Abstract
Oral carcinogenesis is a complex and multifactorial process that involves cumulative genetic and molecular alterations, leading to uncontrolled cell proliferation, impaired DNA repair and defective cell death. At the early stages, the onset of potentially malignant lesions in the oral mucosa, or oral dysplasia, is associated with higher rates of malignant progression towards carcinoma in situ and invasive carcinoma. Efforts have been made to get insights about signaling pathways that are deregulated in oral dysplasia, as these could be translated into novel markers and might represent promising therapeutic targets. In this context, recent evidence underscored the relevance of the Wnt/β-catenin signaling pathway in oral dysplasia, as this pathway is progressively “switched on” through the different grades of dysplasia (mild, moderate and severe dysplasia), with the consequent nuclear translocation of β-catenin and expression of target genes associated with the maintenance of representative traits of oral dysplasia, namely cell proliferation and viability. Intriguingly, recent studies provide an unanticipated connection between active β-catenin signaling and deregulated endosome trafficking in oral dysplasia, highlighting the relevance of endocytic components in oral carcinogenesis. This review summarizes evidence about the role of the Wnt/β-catenin signaling pathway and the underlying mechanisms that account for its aberrant activation in oral carcinogenesis.
1. Introduction
Oral cancer is a subtype of head and neck cancer, representing the sixth most common malignancy in the world [1,2]. Following diagnosis, 40–50% of patients have a five-year survival, and, hence, oral cancer embraces a main problem for global public health [3,4,5,6,7,8]. With the goal of improving patient survival, emphasis has been made on the early detection, diagnosis and treatment of potentially malignant lesions, in order to prevent their progression towards oral cancer. In this context, efforts are focused on unraveling signaling pathways that are deregulated in potentially malignant lesions and oral cancer, as these could be translated into novel therapeutic targets [9,10,11,12,13]. However, unlike the extensive information available for other epithelial cancers, little is known about the molecular mechanisms accounting for the progression of early lesions and oral cancer. Specifically, studies have reported upregulation of the Wnt/β-catenin signaling pathway in oral cancer and in potentially malignant oral lesions, although the activation extent of this pathway varies according to the stage of oral carcinogenesis [11,14,15,16]. Here, we will review the literature that demonstrate the aberrant activation of Wnt/β-catenin in oral carcinogenesis, by first describing the components of the canonical Wnt signaling pathway and its transcriptional role over genes involved in proliferation and cell survival. Then, we will summarize the evidence that show the involvement of the Wnt/β-catenin pathway in oral cancer and potentially malignant lesions. We will also discuss the role of increased Wnt ligand secretion in oral carcinogenesis. Finally, we will propose an endocytosis-dependent mechanism that would explain the upregulation of the Wnt/β-catenin pathway in oral dysplasia. Understanding the role of the Wnt/β-catenin signaling pathway in oral carcinogenesis, especially in oral dysplasia, will be essential to providing alternative therapeutic approaches to improve the outcomes of the patients.
2. Canonical Wnt Pathway
Wnt signaling pathways encompass both canonical and the different noncanonical pathways and are involved in a variety of biological functions, such as cell differentiation, migration and proliferation [17,18]. Unlike the noncanonical pathway, the canonical Wnt pathway has been extensively studied in the context of cancer cell biology, because it is altered in several malignancies, including colon, melanoma, breast, lung and oral cancers [19,20,21,22,23]. Specifically, in the last decade, a body of evidence has shown that this pathway is upregulated in head and neck malignancies, namely oral cancer and premalignant oral lesions, although the mechanisms accounting for such alterations remain poorly understood [16,20,24,25,26,27]. Particularly, it is intriguing that the alterations of this pathway in oral cancer are unlikely due to mutations in the components of this pathway, but rather, due to increased ligand production, as it will be detailed in the following sections of this review [11,15,28,29,30,31,32,33].
The canonical Wnt pathway or Wnt/β-catenin signaling pathway is highly conserved in mammals and is activated by the binding of extracellular Wnt ligands to a membrane receptor in an autocrine/paracrine manner. Once activated, the canonical Wnt pathway induces the stabilization and nuclear translocation of β-catenin, which ultimately assists in the expression of genes involved in cell proliferation, viability, differentiation and migration [34,35]. In the absence of Wnt ligands, β-catenin is phosphorylated on residues Ser33/Ser37/Thr41 by a multiprotein complex, referred to as a “destruction complex”, which is formed by the enzyme glycogen synthase kinase 3β (GSK3β), casein kinase 1α (CK1α), the tumor-suppressor protein adenomatous polyposis coli (APC) and axin [36,37]. Phosphorylation of β-catenin leads to its degradation via proteasome and, subsequently, a decrease in cytoplasmic levels of β-catenin (Figure 1, left panel). As mentioned, activation of the Wnt/β-catenin pathway is initiated by the binding of the Wnt ligands to membrane receptors, namely the seven transmembrane receptors Frizzled (FZ) and the coreceptors LDL (low-density lipoprotein) receptor related protein 5/6 (LRP5/6) [36,38]. Then, this trimeric complex recruits the cytoplasmic protein Dishevelled (Dvl), which, in turn, sequesters the destruction complex, leading to the cytoplasmic stabilization of β-catenin [18,39,40]. Stabilized β-catenin in the cytoplasm is then able to translocate into the nucleus, forming a complex with T-cell factor/lymphoid enhancer factor (TCF/LEF) proteins to induce the transcription of genes involved in cell growth and proliferation, such as c-myc, cyclin D1 and survivin, among others (Figure 1, right panel) [18,34,41]. Hence, as it might be anticipated, several of these target genes are upregulated in different cancers, including oral malignancies and potentially malignant lesions, as will be detailed in the upcoming sections.
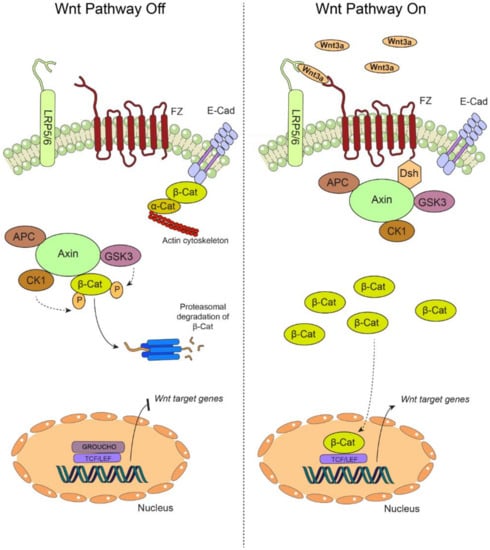
Figure 1.
The canonical Wnt pathway. (Left) In the absence of extracellular Wnt ligands, the transmembrane receptors Frizzled (FZ) and the coreceptors LDL (low-density lipoprotein) receptor related protein 5/6 (LRP 5/6) are unable to associate at the plasma membrane, yielding an “off” state of the pathway. During this off state, β-catenin (β-Cat) is mainly found at cell-cell adhesion complexes, bridging together the intercellular adhesion molecule E-Cadherin (E-cad) and the actin cytoskeleton via interaction with α-catenin (α-Cat). In the cytoplasm, β-catenin is rapidly targeted for proteasomal degradation by the so-called “destruction complex”, which is composed by adenomatous polyposis coli (APC), axin, casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β). The targeting of β-catenin for degradation is based on sequential phosphorylation by CK1 and GSK3β. In these conditions, β-catenin cannot translocate to the nucleus, and the transcription of the target genes is repressed by GROUCHO, which is bound to the TCF/LEF promoters. (Right) Secreted Wnt ligands, such as Wnt3a, are recognized by both FZ and LRP5/6, switching “on” the pathway. The destruction complex is then recruited to the plasma membrane via interaction with the FZ receptor, allowing the cytoplasmic accumulation of β-catenin, which is now available for translocation to the nucleus, where it binds the TCF/LEF promoter by displacing GROUCHO, allowing the transcription of Wnt target genes. Particularly, in oral carcinogenesis, this pathway is “switched on” by the increased secretion of Wnt3a, stabilization of β-catenin and the expression of target genes such as cyclin D1 and survivin (see main text for details).
3. Altered Wnt/β-Catenin Signaling in Oral Carcinogenesis
Aberrant activation of the Wnt/β-catenin signaling pathway is observed in different human cancers [17,40,42], and the most common alterations of this pathway are associated with the aberrant stabilization and nuclear translocation of β-catenin, as a consequence of inactivating mutations in APC or axin, as well as direct mutations in β-catenin [43,44,45,46,47,48], or the overexpression of Wnt ligands [11,49,50]. In this context, mutations in APC, axin and β-catenin are recurrent in colorectal cancer [21,51], whereas mutations in axin have been reported in esophagus squamous cell carcinoma and hepatocellular carcinoma [47,52,53]. In breast cancer, more than 50% of patients depict activated Wnt/β-catenin [23,54,55,56], which is associated with elevated incidences of metastasis [57]. On the other hand, the overexpression of Wnt ligands has been reported in different cancers—for instance, Wnt1 and Wnt5 in hepatocellular, colon or stomach cancers [58,59,60,61], Wnt5 in lung cancer [62,63,64] and Wnt3 in prostate tumors [65,66,67], as well as Wnt3 and Wnt5 in oral cancer [68,69,70,71].
Contrasting the extensive knowledge in other cancers, limited information is available about alterations in components of the Wnt pathway in oral carcinogenesis, and the mechanisms accounting for such alterations are just beginning to be elucidated. For instance, seemingly, and unlike other malignancies, no mutations in components of the Wnt/β-catenin pathway have been identified in oral cancer [28,30,31,33], and current explanations that support the deregulation of this pathway in oral cancer are based on the overproduction of Wnt ligands. This topic will be discussed later in this review. In the following section, we will review the literature that describes aberrant Wnt/β-catenin signaling in oral malignancies—first, by briefly introducing the process of oral carcinogenesis, then the upregulation of β-catenin in oral dysplasia, followed by its deregulation in oral cancer, and, finally, the role of Wnt inhibitors in oral carcinogenesis. In subsequent sections of this review, we will discuss current mechanisms proposed for β-catenin upregulation in oral cancer and oral dysplasia.
