HDAC8 Inhibition Reduces Lesional Iba-1+ Cell Infiltration after Spinal Cord Injury without Effects on Functional Recovery


Abstract
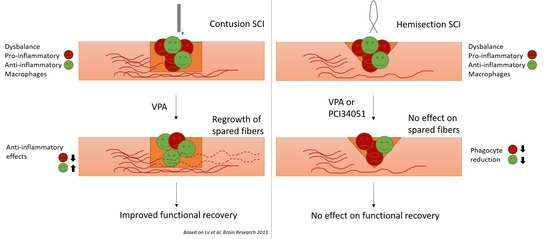
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
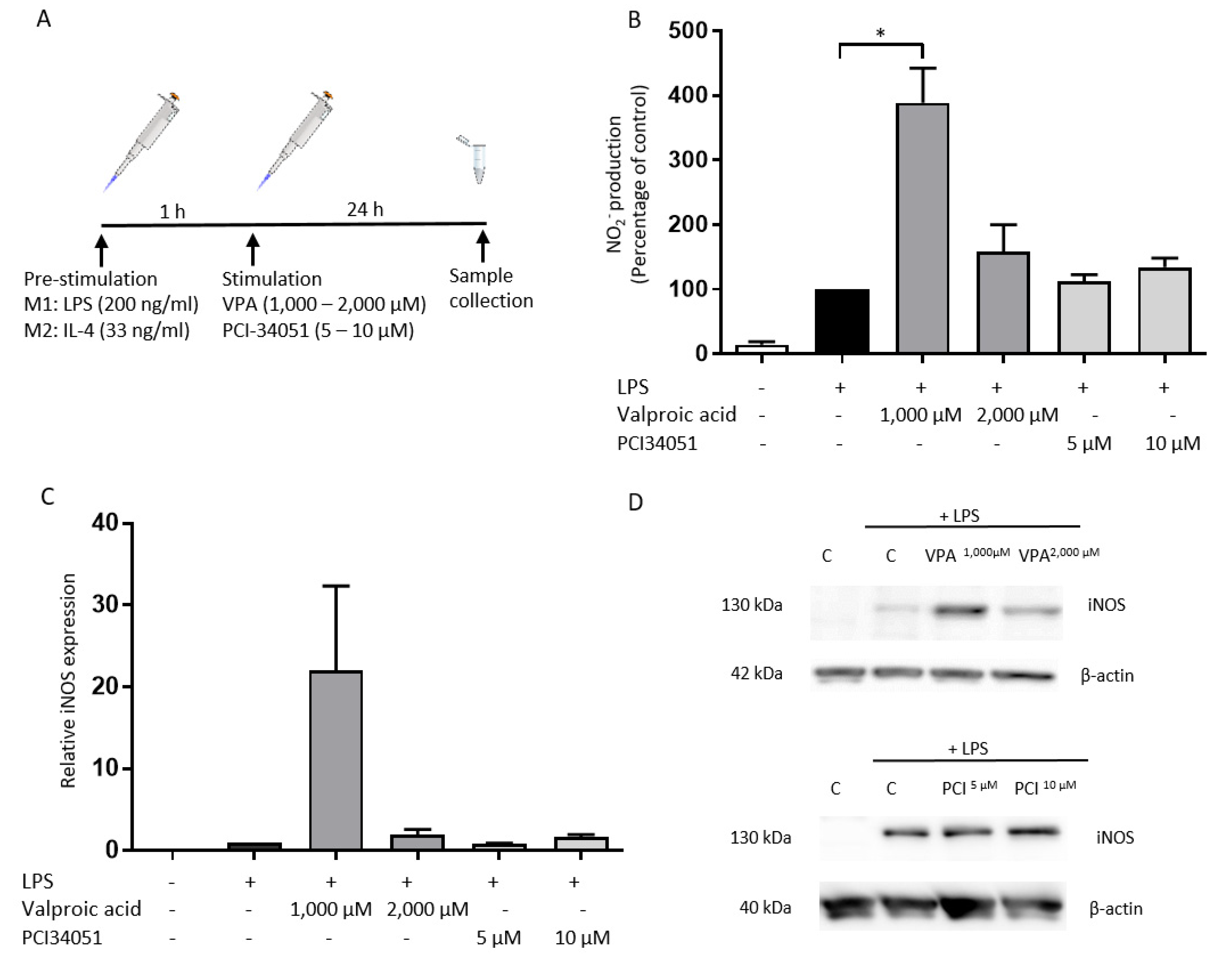
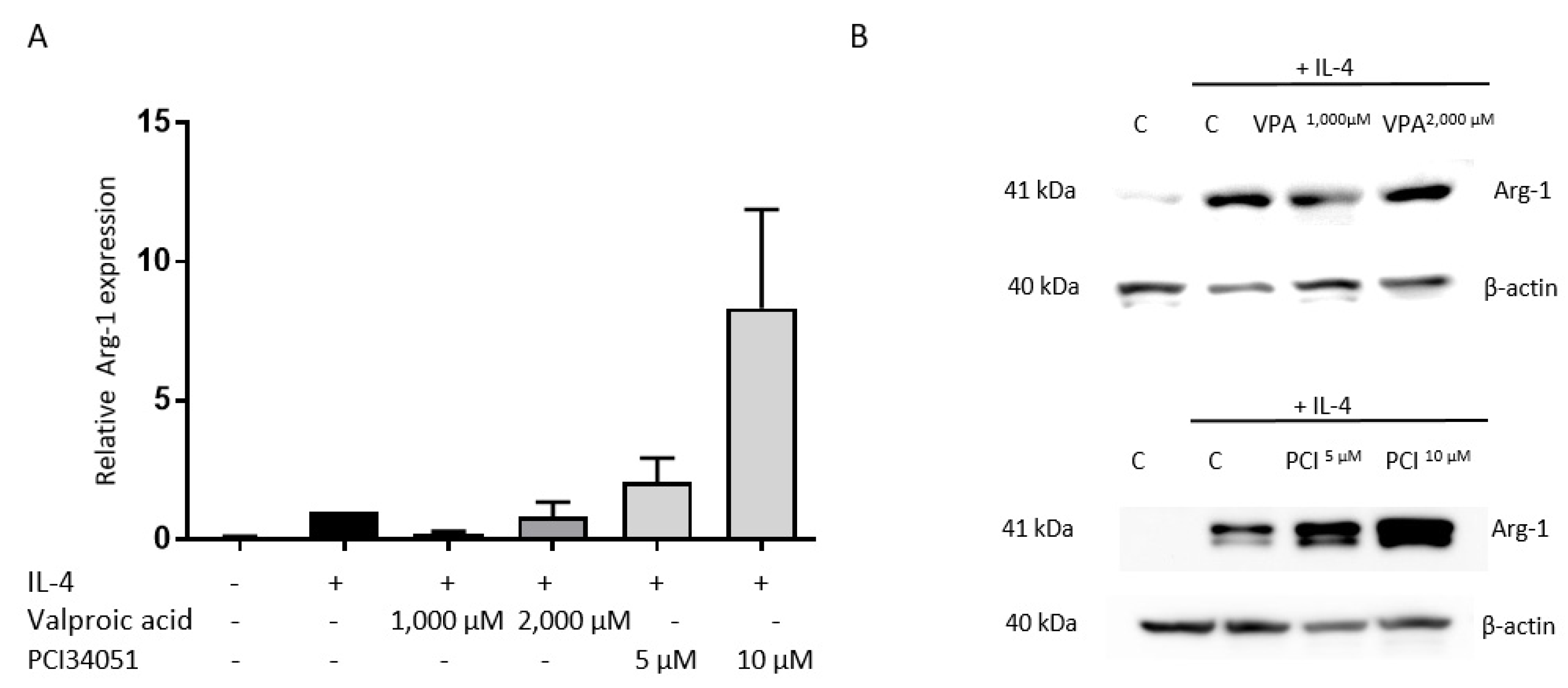
2.1. PCI-34051 Has No Effects on Macrophage Phenotype, whereas VPA Significantly Increases NO2− Production after LPS Stimulation In Vitro
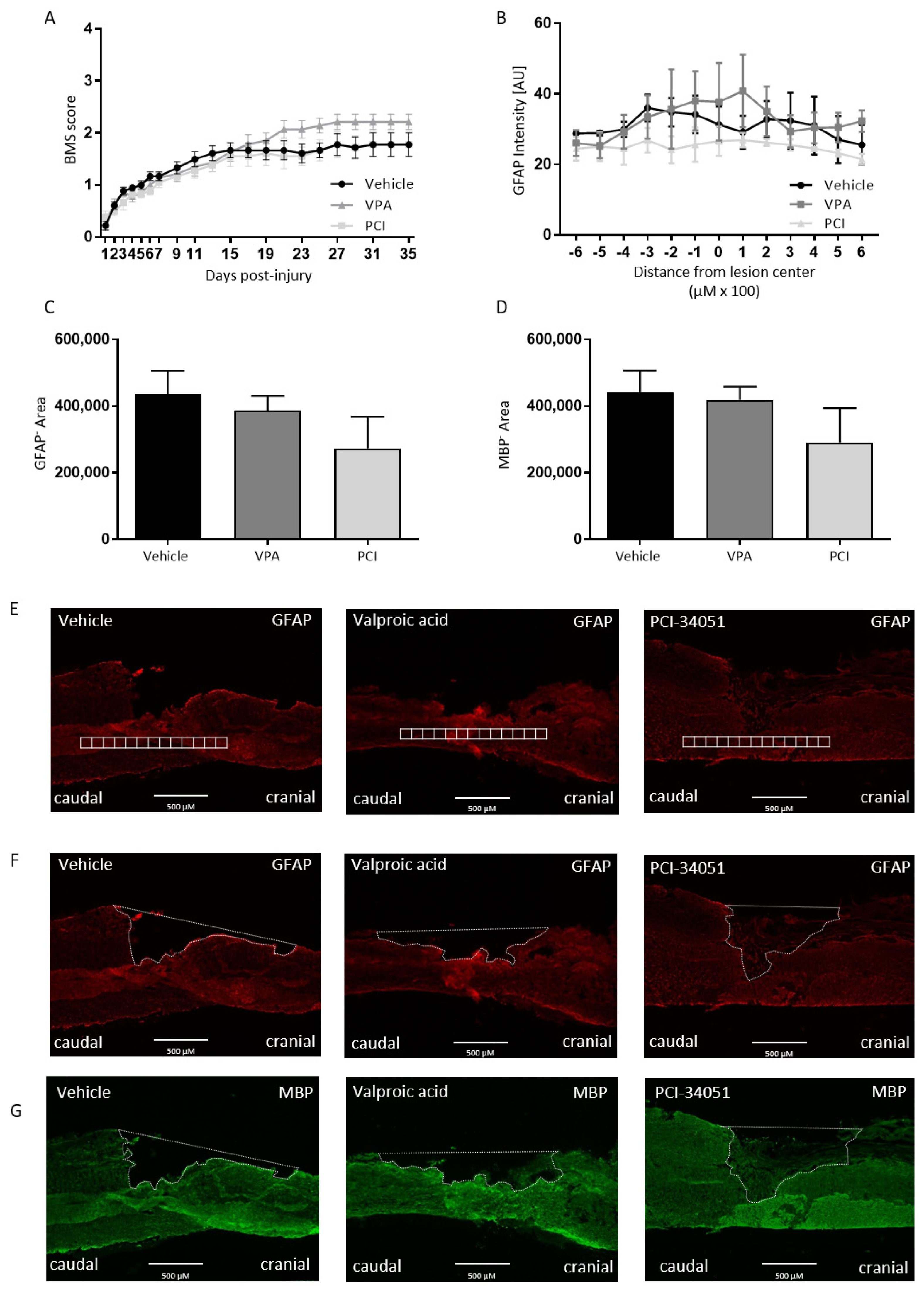
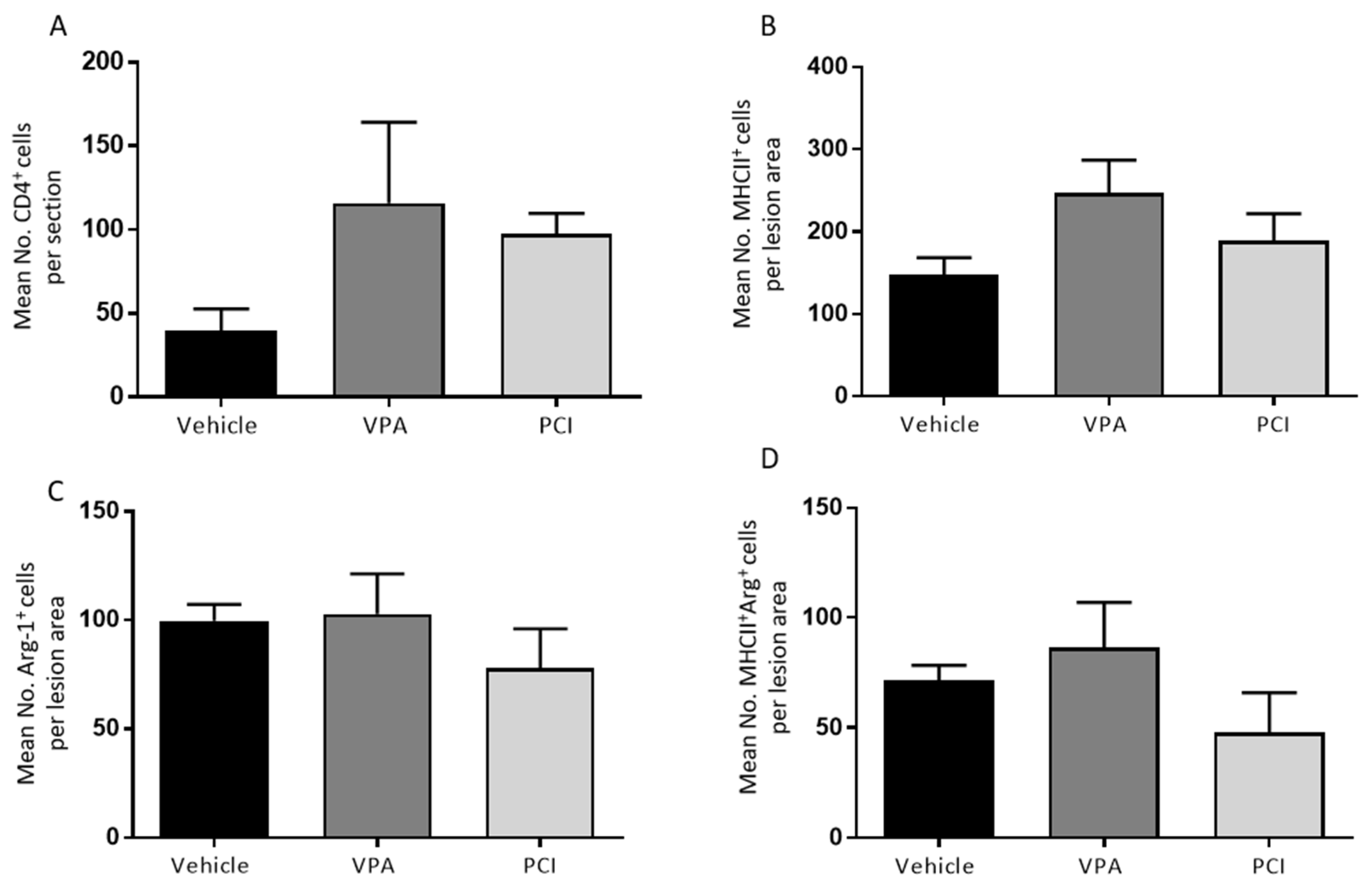
2.2. PCI-34051 Reduces Iba-1+ Cell Infiltration whithout Effects on Functional Recovery, whereas VPA Has No Effects on Histopathological or Functional Recovery after SCI
3. Discussion
4. Materials and Methods
4.1. Isolation and Polarization of Bone Marrow-Derived Macrophages
4.2. Griess Assay
4.3. Western Blot Analysis
4.4. MTT Assay
4.5. Experimental Spinal Cord Injury and HDAC Inhibitor Treatment
4.6. Immunohistochemistry and Quantitative Image Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC-H3 | acetylation histone 3 |
| Arg-1 | arginase-1 |
| BMDMs | bone marrow derived macrophages |
| BMS | Basso mouse scale |
| BSCB | brain spinal cord barrier |
| CD4 | cluster of differentiation 4 |
| GFAP | glial fibrillary protein |
| CNS | central nervous system |
| HAT | histone acetyl transferase |
| H3K9 | histone 3 lysine 9 |
