Kagoshima, T.; Uemura, S.; Saito, Y. Gender differences in coronary plaque characteristics in patients with stable angina: A virtual histology intravascular ultrasound study. Cardiovasc. Interv. 2010, 25, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Jeon, G.H.; Kim, S.H.; Yun, S.C.; Chae, H.D.; Kim, C.H.; Kang, B.M. Association between serum estradiol level and coronary artery calcification in postmenopausal women. Menopause 2010, 17, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ogita, M.; Ako, J.; Momomura, S.I. Gender differences of plaque characteristics in elderly patients with stable angina pectoris: An intravascular ultrasonic radiofrequency data analysis. Int. J. Vasc. Med. 2010, 2010, 134692. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Travison, T.G.; O’Donnell, C.J.; Bhasin, S.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; D’Agostino, R.B.; Basaria, S. Circulating Sex Steroids and Vascular Calcification in Community-Dwelling Men: The Framingham Heart Study. J. Clin. Endocrinol. Metab. 2016, 101, 2160–2167. [Google Scholar] [CrossRef]
- Balica, M.; Bostrom, K.; Shin, V.; Tillisch, K.; Demer, L.L. Calcifying subpopulation of bovine aortic smooth muscle cells is responsive to 17 beta-estradiol. Circulation 1997, 95, 1954–1960. [Google Scholar] [CrossRef]
- McRobb, L.S.; McGrath, K.C.Y.; Tstralis, T.; Liong, E.C.; Tan, J.T.M.; Hughes, G.; Handelsman, D.J.; Heather, A.K. Estrogen Receptor Control of Atherosclerotic Calcification and Smooth Muscle Cell Osteogenic Differentiation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1127–1137. [Google Scholar] [CrossRef]
- Peng, Y.Q.; Xiong, D.; Lin, X.; Cui, R.R.; Xu, F.; Zhong, J.Y.; Zhu., T.; Wu, F.; Mao, M.Z.; Liao, X.B.; et al. Oestrogen Inhibits Arterial Calcification by Promoting Autophagy. Sci. Rep. 2017, 7, 3549. [Google Scholar] [CrossRef]
- Rzewuska-Lech, E.; Jayachandran, M.; Fitzpatrick, L.A.; Miller, V.M. Differential effects of 17beta-estradiol and raloxifene on VSMC phenotype and expression of osteoblast-associated proteins. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E105–E112. [Google Scholar] [CrossRef]
- Tse, J.; McNaulty, M.; Halks-Miller, M.; Kauser, K.; DelVecchio, V.; Vergona, R.; Sullivan, M.E.; Rubanyi, G.M. Accelerated atherosclerosis and premature calcified cartilaginous metaplasia in the aorta of diabetic male Apo E knockout mice can be prevented by chronic treatment with 17 beta-estradiol. Atherosclerosis 1999, 144, 303–313. [Google Scholar] [CrossRef]
- Meir, K.S.; Leitersdorf, E. Atherosclerosis in the apolipoprotein-E-deficient mouse: A decade of progress. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1006–1014. [Google Scholar] [CrossRef]
- Choy, K.; Beck, K.; Png, F.Y.; Wu, B.J.; Leichtweis, S.B.; Thomas, S.R.; Hou, J.Y.; Croft, K.D.; Mori, T.A.; Stocker, R.D. Processes Involved in the Site-Specific Effect of Probucol on Atherosclerosis in Apolipoprotein E Gene Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1684–1690. [Google Scholar] [CrossRef]
- Rattazzi, M.; Bennett, B.J.; Bea, F.; Kirk, E.A.; Ricks, J.L.; Speer, M.; Schwatz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: Potential role of chondrocyte-like cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1420–1425. [Google Scholar] [CrossRef]
- Cheng, C.; Tempel, D.; van Hapern, R.; van der Baan, A.; Grosveld, F.; Daemen, M.J.A.P.; Krams, R.; de Crom, R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 2006, 113, 2744–2753. [Google Scholar] [CrossRef] [PubMed]
- Barth, R.F.; Kellough, D.A.; Allenby., P.; Blower, L.E.; Hammond, S.H.; Allenby, G.M.; Buja, L.M. Assessment of atherosclerotic luminal narrowing of coronary arteries based on morphometrically generated visual guides. Cardiovasc. Pathol. 2017, 29, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E., Jr.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. Circulation 2010, 121, e266–e369. [CrossRef]
- Paulin, S. Grading and measuring coronary artery stenoses. Catheter. Cardiovasc. Diagn. 1979, 5, 213–218. [Google Scholar] [CrossRef]
- Emini Veseli, B.; Perrotta, P.; DeMyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharm. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhandg, W.; Li, X.; Han, J.; Chen, T.; Duan, Y. Impact of age and sex on the development of atherosclerosis and expression of the related genes in apoE deficient mice. Biochem. Biophys. Res. Comm. 2016, 469, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Natural history and histological classification of atherosclerotic lesions: An update. Arterioscler. Thromb. Vasc Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef]