3.1. Oral Carcinogenesis
About 90% of oral cancers originate in the stratified nonkeratinized epithelium of the oral mucosa, which is the reason for its denomination as oral squamous cell carcinoma (OSCC), whose main risk factors include the consumption of tobacco and alcohol [2]. Oral carcinogenesis involves the accumulation of discrete and irreversible genetic alterations, as well as epigenetic changes that lead to altered expression/function in proteins, including p53, NOTCH1, EGFR, CDKN2A, STAT3, Cyclin D1, pRb and components of the Wnt/β-catenin pathway, among others [72,73,74,75,76,77,78]. The development of OSCC originates with the exposure towards a carcinogen that produces early genetic and molecular alterations in oral keratinocytes in all areas of tissue exposed to the carcinogen, which is followed by epithelial dysplasia in varying degrees of evolution, ending up with its malignant transformation to OSCC and metastasis (Figure 2) [6,79,80,81,82]. Initially, the molecular alterations in oral keratinocytes may not be expressed as clinical or histological lesions, increasing the risk of malignant transformation, which has been referred to as field cancerization, as proposed in 1953 by Slaughter et al. [83]. Field cancerization is clinically relevant in the prevention of groups of patients at high risk of developing oral cancer [84,85], because early genomic alterations, including microsatellite alterations, mutations in p53 and chromosomal instability, have been evidenced in otherwise histologically normal epithelium, adjacent to oral carcinomas [86,87]. The detection of genetically altered cells from clonal populations with increased growth and high proliferative rates indicate that lateral clonal extension is frequent in potentially malignant or invasive lesions. Therefore, early detection in asymptomatic stages is relevant, not only to permit an increase in survival rates, but also, to improve the quality of life as a consequence of using less aggressive and mutilating treatments, such as chemoprevention [88,89].
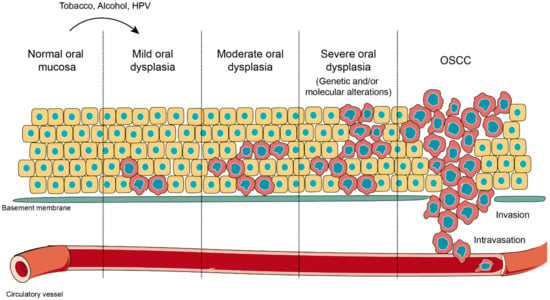
Figure 2.
Oral carcinogenesis. Normal oral mucosa is a stratified layer of epithelial cells arranged over a basement membrane that separates epithelial cells from connective tissue and blood vessels. When oral mucosa is challenged with external stressors, such as tobacco, alcohol or human papilloma virus (HPV) infection, cells in the deepest layers undergo morphological alterations in shape and size. This novel state represents an adaptation response against a harmful stimulation, which is known as oral dysplasia. Oral dysplasia might be categorized as mild, moderate or severe, according with the extension of the lesion and the presence of molecular markers induced as result of the altered genetic expression. Oral dysplasia is considered the previous stage before oral squamous cell carcinoma (OSCC) and the strongest predictor of malignant transformation to cancer. During OSCC, massive phenotypic changes affect all epithelial layers, and it is extended over the tissue border, with ruptures of the basement membrane, in a process that allows the invasion of the connective tissue and incorporation into blood vessels (intravasation).
As mentioned, OSCC is preceded by lesions that are potentially malignant and histopathologically diagnosed as oral epithelial dysplasia, a stage that encompasses cellular and tissue alterations in the oral epithelium. Following the World Health Organization’s recommendations, two ways for classifying oral epithelial dysplasia have been proposed: one is the grading system for dysplasia, which has three categories: mild, moderate and severe dysplasia [7,90,91], whereas a more recent proposal is based on a binary categorization that considers only two states: low-grade dysplasia and high-grade dysplasia [92]. However, the first grading system remains the most widely used (Figure 2) [3,93,94]. Oral dysplasia is characterized by a modification in cell maturation within the epithelia, along with an increased proliferative activity, and, therefore, its diagnosis is the most important indicator of malignant potential [3,8,93,95]. Despite the knowledge that oral dysplasia is associated with high rates of progression towards invasive oral cancer, the mechanisms underlying the evolution of these lesions towards cancer are not fully understood. In this context, several studies have proposed the involvement of the Wnt/β-catenin pathway at the different stages of oral carcinogenesis, which expanded the knowledge about the molecular events involved in the progression from dysplasia towards oral cancer.
3.2. Wnt/β-Catenin Signaling in Oral Dysplasia
Early studies by Sato et al. detected β-catenin at the nucleus in samples of oral dysplasia obtained from a murine model of carcinogenesis [16]. In accordance with these findings, our group found a high nuclear accumulation of β-catenin in biopsies obtained from patients diagnosed with oral dysplasia. In contrast, biopsies obtained from human donors with healthy oral mucosa depicted membranous localization only (Figure 3) [27], which is in accordance with the structural role that β-catenin plays in E-cadherin-mediated cell-cell junctions and epithelial cytoarchitecture, in the context that Wnt signaling is kept off. These data reconciled earlier studies, because they were consistent with reports showing that, in oral dysplasia, β-catenin is detected at the nucleus [15,26,96], while, at the oral carcinoma stage, this protein is mostly accumulated in the cytoplasm, with minimal detection in the nucleus [20,97]. Indeed, immunofluorescence and subcellular fractionation in vitro showed high levels of both total and non-phosphorylated (transcriptionally active) β-catenin in the nucleus of dysplastic oral keratinocytes when compared with nondysplastic oral keratinocytes and OSCC cells [11]. These observations are intriguing, as they are counterintuitive with those reported for other malignancies, where the progressive accumulation of nuclear β-catenin is observed throughout the whole process of carcinogenesis. However, interpretations should be carefully drawn from these observations, because little amounts of nuclear β-catenin might activate the transcription of target genes in OSCC, which is an issue that requires further exploration with appropriate methodologies, such as gene reporter assays and mRNA profiling. Additionally, at the cancer stage, routes other than Wnt/β-catenin, such as those triggered by the epidermal growth factor receptor via MAPK, might account not only for maintaining enhanced cell proliferation but, also, in the acquisition of the migration and invasion capabilities in OSCC cells [13,98,99]. Besides, we emphasize that the nuclear accumulation of β-catenin at the early stages of oral carcinogenesis is a phenomenon that might play a key role in the proliferation of dysplastic cells as a central event in oral dysplasia.
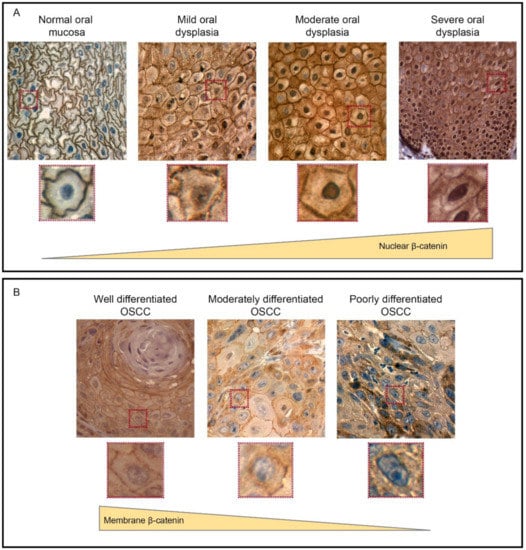
Figure 3.
The expression and localization of β-catenin during oral carcinogenesis. (A) Immunohistochemistry of β-catenin in human oral samples, which revealed that β-catenin is mainly found at the plasma membrane of epithelial cells in normal oral mucosa samples. However, in oral dysplasia, β-catenin is mostly accumulated at the cytoplasm and nucleus of epithelial cells. Importantly, the nuclear detection of β-catenin is progressively increased according with the degree of dysplasia, with the strongest detection in severe and moderate oral dysplasia (zoomed images are shown in lower panels). (B) In OSCC, nuclear β-catenin levels are lower than oral dysplasia. On the other hand, plasma membrane-associated β-catenin decreases when shifting from well-differentiated to moderate and poorly differentiated OSCC (zoomed images are shown in the lower panels). This figure was modified with permission from “Increased nuclear β-catenin expression in oral potentially malignant lesions: A marker of epithelial dysplasia”, Reyes M. et al. 2015, Med Oral Patol Oral Cir Bucal.
Unlike other malignancies, no mutations in components of the Wnt/β-catenin pathway, such as APC, axin or β-catenin itself, have been documented at the different stages of oral carcinogenesis [28,29,30,31], which suggests that mutations in these genes are unlikely to account for the nuclear accumulation of β-catenin in oral dysplasia and, hence, oral cancer. In this scenario, an interesting possibility to explain the aberrant activation of Wnt/β-catenin in oral dysplasia is the overproduction of Wnt ligands. In fact, immunohistochemical data showed the augmented expression of Wnt3a in oral leukoplakia [15]. In accordance with these observations, tissue coimmunofluorescence analysis showed a parallel and progressive increment in both Wnt3a expression and nuclear β-catenin when comparing normal mucosa with mild, moderate and severe dysplasia biopsies [11]. The same study showed that Wnt3a is the main Wnt ligand secreted by dysplastic oral keratinocytes and that, following secretion in cell cultures, this ligand is enough to promote the activation of Wnt/β-catenin in normal keratinocytes. In addition, the pharmacological inhibition of Wnt3a secretion by dysplastic oral keratinocytes interfered with the nuclear translocation of β-catenin and the induction of Wnt/β-catenin target genes, including survivin and cyclin D1, which raises the possibility of new therapeutic approaches for patients with oral dysplasia [11]. Altogether, during oral dysplasia, an exacerbation of Wnt/β-catenin activity might be attributed to an increased release of Wnt ligands. Moreover, as it will be discussed later, the epigenetic silencing of Wnt inhibitors, especially those molecules that interfere with ligand-receptor binding, might also explain the augmented activation of Wnt/β-catenin in oral dysplasia (see Section 3.4). Whether additional mechanisms contribute to β-catenin signaling in oral dysplasia remains unexplored—for instance, the PI3K-Akt pathway, which is known to promote the nuclear translocation of β-catenin in OSCC via the Akt-mediated phosphorylation of GSK3β [99,100,101]. On the other hand, studies in OSCC showed that EGFR overexpression [13,98] and subsequent activation promotes the nuclear translocation of β-catenin in oral cancer cells [102,103]; however, similar studies are not available in oral dysplasia, and further research is necessary to confirm a possible contribution of such pathways in β-catenin signaling during oral dysplasia. Collectively, events that lead to the upregulation of Wnt/β-catenin and consequent expression of target genes involved in cell viability and proliferation contribute to the acquisition of traits that are relevant to the early stages of oral carcinogenesis.
3.3. Wnt/β-Catenin Signaling in Oral Cancer
Unlike oral dysplasia, literature about the localization and function of β-catenin in oral cancer remains a matter of debate, although most studies suggest that β-catenin is mainly cytoplasmic and correlated with poor histological differentiation [104,105]. First, it should be remarked that, in normal oral mucosa, β-catenin is mainly found associated with E-cadherin, maintaining cell-to-cell interactions and preserving the architecture of the epithelium [104,105] (Figure 3). Conversely, in injured oral mucosa, non-membranous pools of β-catenin can be observed during the wound-healing process, which is associated with Axin2 positivity in proliferative Wnt-responsive re-epithelializing cells [106]. Besides, little is known about the role of Wnt/β-catenin in healthy oral mucosa, contrary to its well-described role in oral epithelial development [107].