| H3K27 | histone 3 lysine 27 |
| HDACs | histone deacetylases |
| IL-4 | interleukin-4 |
| Iba-1 | ionized calcium-binding adapter molecule 1 |
| iNOS | nitric oxide synthase |
| LPS | lipopolysaccharide |
| MBP | myelin basic protein |
| MHCII | major histocompatibility complex 2 |
| MMP-9 | matrix metalloproteinase-9 |
| PCI | PCI-34051 |
| SCI | spinal cord injury |
| TBI | traumatic brain injury |
| VPA | valproic acid |
References
- Ramer, L.M.; Ramer, M.S.; Bradbury, E.J. Restoring function after spinal cord injury: Towards clinical translation of experimental strategies. Lancet Neurol. 2014, 13, 1241–1256. [Google Scholar] [CrossRef]
- Rowland, J.W.; Hawryluk, G.W.; Kwon, B.; Fehlings, M.G. Current status of acute spinal cord injury pathophysiology and emerging therapies: Promise on the horizon. Neurosurg. Focus 2008, 25, E2. [Google Scholar] [CrossRef] [PubMed]
- Oyinbo, C.A. Secondary injury mechanisms in traumatic spinal cord injury: A nugget of this multiply cascade. Acta Neurobiol. Exp. 2011, 71, 281–299. [Google Scholar]
- David, S.; Greenhalgh, A.D.; Kroner, A. Macrophage and microglial plasticity in the injured spinal cord. Neuroscience 2015, 307, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.A.; Hamilton, J.A.; Horn, K.P.; Cuascut, F.X.; Cutrone, R.; Lehman, N.; Deans, R.J.; Ting, A.E.; Mays, R.W.; Silver, J. Multipotent Adult Progenitor Cells Prevent Macrophage-Mediated Axonal Dieback and Promote Regrowth after Spinal Cord Injury. J. Neurosci. 