- Abedin, M.; Tintut, Y.; Demer, L.L. Vascular calcification: Mechanisms and clinical ramifications. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Karwowski, W.; Naumnik, B.; Szczepanski, M.; Mysliwiec, M. The mechanism of vascular calcification—A systematic review. Med. Sci. Monit. 2012, 18, RA1. [Google Scholar] [CrossRef]
- Coleman, R.; Hayek, T.; Keidar, S.; Aviram, M. A mouse model for human atherosclerosis: Long-term histopathological study of lesion development in the aortic arch of apolipoprotein E-deficient (E0) mice. Acta Histochem. 2006, 108, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chen, M.; Li, Y.; Wong, F.H.S.; Thiam, C.W.; Houssain, M.Z.; Poh, K.K.; Hirohata, S.; Ogawa, H.; Angeli, V.; et al. Loss of ADAMTS4 reduces high fat diet-induced atherosclerosis and enhances plaque stability in ApoE(-/-) mice. Sci. Rep. 2016, 6, 31130. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Ikeda, M.; Umegaki, K.; Tomita, T. Estrogen-dependent activation of neutral cholesterol ester hydrolase underlying gender difference of atherogenesis in apoE-/- mice. Atherosclerosis 2011, 219, 545–551. [Google Scholar] [CrossRef]
- Freudenberger, T.; Oppermann, M.; Heim, H.K.; Mayer, P.; Kojda, G.; Schror, K.; Fischer, W. Proatherogenic effects of estradiol in a model of accelerated atherosclerosis in ovariectomized ApoE-deficient mice. Basic Res. Cardiol. 2010, 105, 479–486. [Google Scholar] [CrossRef]
- Cann, J.A.; Register, T.C.; Adams, M.R.; Clair, R.W.S.; Espeland, M.A.; Williams, K. Timing of estrogen replacement influences atherosclerosis progression and plaque leukocyte populations in ApoE-/- mice. Atherosclerosis 2008, 201, 43–52. [Google Scholar] [CrossRef]
- Bourassa, P.A.; Milos, P.M.; Gaynor, B.J.; Breslow, J.L.; Aiello, R.J. Estrogen reduces atherosclerotic lesion development in apolipoprotein E-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10022–10027. [Google Scholar] [CrossRef]
- Carneiro, S.S.; Carminati, R.Z.; Freitas, F.P.S.; Podratz, P.L.; Balarini, C.M.; Graceli, J.B.; Meyrelles, S.S.; Vasquez, E.C.; Gavab, A.L. Endogenous female sex hormones delay the development of renal dysfunction in apolipoprotein E-deficient mice. Lipids Health Dis. 2014, 13, 176. [Google Scholar] [CrossRef]
- Koebele, S.V.; Bimonte-Nelson, H.A. Modeling menopause: The utility of rodents in translational behavioral endocrinology research. Maturitas 2016, 87, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Brooks, H.L.; Pollow, D.P.; Hoyer, P.B. The VCD Mouse Model of Menopause and Perimenopause for the Study of Sex Differences in Cardiovascular Disease and the Metabolic Syndrome. Physiology 2016, 31, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Do the Apoe-/- and Ldlr-/- Mice Yield the Same Insight on Atherogenesis? Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, J.; Gamble, M. Theory and Practice of Histological Techniques, 6th ed.; Churchill Livingstone: London, UK, 2008. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]






| Stenosis Measures | Actively-Growing | Established Advanced | ||
|---|---|---|---|---|
| Vehicle | E2 | Vehicle | E2 | |
| Average stenosis (%) | 51 ± 5 | 54 ± 5 | 61 ± 3 | 66 ± 2 |
| Maximum stenosis (%) | 75 ± 4 | 83 ± 3 | 81 ± 3 | 79 ± 3 |
| Location of max stenosis (µm) | 400 ± 56 | 513 ± 65 | 575 ± 93 | 375 ± 82 |
| Group | Total Score |
|---|---|
| Actively-growing lesion vehicle | 11 ± 0.6 |
| Actively-growing lesion E2 | 12 ± 0.8 |
| Established-advanced lesion vehicle | 13 ± 0.6 |
| Established-advanced lesion E2 | 13 ± 0.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebenebe, O.V.; Ashley, Z.; Erickson, J.R.; Heather, A.K. A Timing Effect of 17-β Estradiol on Atherosclerotic Lesion Development in Female ApoE−/− Mice. Int. J. Mol. Sci. 2020, 21, 4710. https://doi.org/10.3390/ijms21134710
Ebenebe OV, Ashley Z, Erickson JR, Heather AK. A Timing Effect of 17-β Estradiol on Atherosclerotic Lesion Development in Female ApoE−/− Mice. International Journal of Molecular Sciences. 2020; 21(13):4710. https://doi.org/10.3390/ijms21134710
Chicago/Turabian StyleEbenebe, Obialunanma V., Zoe Ashley, Jeffrey R. Erickson, and Alison K. Heather. 2020. "A Timing Effect of 17-β Estradiol on Atherosclerotic Lesion Development in Female ApoE−/− Mice" International Journal of Molecular Sciences 21, no. 13: 4710. https://doi.org/10.3390/ijms21134710
APA StyleEbenebe, O. V., Ashley, Z., Erickson, J. R., & Heather, A. K. (2020). A Timing Effect of 17-β Estradiol on Atherosclerotic Lesion Development in Female ApoE−/− Mice. International Journal of Molecular Sciences, 21(13), 4710. https://doi.org/10.3390/ijms21134710