The membranous expression of β-catenin exerts a restrictive effect on cell proliferation and migration, and, hence, the loss of β-catenin at this location is associated with increased cell motility and proliferation, correlating with the tumor grade and histological differentiation in OSSC [104,105]. In this context, focal β-catenin positivity has been observed in some studies [20,29,32,108,109]. However, unlike dysplasia, only 27% of samples with OSCC depicted nuclear β-catenin [11,27]. This phenomenon could be explained on the basis that the low nuclear localization of β-catenin is not necessarily translated into less transcriptional activity, because minimal amounts of β-catenin might be sufficient to allow transcriptional effects. To address this possibility, sophisticated approaches other than immunohistochemical analyses will be required. Conversely, most literature has been focused on demonstrating the tissue localization of this protein, rather than its transcriptional activity. Besides, it remains possible that the activation of Wnt/β-catenin is necessary only at the early stages of oral carcinogenesis, contributing to the increased rates of cell proliferation that are characteristic in oral dysplasia but following the onset of invasive OSCC; other signaling pathways become relevant to promote or sustain these effects.
Despite that several studies have explored the possibility of mutations in the components of the Wnt/β-catenin pathway in OSCC, no such mutations have been described so far [28,29,30,31]. Hence, it has been suggested that mutations in the components downstream of this pathway do not account for either cytoplasmic or sporadic nuclear localizations of β-catenin in OSCC cells, raising the possibility that Wnt ligands are involved. In this context, several Wnt ligands are overexpressed in oral cancer. For instance, high levels of Wnt3a are correlated with augmented nuclear β-catenin at the invasive front of OSCC [32]. Wnt5a, which is barely detected in normal oral mucosa, depicts a progressive increase at the oral dysplasia phase, reaching a maximal expression at the oral carcinoma stage [69,70]. On the other hand, Wnt7b has also been found overexpressed in OSCC, and it accounts for the activation of Wnt/β-catenin and tumor cell proliferation and invasion in vitro [110]. Finally, Wnt7a has been proposed as a promoter of OSCC progression by increasing the MMP-9 expression in a β-catenin-dependent manner [111]. Collectively, these evidences support the notion that Wnt ligands are relevant players in oral cancer progression.
In relation to Wnt/β-catenin target genes, reports have explored their expressions in oral cancer, with data showing abnormal expressions in a subset of those genes [112,113,114,115]. For example, high levels of cyclin D1, survivin and c-myc, which are classical Wnt/β-catenin targets, have been detected in OSCC samples when compared with control groups [76,116,117,118,119,120], indicating that Wnt/β-catenin target genes might be upregulated in oral cancer, contributing to cell migration and invasion.
Despite that most evidence has shown a low detection of membranous β-catenin in OSCC, and that augmented cytoplasmic β-catenin in invasive cancers is observed at the expense of decreased detection at the cell-cell junctions, this protein appears only sporadically located in the nucleus of OSCC. Hence, the assessment of functionality in this pathway becomes relevant, namely by exploring downstream transcriptions and their consequences in cell fates. This is due, in part, to the limited use of methodologies that are necessary to assess the status of Wnt/β-catenin-dependent events, such as gene expression. In this context, recent studies have explored the mechanistic aspects of β-catenin-dependent events in early lesions of the oral mucosa based on both in vitro models and in clinical samples (see the following sections).
3.4. Wnt Inhibitors in Oral Carcinogenesis
The Wnt/β-catenin signaling pathway is subject to regulation by Wnt antagonists, which include members of the Dickkopf family (Dkk), Wnt inhibitory factor 1 (WIF-1) and secreted frizzled-related proteins (SFRPs). Thus, a balanced function between the Wnt ligands and their inhibitors contributes to cell proliferation in normal tissues [37,121]. In different cancers, decreased expressions of these antagonists have been shown via epigenetic transcriptional silencing [35,122,123,124,125], and, in oral carcinogenesis, it has been reported that the silencing of these inhibitors (e.g., via DNA methylation) leads to cytoplasmic accumulation and the nuclear translocation of β-catenin with the concomitant activation of target genes such as c-myc and cyclin D1 [71,108,126]. These events become noticeable when compared with healthy oral mucosa, where epigenetic silencing of these inhibitors is not frequent, β-catenin depicts a membranous expression and Wnt ligands are poorly expressed [71,108,126], despite the highly proliferative nature of oral mucosa.
In OSCC, members of the SFRP gene family, which prevent the binding of Wnt to frizzled receptors, are epigenetically downregulated via hypermethylation in their promoters [71]. Additionally, the role of SFRP2 in OSCC has been investigated by using in vivo and in vitro models, observing that the methylation of the SFRP2 promoter occurred more frequently in the tumor region than in the nontumor adjacent tissue and that the mRNA encoding for SFRP2 was significantly inhibited in OSCC biopsies [126]. In the same line, it has been suggested that changes in β-catenin localization are likely due to epigenetic modifications that affect the expression of SFRP and WIF-1 in OSCC [108], raising the possibility that methylation levels serve as possible prognostic approaches in OSCC [127]. In order to have a better understanding of the relevance of Wnt antagonists in oral carcinogenesis, new studies will be required. Likewise, the roles that these antagonists might play in premalignant lesions remain unexplored yet.
4. Mechanisms Involved in the Aberrant Activation of β-Catenin in Oral Cancer
4.1. Endosomal Trafficking and Wnt/β-Catenin Signaling
For years, the endocytic trafficking was considered as a mechanism of downregulation in signaling pathways, based on the fact that the internalization of the cell surface receptors and their subsequent transport from early endosomes to late endosomes and lysosomes leads to their degradation [128]. However, endocytosis plays a key role in the activation of signaling pathways, including the canonical Wnt pathway, since the internalization of the trimeric complex, formed by the Wnt ligand, Frizzled and LRP6, along with the β-catenin destruction complex, in endosomal compartments is necessary for the activation of the Wnt/β-catenin pathway [129].
Specifically, the binding of Wnt ligands causes phosphorylation of the cytoplasmic tail of LRP5/6 by GSK3β and CK1α. This phosphorylation enables the binding of the remaining proteins of the destruction complex at the plasma membrane, forming a structure known as “LRP6 signalosome”, which contains aggregates of Frizzled, phosphorylated LRP5/6, Dvl, axin, APC and GSK3β and whose complex is internalized in endosomal structures [129,130,131,132,133]. The inactivation of GSK3β has been considered as a key step in Wnt/β-catenin signaling, which begins with its binding to the LPR5/6 cytoplasmic tail and continues after its sequestration in early endosomes and multivesicular bodies. In this way, β-catenin cannot be phosphorylated by GSK3β, thus allowing its stabilization in the cytoplasm and subsequent translocation to the nucleus, where it activates the transcription of the target genes [134]. Although the mechanism of sequestration of the β-catenin destruction complex has been described, the role that components of the endocytic machinery play in this phenomenon has not been fully explored. In this scenario, recent studies have shown that endocytic proteins are relevant for Wnt/β-catenin signaling, thus providing insights into the mechanisms involved in β-catenin upregulation in oral cancer. In the following sections, we will discuss novel connections between endocytic trafficking and Wnt/β-catenin signaling in oral cancer by, firstly, introducing the role of endocytic trafficking in cancer and, then, its relevance in oral carcinogenesis via modulation of the Wnt/β-catenin pathway.
4.2. Deregulated Endocytosis in Cancer
A body of evidence accumulated in the past two decades supported the notion that uncontrolled endocytic trafficking is a recurrent phenomenon in malignancy, which is due, in part, to the fact that endosome trafficking controls a plethora of cellular processes [135,136]. In this context, it has been demonstrated that endocytic trafficking impacts at different aspects of the cell function, namely the availability of cell surface molecules that sustain cell signaling and cell interactions with the surrounding environment and by providing signaling platforms at the so-called signaling endosomes [129,137]. On the one hand, the endocytosis of cell adhesion molecules, such as E-cadherin, contribute to the epithelial-to-mesenchymal transition during tumor progression, whereas endocytic trafficking of cell-extracellular adhesion molecules, specifically integrins, along with the turnover of integrin-based focal adhesions, represent key events to sustain tumor cell migration, invasion and metastasis [137]. On the other hand, compartmentalization at the so-called “signaling endosomes” provides signaling platforms that orchestrate the activation of molecules involved in tumor cell proliferation, migration and metastasis, such as Rac1, MAPK and tyrosine kinases [129]. However, another layer of complexity is added by the fact that endosomes are also known to “switch off” negative regulators of signaling pathways, such as the Wnt/β-catenin pathway, for which sequestration of the destruction complex within multivesicular bodies allows β-catenin stabilization, nuclear translocation and the transcription of target genes [129,134]. Consequently, increasing studies have reported a deregulation of endocytic proteins in cancer, such as small GTPases, tethering molecules, effectors and adaptor proteins [135,137]. This is particularly observed for small GTPases of the Rab family of proteins, because they are master regulators of endosome trafficking and dynamics [138]. Specifically, Rab proteins are known to control cell adhesion, viability, anchorage independency, tumor cell migration, invasion and metastasis by a plethora of mechanisms, including enhanced integrin traffic [139,140,141,142], focal adhesion disassembly [143,144,145], Rho-GTPase balance [146,147,148] and mitosis [149], among many other events.
In oral cancer, alterations in endosomal proteins and regulators of intracellular trafficking have been reported, and these include the overexpression of Rab proteins [150,151], caveolin-1 [152] and the large GTPase GBP1 [153], whereas epigenetic silencing of the endocytic recycling regulator, Rab25, is associated with lymph node metastasis in oral and oropharyngeal squamous cell carcinomas [154,155]. Alternatively, the amplification of chromosomal regions encompassing genes that encode for Rab5, Rab7 and Rab11 has been associated with the upregulation of these GTPases in clinical samples of metastatic OSCC, and their expressions are associated with increased tumor cell migration and poor patient survival [151]. In another study, an immunohistochemical analysis showed an overexpression of Rab5 in 50% of cases of OSCC [156].