2011, 31, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Shechter, R.; Miller, O.; Yovel, G.; Rosenzweig, N.; London, A.; Ruckh, J.; Kim, K.W.; Klein, E.; Kalchenko, V.; Bendel, P.; et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 2013, 38, 555–569. [Google Scholar] [CrossRef]
- Alessandro, D.; Puneet, O. The promise and perils of HDAC inhibitors in neurodegeneration. Ann. Clin. Transl. Neurol. 2015, 2, 79–101. [Google Scholar] [CrossRef]
- Cantley, M.D.; Haynes, D.R. Epigenetic regulation of inflammation: Progressing from broad acting histone deacetylase (HDAC) inhibitors to targeting specific HDACs. Inflammopharmacology 2013, 21, 301–307. [Google Scholar] [CrossRef]
- Wang, G.; Jiang, X.; Pu, H.; Zhang, W.; An, C.; Hu, X.; Liou, A.K.; Leak, R.K.; Gao, Y.; Chen, J. Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway: Scriptaid protects against TBI via AKT. Neurotherapeutics 2013, 10, 124–142. [Google Scholar] [CrossRef]
- Faraco, G.; Pittelli, M.; Cavone, L.; Fossati, S.; Porcu, M.; Mascagni, P.; Fossati, G.; Moroni, F.; Chiarugi, A. Histone deacetylase (HDAC) inhibitors reduce the glial inflammatory response in vitro and in vivo. Neurobiol. Dis. 2009, 36, 269–279. [Google Scholar] [CrossRef]