Based on these observations, it is relevant to explore possible consequences derived from Rab upregulation in OSCC, especially traits associated with malignancy. Intriguingly, whether these GTPases are altered at the early stages of oral mucosal carcinogenesis remains largely unknown. In this context, our recent studies indicate that a subset of Rab-GTPases are upregulated in oral dysplasia [14]. Specifically, the activity of Rab5, but not Rab7 or Rab11, is increased in dysplastic oral keratinocytes when compared with nondysplastic oral keratinocytes. In accordance with this, the progressive enlargement of early endosomes is detected through the different grades of oral dysplasia, as shown in biopsies of mild, moderate and severe dysplasia (Figure 4) [14]. Consequences of such upregulation are yet to be fully understood, but augmented Rab5 activity in oral dysplasia is known to be associated with the aberrant localization of β-catenin to the nucleus of dysplastic oral keratinocytes due to the augmented sequestration of the β-catenin destruction complex in endosomes (Figure 4), as it will be described in the following section.
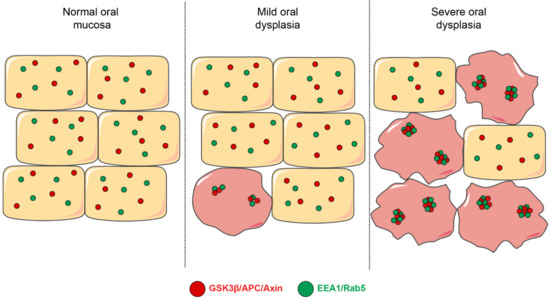
Figure 4.
Early endosomes and their colocalization with components of the β-catenin destruction complex. This figure depicts a model that summarizes different aspects related to the mechanisms involved in Wnt/β-catenin signaling in oral dysplasia. First, early endosomes (shown in green, EEA1- and Rab5-positive) are progressively enlarged throughout the different stages of oral dysplasia in a manner that their co-localization with the components of the β-catenin destruction complex is also augmented during the progression of oral dysplasia. Specifically, the proteins APC, axin and GSK3β have been shown to increasingly colocalize with EEA1- and Rab5-positive early endosomes.
4.3. Endosomal Sequestration of the Destruction Complex in Oral Dysplasia
Despite that the nuclear accumulation of β-catenin is recognized as a recurrent event in oral dysplasia, mechanisms accounting for such phenomenon have remained elusive [11,15,16,27]. Recently, a link was established between this phenomenon and components of the endosomal machinery in a model that proposes that augmented Rab5 activity accounts for early endosome enlargement in dysplastic oral keratinocytes [14]. This event is followed by the increased sequestration of components of the β-catenin destruction complex, including APC, axin and GSK3β, within EEA1- and Rab5-positive compartments (Figure 4). Although this study did not explore the fate of the β-catenin destruction complex, data suggested that a subset of proteins forming this complex are targeted en route to the endo-lysosomal degradative pathway in dysplastic oral keratinocytes, since both proteins GSK3β and axin appeared enriched in fractions that were resistant against the proteinase K treatment, suggesting their accumulation in multivesicular bodies [14] (see proposed model, Figure 5). Confirmation of these possibilities will require further assessments by alternative approaches, including colocalization analyses of components of the destruction complex and late endosome/lysosome markers, as well as electron microscopy for ultrastructural assessments, as previously described [134]. Alternatively, causal-effect studies in cell culture models would permit evaluating whether the inhibition of the endo-lysosomal system is followed by a diminished downregulation of the destruction complex, hence preventing the aberrant stabilization of β-catenin in dysplastic oral keratinocytes.
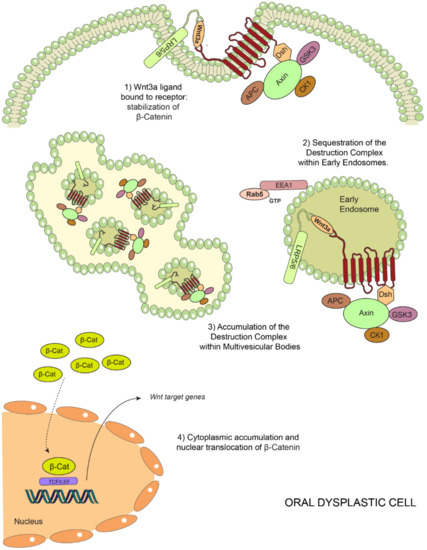
Figure 5.
Model of endosomal sequestration of the destruction complex in oral dysplasia. Binding of Wnt3a to Frizzled (FZ) and LRP5/6 leads to the recruitment of the destruction complex to the FZ-LRP5/6 receptor complex at the plasma membrane. This is followed by a decreased proteasomal degradation of β-catenin (1). Ligand binding and subsequent posttranslational modifications (not described in this scheme) lead to endocytosis of this supramolecular complex, also known as the “Wnt signalosome complex”, and subsequent trafficking en route to early endosomes and multivesicular bodies in a Rab5-dependent manner (2). In oral dysplasia, high Wnt3a levels and increased Rab5 activity lead to the enhanced sequestration of the destruction complex within EEA1-positive early endosomes. Consequently, higher levels of components of the destruction complex are detected in multivesicular bodies (3). These events ultimately lead to a more robust stabilization of β-catenin in the cytoplasm and a consequent nuclear translocation in order to bind TCF/LEF factors, activating the transcription of the target genes (4).
Although further research is still required, and that more mechanistic insights should be drawn by in vitro approaches, it must be emphasized that current evidence showing the endosomal sequestration of components of the β-catenin destruction complex in patient biopsies represents a landmark finding to understand the “deregulation” of this pathway during oral carcinogenesis [11,14]. Intriguingly, whether this mechanism remains upregulated at late stages of carcinogenesis, namely carcinoma in situ and frank invasive OSCC, is an issue that needs to be explored in order to identify potential markers for predicting oral cancer progression.
5. Therapeutic Approaches Based on Targeting Wnt Secretion
As discussed, the Wnt/β-catenin pathway appears to be relevant in several malignancies, and, hence, therapies aiming to target Wnt signaling represent attractive therapeutic approaches. For instance, recombinant proteins that minimize Wnt-Frizzled interactions or Wnt inhibition have been proposed and thought to impact the outcomes of cancers with deregulated Wnt ligand secretions [157,158,159,160]. In oral cancer, little has been explored about possible Wnt-based therapies, which is mainly due to the lack of understanding about the mechanisms involved in Wnt secretion, cellular targets and downstream signaling. As previously explained, the ligand-mediated activation of Wnt/β-catenin is the best-known mechanism associated with β-catenin stabilization and nuclear translocation in oral cancer cells and dysplastic keratinocytes, and, hence, the remaining paragraphs will be dedicated to discussing some possibilities in this scenario.
Following their translation at the endoplasmic reticulum, Wnt ligands are posttranslationally modified by palmitoylation and glycosylation [34,38,161]. Palmitoylation is achieved by the enzyme o-acyl-transferase porcupine (PORCN) in a step that is essential for Wnt ligand secretion [161,162,163]. With this information, studies have shown that the inhibition of PORCN with the compound Wnt-C59 delays tumor growth, as observed in a model of breast cancer in transgenic mice, further indicating that the use of such inhibitors is safe and feasible in preclinical models [49]. Likewise, Cheng et al. reported that Wnt-C59 suppresses the growth of tumors derived from nasopharyngeal carcinoma in a murine model [50]. In the same line, it has been shown that compound LGK974, which is a potent inhibitor of Wnt signaling, is highly effective in decreasing tumor growth in mice [164]. Collectively, these evidences indicate that the inhibition of Wnt secretion is efficient in decreasing tumor growth when facing the aberrant activation of the Wnt pathway.
Conversely to these descriptions, which have been focused on the cancer stage itself, no study is available about a potential use of these pharmacological inhibitors at earlier stages of oral carcinogenesis, whose targeting should be significant to preclude tumor progression. To this end, a more comprehensive understanding of the mechanisms involved in Wnt activation during oral carcinogenesis will be required to rationally propose effective therapies in oral cancer.
6. Conclusions and Perspectives
Consequences of uncontrolled Wnt/β-catenin signaling in oral cancer are becoming elucidated, with special emphasis on the early events of oral carcinogenesis. Particularly, an increase in the secretion of Wnt ligands has a fundamental role in the progression of oral cancer through its effect on the processes of tumorigenesis and metastasis. In this context, therapeutic approaches aiming to use Wnt inhibitors appear advantageous for cancers associated with the overexpression of Wnt ligands, since the evidence supports their safety and feasibility in preclinical models, with a powerful inhibitory effect on Wnt signaling. This is expected to be impactful, not only by reducing the growth of tumors caused by the aberrant activation of this pathway, but also, by bringing promising therapies of chemoprevention in order to manage early malignant lesions and prevent their progression towards cancer.
Recent studies indicate the involvement of endocytic proteins in promoting Wnt/β-catenin signaling during oral carcinogenesis, an effect namely attributed to their effects in the endosomal sequestration of the β-catenin destruction complex. Although these mechanisms are not completely understood, current models propose that augmented Rab5 activity accounts for early endosome enlargement, followed by increased sequestration of the components of the β-catenin destruction complex and the nuclear localization of β-catenin, which represents a landmark finding to understand the “deregulation” of this pathway during oral carcinogenesis (Figure 6). Further research is needed to have a comprehensive understanding of this mechanism and whether it remains upregulated at the late stages of oral carcinogenesis. This will also permit the identification and validation of possible markers to predict the progression of oral cancer and to propose potential, new and effective therapeutic objectives that favor both the quality of life and the survival of patients in the process of developing carcinoma in situ and frank invasive OSCC.
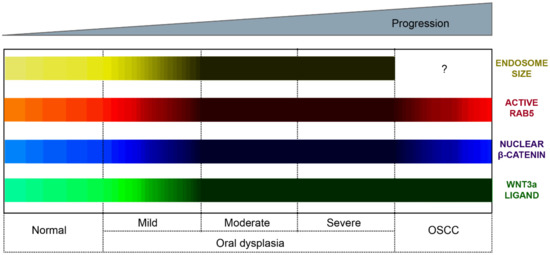
Figure 6.