- Shein, N.A.; Shohami, E. Histone deacetylase inhibitors as therapeutic agents for acute central nervous system injuries. Mol. Med. 2011, 17, 448–456. [Google Scholar] [CrossRef]
- Pomeshchik, Y.; Kidin, I.; Korhonen, P.; Savchenko, E.; Jaronen, M.; Lehtonen, S.; Wojciechowski, S.; Kanninen, K.; Koistinaho, J.; Malm, T. Interleukin-33 treatment reduces secondary injury and improves functional recovery after contusion spinal cord injury. Brain Behav. Immun. 2015, 44, 68–81. [Google Scholar] [CrossRef]
- Lv, L.; Han, X.; Sun, Y.; Wang, X.; Dong, Q. Valproic acid improves locomotion in vivo after SCI and axonal growth of neurons in vitro. Exp. Neurol. 2012, 233, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Then, F.; Melia, T.J.; Mazzulli, J.R.; Cui, L.; Savas, J.N.; Voisine, C.; Paganetti, P.; Tanese, N.; Hart, A.C.; et al. Acetylation Targets Mutant Huntingtin to Autophagosomes for Degradation. Cell 2009, 137, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Leus, N.G.; Zwinderman, M.R.; Dekker, F.J. Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-kappaB-mediated inflammation. Curr. Opin. Chem. Biol. 2016, 33, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Das Gupta, K.; Shakespear, M.R.; Iyer, A.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases in monocyte/macrophage development, activation and metabolism: Refining HDAC targets for inflammatory and infectious diseases. Clin. Transl. Immunol. 2016, 5, e62. [Google Scholar] [CrossRef]
- Dietz, K.C.; Casaccia, P. HDAC inhibitors and neurodegeneration: At the edge between protection and damage. Pharmacol. Res. 2010, 62, 11–17. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Sanchez, S.; Lemmens, S.; Baeten, P.; Sommer, D.; Dooley, D.; Hendrix, S.; Gou Fabregas, M. HDAC3 Inhibition Promotes Alternative Activation of Macrophages but Does Not Affect Functional Recovery after Spinal Cord Injury. Exp. Neurobiol. 2018, 27, 437–452. [Google Scholar] [CrossRef]
- Guo, L.; Guo, H.; Gao, C.; Mi, Z.; Russell, W.B.; Kuo, P.C. Stat1 acetylation inhibits inducible nitric oxide synthase expression in interferon-gamma-treated RAW264.7 murine macrophages. Surgery 2007, 142, 156–162. [Google Scholar] [CrossRef][Green Version]
- Serrat, N.; Sebastian, C.; Pereira-Lopes, S.; Valverde-Estrella, L.; Lloberas, J.; Celada, A. The response of secondary genes to lipopolysaccharides in macrophages depends on histone deacetylase and phosphorylation of C/EBPbeta. J. Immunol. 2014, 192, 418–426. [Google Scholar] [CrossRef]
- Zhang, K.; Zheng, J.; Bian, G.; Liu, L.; Xue, Q.; Liu, F.; Yu, C.; Zhang, H.; Song, B.; Chung, S.K.; et al. Polarized Macrophages Have Distinct Roles in the Differentiation and Migration of Embryonic Spinal-cord-derived Neural Stem Cells After Grafting to Injured Sites of Spinal Cord. Mol. Ther. 2015, 23, 1077–1091. [Google Scholar] [CrossRef]