Subcellular and molecular events in oral carcinogenesis. This scheme provides a summary of subcellular and molecular changes observed during oral carcinogenesis. These changes include the Wnt3a expression, nuclear detection of β-catenin, endosome enlargement and the activation status of Rab5 GTPase. We propose a model whereby oral carcinogenesis is associated with the progressive expression of Wnt3a and the consequent stabilization and nuclear translocation of β-catenin. These events are accompanied by the continuous activation of Rab5-GTPase, endosome enlargement and the increased sequestration of the destruction complex within endosomes (see main text for details). Color codes represent the Wnt3a expression (green bar), nuclear β-catenin detection (blue bar), Rab5 activity (red bar) and early endosome size (yellow bar). The lighter the color, the lower the expression/detection/activity/size. The darker the color, the higher the expression/detection/activity/size for each case. The Wnt3a representation summarizes evidence obtained from immunohistochemical analyses in clinical samples and in vitro measurements of ligand secretions in cell culture models. The nuclear β-catenin representation summarizes evidence obtained in both clinical samples and cell culture models. The nuclear detection progressively increases from normal oral mucosa through the different stages of oral dysplasia (mild, moderate and severe); however, the nuclear detection of this protein decreases in OSCC. The Rab5 activity (Rab5-GTP levels) has been measured in cell culture models and is shown substantially increased in dysplastic oral keratinocytes (models of moderate/severe dysplasia) in comparison with nondysplastic oral keratinocytes and OSCC cells. The endosomal size (early endosomes) has been measured by tissue immunofluorescence in clinical samples of normal mucosa and mild, moderate and severe dysplasia, as well as in cell culture models, using different markers of early endosomes (the interrogation symbol indicates that this has not been explored in OSCC).
Author Contributions
Conceptualization, writing—original draft preparation and review and editing, M.R., T.F., D.B., D.P.-O. and V.A.T.; project administration and funding acquisition, M.R. and V.A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Fund for Scientific and Technological Development (FONDECYT) 1180495 (to V.A.T.), the U-Inicia Program at Universidad de Chile UI-024/19 (to M.R.) and the Advanced Center for Chronic Diseases, FONDAP ACCDiS 15130011 (to V.A.T.).
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript or in the decision to publish the results.
References
- Haddad, R.I.; Shin, D.M. Recent advances in head and neck cancer. N. Engl. J. Med. 2008, 359, 1143–1154. [Google Scholar] [CrossRef]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Neville, B.W.; Day, T.A. Oral cancer and precancerous lesions. CA. Cancer J. Clin. 2002, 52, 195–215. [Google Scholar] [CrossRef]
- Polanska, H.; Raudenska, M.; Gumulec, J.; Sztalmachova, M.; Adam, V.; Kizek, R.; Masarik, M. Clinical significance of head and neck squamous cell cancer biomarkers. Oral Oncol. 2014, 50, 168–177. [Google Scholar] [CrossRef]
- Genden, E.M.; Ferlito, A.; Silver, C.E.; Takes, R.P.; Suarez, C.; Owen, R.P.; Haigentz, M.; Stoeckli, S.J.; Shaha, A.R.; Rapidis, A.D.; et al. Contemporary management of cancer of the oral cavity. Eur. Arch. Otorhinolaryngol. 2010, 267, 1001–1017. [Google Scholar] [CrossRef]
- Dost, F.; Le Cao, K.; Ford, P.J.; Ades, C.; Farah, C.S. Malignant transformation of oral epithelial dysplasia: A real-world evaluation of histopathologic grading. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2014, 117, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S.; Reibel, J.; Bouquot, J.; Dabelsteen, E. Oral epithelial dysplasia classification systems: Predictive value, utility, weaknesses and scope for improvement. J. Oral. Pathol. Med. 2008, 37, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Awadallah, M.; Idle, M.; Patel, K.; Kademani, D. Management update of potentially premalignant oral epithelial lesions. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2018, 125, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Dietel, M.; Johrens, K.; Laffert, M.V.; Hummel, M.; Blaker, H.; Pfitzner, B.M.; Lehmann, A.; Denkert, C.; Darb-Esfahani, S.; Lenze, D.; et al. A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: A review focussing on clinical relevance. Cancer Gene Ther. 2015, 22, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulou, E.A.; Stebbing, J.; Scully, C. Targeted cancer therapies. J. Am. Dent. Assoc. 2018, 149, 100–111. [Google Scholar] [CrossRef]
- Reyes, M.; Pena-Oyarzun, D.; Maturana, A.; Torres, V.A. Nuclear localization of beta-catenin and expression of target genes are associated with increased Wnt secretion in oral dysplasia. Oral. Oncol. 2019, 94, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Sha, J.; Kanno, T. The role of carcinogenesis-related biomarkers in the wnt pathway and their effects on Epithelial–Mesenchymal Transition (EMT) in oral squamous cell carcinoma. Cancers 2020, 12, 555. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.A.P.; Noguti, J.; Oshima, C.T.F.; Ribeiro, D.A. Effective targeting of the epidermal growth factor receptor (EGFR) for treating oral cancer: A promising approach. Anticancer Res. 2014, 34, 1547–1552. [Google Scholar] [PubMed]
- Reyes, M.; Pena-Oyarzun, D.; Silva, P.; Venegas, S.; Criollo, A.; Torres, V.A. Nuclear accumulation of beta-catenin is associated with endosomal sequestration of the destruction complex and increased activation of Rab5 in oral dysplasia. Faseb J. 2020, 34, 4009–4025. [Google Scholar] [CrossRef]
- Ishida, K.; Ito, S.; Wada, N.; Deguchi, H.; Hata, T.; Hosoda, M.; Nohno, T. Nuclear localization of beta-catenin involved in precancerous change in oral leukoplakia. Mol. Cancer 2007, 6, 62. [Google Scholar] [CrossRef]
- Sato, K.; Okazaki, Y.; Tonogi, M.; Tanaka, Y.; Yamane, G.-Y. Expression of beta-catenin in rat oral epithelial dysplasia induced by 4-nitroquinoline 1-oxide. Oral Oncol. 2002, 38, 772–778. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Boil. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Boil. 2012, 4, a011163. [Google Scholar] [CrossRef]
- Balint, K.; Xiao, M.; Pinnix, C.C.; Soma, A.; Veres, I.; Juhasz, I.; Brown, E.J.; Capobianco, A.J.; Herlyn, M.; Liu, Z.-J. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J. Clin. Investig. 2005, 115, 3166–3176. [Google Scholar] [CrossRef]
- Santoro, A.; Pannone, G.; Papagerakis, S.; McGuff, H.S.; Cafarelli, B.; Lepore, S.; De Maria, S.; Rubini, C.; Mattoni, M.; Staibano, S.; et al. Beta-catenin and epithelial tumors: A study based on 374 oropharyngeal cancers. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Zhang, B.; Xi, G.-M.; Wu, Y.; Liu, H.-B.; Liu, Y.-F.; Xu, W.-J.; Zhu, Q.-Q.; Cai, F.; Zhou, Z.-J.; et al. PRC1 contributes to tumorigenesis of lung adenocarcinoma in association with the Wnt/β-catenin signaling pathway. Mol. Cancer 2017, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Khalil, S.; Tan, G.A.; Giri, D.D.; Zhou, X.K.; Howe, L. Activation status of Wnt/ß-catenin signaling in normal and neoplastic breast tissues: Relationship to HER2/NEU expression in human and mouse. PLoS ONE 2012, 7, e33421. [Google Scholar] [CrossRef]
- Ravindran, G.; Devaraj, H. Aberrant expression of beta-catenin and its association with DeltaNp63, Notch-1, and clinicopathological factors in oral squamous cell carcinoma. Clin. Oral. Investig. 2012, 16, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Marimuthu, M.; Andiappan, M.; Wahab, A.; Muthusekhar, M.R.; Balakrishnan, A.; Shanmugam, S. Canonical Wnt pathway gene expression and their clinical correlation in oral squamous cell carcinoma. Indian J. Dent. Res. 2018, 29, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Sawhney, M.; DattaGupta, S.; Shukla, N.K.; Srivastava, A.; Walfish, P.G.; Ralhan, R. Clinical significance of altered expression of beta-catenin and E-cadherin in oral dysplasia and cancer: Potential link with ALCAM expression. PLoS ONE 2013, 8, e67361. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.; Rojas-Alcayaga, G.; Maturana, A.; Aitken, J.P.; Rojas, C.; Ortega, A.V. Increased nuclear beta-catenin expression in oral potentially malignant lesions: A marker of epithelial dysplasia. Med. Oral. Patol. Oral. Cir. Bucal. 2015, 20, e540. [Google Scholar] [CrossRef]
- Iwai, S.; Katagiri, W.; Kong, C.; Amekawa, S.; Nakazawa, M.; Yura, Y. Mutations of the APC, beta-catenin, and axin 1 genes and cytoplasmic accumulation of beta-catenin in oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2005, 131, 773–782. [Google Scholar] [CrossRef]
- Gasparoni, A.; Chaves, A.; Fonzi, L.; Johnson, G.K.; Schneider, G.B.; Squier, C.A. Subcellular localization of beta-catenin in malignant cell lines and squamous cell carcinomas of the oral cavity. J. Oral Pathol. Med. 2002, 31, 385–394. [Google Scholar] [CrossRef]
- Tsuchiya, R.; Yamamoto, G.; Nagoshi, Y.; Aida, T.; Irié, T.; Tachikawa, T. Expression of adenomatous polyposis coli (APC) in tumorigenesis of human oral squamous cell carcinoma. Oral Oncol. 2004, 40, 932–940. [Google Scholar] [CrossRef]
- Yeh, K.-T.; Chang, J.-G.; Lin, T.-H.; Wang, Y.-F.; Chang, J.-Y.; Shih, M.-C.; Lin, C.-C. Correlation between protein expression and epigenetic and mutation changes of Wnt pathway-related genes in oral cancer. Int. J. Oncol. 2003, 23, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Uraguchi, M.; Morikawa, M.; Shirakawa, M.; Sanada, K.; Imai, K. Activation of WNT family expression and signaling in squamous cell carcinomas of the oral cavity. J. Dent. Res. 2004, 83, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Odajima, T.; Sasaki, Y.; Tanaka, N.; Kato-Mori, Y.; Asanuma, H.; Ikeda, T.; Satoh, M.; Hiratsuka, H.; Tokino, T.; Sawada, N. Abnormal beta-catenin expression in oral cancer with no gene mutation: Correlation with expression of cyclin D1 and epidermal growth factor receptor, Ki-67 labeling index, and clinicopathological features. Hum. Pathol. 2005, 36, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Nusse, R. Wnt Proteins. Cold Spring Harb. Perspect. Boil. 2012, 4, a007864. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.-M.; Niehrs, C. Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harb. Perspect. Boil. 2012, 5, a015081. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Mikels, A.J.; Nusse, R. Wnts as ligands: Processing, secretion and reception. Oncogene 2006, 25, 7461–7468. [Google Scholar] [CrossRef]
- Gordon, M.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Boil. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Fuentes, R.G.; Arai, M.A.; Ishibashi, M. Natural compounds with Wnt signal modulating activity. Nat. Prod. Rep. 2015, 32, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Nagse, H.; Ando, H.; Horii, A.; Ichii, S.; Nakatsuru, S.; Aoki, T.; Miki, Y.; Mori, T.; Nakamura, Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum. Mol. Genet. 1992, 1, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef]
- Boynton, R.F.; Blount, P.L.; Yin, J.; Brown, V.L.; Huang, Y.; Tong, Y.; McDaniel, T.; Newkirk, C.; Resau, J.H.; Raskind, W.H. Loss of heterozygosity involving the APC and MCC genetic loci occurs in the majority of human esophageal cancers. Proc. Natl. Acad. Sci. USA 1992, 89, 3385–3388. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; Wichen, D.V.; Weger, R.D.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a β-catenin-tcf complex in apc−/−colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef]
- Coste, A.D.L.; Romagnolo, B.; Billuart, P.; Renard, C.-A.; Buendia, M.A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; et al. Somatic mutations of the -catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 1998, 95, 8847–8851. [Google Scholar] [CrossRef]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013, 73, 502–507. [Google Scholar] [CrossRef]
- Cheng, Y.; Phoon, Y.P.; Jin, X.; Chong, S.Y.S.; Ip, J.C.Y.; Wong, B.W.Y.; Lung, M.L. Wnt-C59 arrests stemness and suppresses growth of nasopharyngeal carcinoma in mice by inhibiting the Wnt pathway in the tumor microenvironment. Oncotarget 2015, 6, 14428–14439. [Google Scholar] [CrossRef][Green Version]
- Vermeulen, L.; Melo, F.D.S.E.; Van Der Heijden, M.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Roberts, L.R.; Aderca, I.N.; Dong, X.; Qian, C.; Murphy, L.M.; Nagorney, D.M.; Burgart, L.J.; Roche, P.C.; I Smith, D.; et al. Mutational spectrum of β-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002, 21, 4863–4871. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Fukuchi, M.; Miyazaki, T.; Masuda, N.; Kato, H.; Kuwano, H. Reduced expression of Axin correlates with tumour progression of oesophageal squamous cell carcinoma. Br. J. Cancer 2003, 88, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Pukrop, T.; Klemm, F.; Hagemann, T.; Gradl, D.; Schulz, M.; Siemes, S.; Trumper, L.; Binder, C. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 5454–5459. [Google Scholar] [CrossRef]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Xia, W.; Wang, J.C.; Kwong, K.Y.; Spohn, B.; Wen, Y.; Pestell, R.G.; Hung, M.-C. β-catenin, a novel prognostic marker for breast cancer: Its roles in cyclin D1 expression and cancer progression. Proc. Natl. Acad. Sci. USA 2000, 97, 4262–4266. [Google Scholar] [CrossRef]
- Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Rothberg, B.E.G.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; McMahon, M.; et al. β-Catenin signaling controls metastasis in braf-activated pten-deficient melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef]
- Hanaki, H.; Yamamoto, H.; Sakane, H.; Matsumoto, S.; Ohdan, H.; Sato, A.; Kikuchi, A. An anti-wnt5a antibody suppresses metastasis of gastric cancer cells in vivo by inhibiting receptor-mediated endocytosis. Mol. Cancer Ther. 2012, 11, 298–307. [Google Scholar] [CrossRef]
- Huang, T.-C.; Lee, P.-T.; Wu, M.-H.; Huang, C.-C.; Ko, C.-Y.; Lee, Y.-C.; Lin, D.-Y.; Cheng, Y.-W.; Lee, K.-H. Distinct roles and differential expression levels of Wnt5a mRNA isoforms in colorectal cancer cells. PLoS ONE 2017, 12, e0181034. [Google Scholar] [CrossRef]
- Wang, T.; Liu, X.; Wang, J. Up-regulation of Wnt5a inhibits proliferation and migration of hepatocellular carcinoma cells. J. Cancer Res. Ther. 2019, 15, 904–908. [Google Scholar] [CrossRef]
- Jia, S.; Qu, T.; Feng, M.; Ji, K.; Li, Z.; Jiang, W.G.; Ji, J. Association of Wnt1-inducible signaling pathway protein-1 with the proliferation, migration and invasion in gastric cancer cells. Tumor Boil. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, K.; Wu, J.; Shi, J.; Xue, J.; Li, J.; Chen, J.; Zhu, Y.; Wei, J.; He, J.; et al. Wnt5a increases properties of lung cancer stem cells and resistance to cisplatin through activation of Wnt5a/PKC signaling pathway. Stem Cells Int. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tang, Z.; Gong, H.; Zhu, L.; Liu, X. Wnt5a promotes epithelial-to-mesenchymal transition and metastasis in non-small-cell lung cancer. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Whang, Y.M.; Jo, U.; Sung, J.S.; Ju, H.J.; Kim, H.K.; Park, K.H.; Lee, J.W.; Koh, I.S.; Kim, Y.H. Wnt5a is associated with cigarette smoke-related lung carcinogenesis via protein kinase C. PLoS ONE 2013, 8, e53012. [Google Scholar] [CrossRef]
- Lu, W.; Tinsley, H.; Keeton, A.; Qu, Z.; Piazza, G.A.; Li, Y. Suppression of Wnt/β-catenin signaling inhibits prostate cancer cell proliferation. Eur. J. Pharmacol. 2008, 602, 8–14. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Lanman, N.A.; Pascuzzi, P.; Gupta, S.; Liu, X. Inhibition of the Wnt/β-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar] [CrossRef]
- Aldahl, J.; Mi, J.; Pineda, A.; Kim, W.K.; Olson, A.; Hooker, E.; He, Y.; Yu, E.-J.; Le, V.; Lee, D.-H.; et al. Aberrant activation of hepatocyte growth factor/MET signaling promotes β-catenin–mediated prostatic tumorigenesis. J. Boil. Chem. 2020, 295, 631–644. [Google Scholar] [CrossRef]
- Prgomet, Z.; Andersson, T.; Lindberg, P. Higher expression of WNT5A protein in oral squamous cell carcinoma compared with dysplasia and oral mucosa with a normal appearance. Eur. J. Oral Sci. 2017, 125, 237–246. [Google Scholar] [CrossRef]
- Prgomet, Z.; Axelsson, L.; Lindberg, P.; Andersson, T. Migration and invasion of oral squamous carcinoma cells is promoted by WNT5A, a regulator of cancer progression. J. Oral Pathol. Med. 2014, 44, 776–784. [Google Scholar] [CrossRef]
- Filho, P.A.; Letra, A.; Cramer, A.; Prasad, J.; Garlet, G.P.; Vieira, A.; Ferris, R.; Menezes, R. Insights from studies with oral cleft genes suggest associations between WNT-pathway genes and risk of oral cancer. J. Dent. Res. 2011, 90, 740–746. [Google Scholar] [CrossRef]
- Sogabe, Y.; Suzuki, H.; Toyota, M.; Ogi, K.; Imai, T.; Nojima, M.; Sasaki, Y.; Hiratsuka, H.; Tokino, T. Epigenetic inactivation of SFRP genes in oral squamous cell carcinoma. Int. J. Oncol. 2008, 32, 1253–1261. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nasser, W.; Flechtenmacher, C.; Holzinger, D.; Hofele, C.; Bosch, F.X. Aberrant expression of p53, p16INK4a and Ki-67 as basic biomarker for malignant progression of oral leukoplakias. J. Oral Pathol. Med. 2011, 40, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Trivedi, T.; Tankshali, R.; Goswami, J.; Shah, J.; Jetly, D.; Kobawala, T.; Patel, K.; Shukla, S.; Shah, P.; et al. Molecular alterations in oral carcinogenesis: Significant risk predictors in malignant transformation and tumor progression. Int. J. Boil. Markers 2007, 22, 132–143. [Google Scholar] [CrossRef]
- Ramakrishna, A.; Shreedhar, B.; Narayan, T.; Mohanty, L.; Shenoy, S.; Jamadar, S. Cyclin D1 an early biomarker in oral carcinogenesis. J. Oral Maxillofac. Pathol. 2013, 17, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Cordell, K.G.; Lee, J.S.; Worden, F.P.; Prince, M.E.; Tran, H.H.; Wolf, G.T.; Urba, S.G.; Chepeha, D.B.; Teknos, T.N.; et al. EGFR, p16, HPV titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J. Clin. Oncol. 2008, 26, 3128–3137. [Google Scholar] [CrossRef]
- Guan, G.; Bakr, M.M.; Firth, N.; Love, R.M. Expression of cyclin D1 correlates with p27KIP1 and regulates the degree of oral dysplasia and squamous cell carcinoma differentiation. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2018, 126, 174–183. [Google Scholar] [CrossRef]
- Ramasubramanian, A.; Ramani, P.; Sherlin, H.J.; Premkumar, P.; Natesan, A.; Thiruvengadam, C. Immunohistochemical evaluation of oral epithelial dysplasia using cyclin-D1, p27 and p63 expression as predictors of malignant transformation. J. Nat. Sci. Boil. Med. 2013, 4, 349–358. [Google Scholar] [CrossRef]
- Shah, N.G.; Trivedi, T.I.; Tankshali, R.A.; Goswami, J.V.; Jetly, D.H.; Shukla, S.N.; Shah, P.M.; Verma, R.J. Prognostic significance of molecular markers in oral squamous cell carcinoma: A multivariate analysis. Head Neck 2009, 31, 1544–1556. [Google Scholar] [CrossRef]
- Edwards, P.C. The natural history of oral epithelial dysplasia: Perspective on Dost et al. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 263–266. [Google Scholar] [CrossRef]
- Speight, P.M. Update on oral epithelial dysplasia and progression to cancer. Head Neck Pathol. 2007, 1, 61–66. [Google Scholar] [CrossRef]
- Van Der Waal, I. Oral potentially malignant disorders: Is malignant transformation predictable and preventable? Med. Oral Patol. Oral Cir. Bucal 2014, 19, e386–e390. [Google Scholar] [CrossRef] [PubMed]
- Cervigne, N.K.; Machado, J.; Goswami, R.; Sadikovic, B.; Bradley, G.; Perez-Ordonez, B.; Galloni, N.N.; Gilbert, R.; Gullane, P.; Irish, J.C.; et al. Recurrent genomic alterations in sequential progressive leukoplakia and oral cancer: Drivers of oral tumorigenesis? Hum. Mol. Genet. 2014, 23, 2618–2628. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Braakhuis, B.J.; Tabor, M.P.; Kummer, J.A.; Leemans, C.R.; Brakenhoff, R.H. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar]
- Kumar, K.U.G.; Sathiasekar, A.C.; Mathew, D.G.; Lal, M.S.J.; Prakash, A.A.A. Oral field cancerization and its clinical implications in the management in potentially malignant disorders. J. Pharm. Bioallied Sci. 2017, 9, S23–S25. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.; Tabor, M.P.; Leemans, C.R.; Van Der Waal, I.; Snow, G.B.; Brakenhoff, R.H. Second primary tumors and field cancerization in oral and oropharyngeal cancer: Molecular techniques provide new insights and definitions. Head Neck 2002, 24, 198–206. [Google Scholar] [CrossRef]
- Francis, G.; Kumar, U.D.; Nalinakumari, K.; Jayasree, K.; Kannan, S. Accumulation of inactive p53 protein in oral squamous cell carcinoma: Stabilization by protein interaction. Eur. J. Oral Sci. 2013, 121, 21–28. [Google Scholar] [CrossRef]
- Tanaka, T.; Tanaka, M.; Tanaka, T. Oral carcinogenesis and oral cancer chemoprevention: A review. Pathol. Res. Int. 2011, 2011, 1–10. [Google Scholar] [CrossRef]
- Bodhade, A.S.; Dive, A.M. Chemoprevention of premalignant and malignant lesions of oral cavity: Recent trends. Eur. J. Dent. 2013, 7, 246–250. [Google Scholar] [CrossRef]
- Warnakulasuriya, S.; Johnson, N.W.; Van Der Waal, I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J. Oral Pathol. Med. 2007, 36, 575–580. [Google Scholar] [CrossRef]
- Fleskens, S.; Slootweg, P.J. Grading systems in head and neck dysplasia: Their prognostic value, weaknesses and utility. Head Neck Oncol. 2009, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, A.K.; Chan, J.K.; Takata, T.; Grandis, J.R.; Slootweg, P.J. The fourth edition of the head and neck World Health Organization blue book: Editors’ perspectives. Hum. Pathol. 2017, 66, 10–12. [Google Scholar] [CrossRef]
- Nag, R.; Das, R.K. Analysis of images for detection of oral epithelial dysplasia: A review. Oral Oncol. 2018, 78, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Reichart, P.; Philipsen, H.P. Oral erythroplakia—A review. Oral Oncol. 2005, 41, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Oliver, R.J.; Macdonald, D.G.; Felix, D.H. Aspects of cell proliferation in oral epithelial dysplastic lesions. J. Oral Pathol. Med. 2000, 29, 49–55. [Google Scholar] [CrossRef]
- Alvarado, C.G.; Maruyama, S.; Cheng, J.; Ida-Yonemochi, H.; Kobayashi, T.; Yamazaki, M.; Takagi, R.; Saku, T. Nuclear translocation of beta-catenin synchronized with loss of E-cadherin in oral epithelial dysplasia with a characteristic two-phase appearance. Histopathology 2011, 59, 283–291. [Google Scholar] [CrossRef]
- Lo Muzio, L.; Lo Russo, L.; Falaschini, S.; Ciavarella, D.; Pentenero, M.; Arduino, P.; Favia, G.; Maiorano, E.; Rubini, C.; Pieramici, T.; et al. beta- and gamma-catenin expression in oral dysplasia. Oral. Oncol. 2009, 45, 501–504. [Google Scholar] [CrossRef]
- Elferink, L.A.; Resto, V.A. Receptor-tyrosine-kinase-targeted therapies for head and neck cancer. J. Signal Transduct. 2011, 2011, 1–11. [Google Scholar] [CrossRef]
- Chang, K.-Y.; Tsai, S.-Y.; Chen, S.-H.; Tsou, H.; Yen, C.-J.; Liu, K.-J.; Fang, H.-L.; Wu, H.-C.; Chuang, B.-F.; Chou, S.-W.; et al. Dissecting the EGFR-PI3K-AKT pathway in oral cancer highlights the role of the EGFR variant III and its clinical relevance. J. Biomed. Sci. 2013, 20, 43. [Google Scholar] [CrossRef]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef]
- Sophia, J.; Kowshik, J.; Mishra, R.; Nagini, S. Nimbolide, a neem limonoid inhibits phosphatidyl inositol-3 kinase to activate glycogen synthase kinase-3β in a hamster model of oral oncogenesis. Sci. Rep. 2016, 6, 22192. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, C. Convergence between Wnt-beta-catenin and EGFR signaling in cancer. Mol. Cancer 2010, 9, 236. [Google Scholar] [CrossRef]
- Lee, C.-H.; Hung, H.-W.; Hung, P.-H.; Shieh, Y.-S. Epidermal growth factor receptor regulates β-catenin location, stability, and transcriptional activity in oral cancer. Mol. Cancer 2010, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Chaw, S.; Majeed, A.A.; Dalley, A.; Chan, A.; Stein, S.; Farah, C.S. Epithelial to mesenchymal transition (EMT) biomarkers–E-cadherin, beta-catenin, APC and Vimentin–in oral squamous cell carcinogenesis and transformation. Oral Oncol. 2012, 48, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Zaid, K.W. Immunohistochemical assessment of E-cadherin and beta-catenin in the histological differentiations of oral squamous cell carcinoma. Asian Pac. J. Cancer Prev. 2014, 15, 8847–8853. [Google Scholar] [CrossRef]
- Yuan, X.; Xu, Q.; Zhang, X.; Van Brunt, L.A.; Ticha, P.; Helms, J. Wnt-responsive stem cell fates in the oral mucosa. iScience 2019, 21, 84–94. [Google Scholar] [CrossRef]
- Lin, C.; Fisher, A.V.; Yin, Y.; Maruyama, T.; Veith, G.M.; Dhandha, M.; Huang, G.J.; Hsu, W.; Ma, L. The inductive role of Wnt-β-Catenin signaling in the formation of oral apparatus. Dev. Boil. 2011, 356, 40–50. [Google Scholar] [CrossRef]
- Pannone, G.; Bufo, P.; Santoro, A.; Franco, R.; Aquino, G.; Longo, F.; Botti, G.; Serpico, R.; Cafarelli, B.; Abbruzzese, A.; et al. WNT pathway in oral cancer: Epigenetic inactivation of WNT-inhibitors. Oncol. Rep. 2010, 24, 1035–1041. [Google Scholar]
- Lyakhovitsky, A.; Barzilai, A.; Fogel, M.; Trau, H.; Huszar, M. Expression of e-cadherin and beta-catenin in cutaneous squamous cell carcinoma and its precursors. Am. J. Dermatopathol. 2004, 26, 372–378. [Google Scholar] [CrossRef]
- Shiah, S.-G.; Hsiao, J.-R.; Chang, W.-M.; Chen, Y.-W.; Jin, Y.-T.; Wong, T.-Y.; Huang, J.-S.; Tsai, S.-T.; Hsu, Y.-M.; Chou, S.-T.; et al. Downregulated miR329 and miR410 promote the proliferation and invasion of oral squamous cell carcinoma by targeting Wnt-7b. Cancer Res. 2014, 74, 7560–7572. [Google Scholar] [CrossRef]
- Xie, H.; Ma, Y.; Li, J.; Chen, H.; Xie, Y.; Chen, M.; Zhao, X.; Tang, S.; Zhao, S.; Zhang, Y.; et al. WNT7A promotes EGF-induced migration of oral squamous cell carcinoma cells by activating β-catenin/MMP9-mediated signaling. Front. Pharmacol. 2020, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Ritta’, M.; De Andrea, M.; Mondini, M.; Mazibrada, J.; Giordano, C.; Pecorari, G.; Garzaro, M.; Landolfo, V.; Schena, M.; Chiusa, L.; et al. Cell cycle and viral and immunologic profiles of head and neck squamous cell carcinoma as predictable variables of tumor progression. Head Neck 2009, 31, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Basnaker, M.; Sp, S.; Bnvs, S. Cyclin d1 gene expression in oral mucosa of tobacco chewers"-an immunohistochemical study. J. Clin. Diagn. Res. 2014, 8, ZC70. [Google Scholar] [CrossRef]
- Perisanidis, B.; Wrba, F.; Brandstetter, A.; El Gazzar, S.; Papadogeorgakis, N.; Kyzas, P.A.; Filipits, M.; Perisanidis, C.; Seemann, R.; Ewers, R. Evaluation of immunohistochemical expression of p53, p21, p27, cyclin D1, and Ki67 in oral and oropharyngeal squamous cell carcinoma. J. Oral Pathol. Med. 2012, 41, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Olimid, D.A.; Simionescu, C.E.; Mărgăritescu, C.; Florescu, A. Immunoexpression of Ki67 and cyclin D1 in oral squamous carcinomas. Rom. J. Morphol. Embryol. 2012, 53 (Suppl. 3), 795–798. [Google Scholar] [PubMed]
- Negi, A.; Puri, A.; Gupta, R.; Nangia, R.; Sachdeva, A.; Mittal, M. Comparison of immunohistochemical expression of antiapoptotic protein survivin in normal oral mucosa, oral leukoplakia, and oral squamous cell carcinoma. Pathol. Res. Int. 2015, 2015, 1–6. [Google Scholar] [CrossRef]
- Deo, P.N.; Deshmukh, R. Expression of survivin in dysplasia and different grades of oral squamous cell carcinoma. Transl. Res. Oral Oncol. 2017, 2. [Google Scholar] [CrossRef]
- Li, S.; Yang, Y.; Ding, Y.; Tang, X.; Sun, Z. Impacts of survivin and caspase-3 on apoptosis and angiogenesis in oral cancer. Oncol. Lett. 2017, 14, 3774–3779. [Google Scholar] [CrossRef]
- Srinivasan, M.; Jewell, S. Quantitative estimation of PCNA, c-myc, EGFR and TGF-α in oral submucous fibrosis—An immunohistochemical study. Oral Oncol. 2001, 37, 461–467. [Google Scholar] [CrossRef]
- Pallavi, N.; Nalabolu, G.R.K.; Hiremath, S.K.S. Bcl-2 and c-Myc expression in oral dysplasia and oral squamous cell carcinoma: An immunohistochemical study to assess tumor progression. J. Oral Maxillofac. Pathol. 2018, 22, 325–331. [Google Scholar] [CrossRef]
- Voronkov, A.; Krauss, S. Wnt/beta-catenin signaling and small molecule inhibitors. Curr. Pharm. Des. 2013, 19, 634–664. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Sasaki, S.; Suzuki, H.; Toyota, M.; Maruyama, R.; Nojima, M.; Yamamoto, H.; Omata, M.; Tokino, T.; Imai, K.; et al. Frequent epigenetic inactivation of SFRP genes in hepatocellular carcinoma. J. Gastroenterol. 2008, 43, 378–389. [Google Scholar] [CrossRef]
- Lee, J.; Yoon, Y.S.; Chung, J.H. Epigenetic silencing of the WNT antagonist DICKKOPF-1 in cervical cancer cell lines. Gynecol. Oncol. 2008, 109, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-W.; Chung, M.-T.; Lai, H.-C.; De Yan, M.; Shih, Y.-L.; Chang, C.-C.; Yu, M.-H. Methylation analysis of SFRP genes family in cervical adenocarcinoma. J. Cancer Res. Clin. Oncol. 2009, 135, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Rawson, J.B.; Manno, M.; Mrkonjic, M.; Daftary, D.; Dicks, E.; Buchanan, D.; Younghusband, H.B.; Parfrey, P.S.; Young, J.; Pollett, A.; et al. Promoter methylation of Wnt antagonists DKK1 and SFRP1 is associated with opposing tumor subtypes in two large populations of colorectal cancer patients. Carcinogenesis 2011, 32, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Wang, L.; Zhu, L.; Zhang, C.; Zhou, J. Secreted frizzled-related protein 2 is epigenetically silenced and functions as a tumor suppressor in oral squamous cell carcinoma. Mol. Med. Rep. 2014, 10, 2293–2298. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Paluszczak, J.; Sarbak, J.; Kostrzewska-Poczekaj, M.; Kiwerska, K.; Jarmuz-Szymczak, M.; Grenman, R.; Mielcarek-Kuchta, D.; Baer-Dubowska, W. The negative regulators of Wnt pathway-DACH1, DKK1, and WIF1 are methylated in oral and oropharyngeal cancer and WIF1 methylation predicts shorter survival. Tumor Boil. 2015, 36, 2855–2861. [Google Scholar] [CrossRef]
- Miaczynska, M.; Pelkmans, L.; Zerial, M. Not just a sink: Endosomes in control of signal transduction. Curr. Opin. Cell Boil. 2004, 16, 400–406. [Google Scholar] [CrossRef]
- Dobrowolski, R.; De Robertis, E.M. Endocytic control of growth factor signalling: Multivesicular bodies as signalling organelles. Nat. Rev. Mol. Cell Boil. 2012, 13, 53–60. [Google Scholar] [CrossRef]
- Blitzer, J.T.; Nusse, R. A critical role for endocytosis in Wnt signaling. BMC Cell Boil. 2006, 7, 28. [Google Scholar] [CrossRef]
- Hagemann, A.I.; Kurz, J.; Kauffeld, S.; Chen, Q.; Reeves, P.M.; Weber, S.; Schindler, S.; Davidson, G.; Kirchhausen, T.; Scholpp, S. In vivo analysis of formation and endocytosis of the Wnt/beta-catenin signaling complex in zebrafish embryos. J. Cell Sci. 2014, 127, 3970–3982. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Yamamoto, H. Regulation of Wnt signalling by receptor-mediated endocytosis. J. Biochem. 2007, 141, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Komekado, H.; Kikuchi, A. caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of β-catenin. Dev. Cell 2006, 11, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Taelman, V.F.; Dobrowolski, R.; Plouhinec, J.-L.; Fuentealba, L.C.; Vorwald, P.P.; Gumper, I.; Sabatini, D.D.; De Robertis, E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 2010, 143, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, Y.; Mills, G.B.; Yarden, Y. Derailed endocytosis: An emerging feature of cancer. Nat. Rev. Cancer 2008, 8, 835–850. [Google Scholar] [CrossRef]
- Lanzetti, L.; Di Fiore, P.P. Behind the scenes: Endo/exocytosis in the acquisition of metastatic traits. Cancer Res. 2017, 77, 1813–1817. [Google Scholar] [CrossRef]
- Mellman, I.; Yarden, Y. Endocytosis and cancer. Cold Spring Harb. Perspect. Boil. 2013, 5, a016949. [Google Scholar] [CrossRef]
- Stenmark, H.A. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Boil. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Binns, D.; Januszewski, T.; Chen, Y.; Hill, J.; Markin, V.S.; Zhao, Y.; Gilpin, C.; Chapman, K.D.; Anderson, R.G.; Goodman, J.M. An intimate collaboration between peroxisomes and lipid bodies. J. Cell Boil. 2006, 173, 719–731. [Google Scholar] [CrossRef]
- Franceschi, N.D.; Hamidi, H.; Alanko, J.; Sahgal, P.; Ivaska, J. Integrin traffic–the update. J. Cell Sci. 2015, 128, 839–852. [Google Scholar] [CrossRef]
- Jones, M.C.; Caswell, P.T.; Norman, J.C. Endocytic recycling pathways: Emerging regulators of cell migration. Curr. Opin. Cell Boil. 2006, 18, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Alanko, J.; Mai, A.; Jacquemet, G.; Schauer, K.; Kaukonen, R.; Saari, M.; Goud, B.; Ivaska, J. Integrin endosomal signalling suppresses anoikis. Nature 2015, 17, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- . Palamidessi, A.; Frittoli, E.; Ducano, N.; Offenhauser, N.; Sigismund, S.; Kajiho, H.; Parazzoli, D.; Oldani, A.; Gobbi, M.; Serini, G.G.; et al. The GTPase-activating protein RN-TRE controls focal adhesion turnover and cell migration. Curr. Boil. 2013, 23, 2355–2364. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, P.; Ortiz, R.; Díaz, J.; Quest, A.F.G.; Leyton, L.; Stupack, D.; Torres, V.A. Rab5 activation promotes focal adhesion disassembly, migration and invasiveness in tumor cells. J. Cell Sci. 2013, 126, 3835–3847. [Google Scholar] [CrossRef]
- Mendoza, P.A.; Silva, P.; Díaz, J.; Arriagada, C.; Canales, J.; Cerda, O.; Torres, V.A. Calpain2 mediates Rab5-driven focal adhesion disassembly and cell migration. Cell Adhes. Migr. 2018, 12, 185–194. [Google Scholar] [CrossRef]
- Palamidessi, A.; Frittoli, E.; Garrè, M.; Faretta, M.; Mione, M.C.; Testa, I.; Diaspro, A.; Lanzetti, L.; Scita, G.; Di Fiore, P.P. Endocytic trafficking of rac is required for the spatial restriction of signaling in cell migration. Cell 2008, 134, 135–147. [Google Scholar] [CrossRef]
- Díaz, J.; Mendoza, P.; Ortiz, R.; Díaz, N.; Leyton, L.; Stupack, D.; Quest, A.F.; Torres, V.A. Rab5 is required in metastatic cancer cells for Caveolin-1-enhanced Rac1 activation, migration and invasion. J. Cell Sci. 2014, 127, 2401–2406. [Google Scholar] [CrossRef]
- Frittoli, E.; Palamidessi, A.; Marighetti, P.; Confalonieri, S.; Bianchi, F.; Malinverno, C.; Mazzarol, G.; Viale, G.; Martin-Padura, I.; Garré, M.; et al. A RAB5/RAB4 recycling circuitry induces a proteolytic invasive program and promotes tumor dissemination. J. Cell Biol. 2014, 206, 307–328. [Google Scholar] [CrossRef]
- Serio, G.; Margaria, V.; Jensen, S.; Oldani, A.; Bartek, J.; Bussolino, F.; Lanzetti, L. Small GTPase Rab5 participates in chromosome congression and regulates localization of the centromere-associated protein CENP-F to kinetochores. Proc. Natl. Acad. Sci. USA 2011, 108, 17337–17342. [Google Scholar] [CrossRef]
- Dey, K.K.; Pal, I.; Bharti, R.; Dey, G.; Kumar, B.N.P.; Rajput, S.; Parekh, A.; Parida, S.; Halder, P.; Kulavi, I.; et al. Identification of RAB2A and PRDX1 as the potential biomarkers for oral squamous cell carcinoma using mass spectrometry-based comparative proteomic approach. Tumor Boil. 2015, 36, 9829–9837. [Google Scholar] [CrossRef]
- Da Silva, S.D.; Marchi, F.A.; Xu, B.; Bijian, K.; Alobaid, F.; Mlynarek, A.; Rogatto, S.R.; Hier, M.; Kowalski, L.P.; Alaoui-Jamali, M.A. Predominant Rab-GTPase amplicons contributing to oral squamous cell carcinoma progression to metastasis. Oncotarget 2015, 6, 21950–21963. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Miyazawa, H.; Kobayashi, H.; Noguchi, N.; Lambert, D.; Kawashiri, S. Caveolin-1 expression at metastatic lymph nodes predicts unfavorable outcome in patients with oral squamous cell carcinoma. Pathol. Oncol. Res. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-J.; Chang, K.-P.; Chang, Y.-J.; Hsu, C.-W.; Liang, Y.; Yu, J.-S.; Chi, L.-M.; Chang, Y.-S.; Wu, C.-C. Identification of guanylate-binding protein 1 as a potential oral cancer marker involved in cell invasion using OMICS-based analysis. J. Proteome Res. 2011, 10, 3778–3788. [Google Scholar] [CrossRef]
- Amornphimoltham, P.; Rechache, K.; Thompson, J.; Masedunskas, A.; Leelahavanichkul, K.; Patel, V.; Molinolo, A.; Gutkind, J.S.; Weigert, R. Rab25 regulates invasion and metastasis in head and neck cancer. Clin. Cancer Res. 2013, 19, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M.J.A.M.; Melchers, L.J.; Mastik, M.F.; Slagter-Menkema, L.; Groen, H.J.M.; Van Der Laan, B.F.A.M.; Van Criekinge, W.; De Meyer, T.; Denil, S.; Van Der Vegt, B.; et al. RAB25 expression is epigenetically downregulated in oral and oropharyngeal squamous cell carcinoma with lymph node metastasis. Epigenetics 2016, 11, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lu, C.; Ai, H. Rab5a is overexpressed in oral cancer and promotes invasion through ERK/MMP signaling. Mol. Med. Rep. 2017, 16, 4569–4576. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.; Keller, T.H. The use of porcupine inhibitors to target Wnt-driven cancers. Bioorg. Med. Chem. Lett. 2015, 25, 5472–5476. [Google Scholar] [CrossRef]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2015, 35, 2197–2207. [Google Scholar] [CrossRef]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef]
- Madan, B.; Virshup, D.M. Targeting Wnts at the source-new mechanisms, new biomarkers, new drugs. Mol. Cancer Ther. 2015, 14, 1087–1094. [Google Scholar] [CrossRef]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Heuvel, M.V.D.; Harryman-Samos, C.; Klingensmith, J.; Perrimon, N.; Nusse, R. Mutations in the segment polarity genes wingless and porcupine impair secretion of the wingless protein. EMBO J. 1993, 12, 5293–5302. [Google Scholar] [CrossRef] [PubMed]
- Herr, P.; Basler, K. Porcupine-mediated lipidation is required for Wnt recognition by Wls. Dev. Boil. 2012, 361, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).