- Chateauvieux, S.; Morceau, F.; Dicato, M.; Diederich, M. Molecular and therapeutic potential and toxicity of valproic acid. J. Biomed. Biotechnol. 2010, 2010, 479364. [Google Scholar] [CrossRef] [PubMed]
- Halili, M.A.; Andrews, M.R.; Labzin, L.I.; Schroder, K.; Matthias, G.; Cao, C.; Lovelace, E.; Reid, R.C.; Le, G.T.; Hume, D.A.; et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukoc. Biol. 2010, 87, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Jurkin, J.; Zupkovitz, G.; Lagger, S.; Grausenburger, R.; Hagelkruys, A.; Kenner, L.; Seiser, C. Distinct and redundant functions of histone deacetylases HDAC1 and HDAC2 in proliferation and tumorigenesis. Cell Cycle 2011, 10, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Vazin, T.; Goodwill, P.W.; Conway, A.; Verma, A.; Saritas, E.U.; Schaffer, D.; Conolly, S.M. Magnetic Particle Imaging tracks the long-term fate of in vivo neural cell implants with high image contrast. Sci. Rep. 2015, 5, 14055. [Google Scholar] [CrossRef]
- Zhan, J.S.; Gao, K.; Chai, R.C.; Jia, X.H.; Luo, D.P.; Ge, G.; Jiang, Y.W.; Fung, Y.W.; Li, L.; Yu, A.C. Astrocytes in Migration. Neurochem. Res. 2017, 42, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Noble, L.J.; Donovan, F.; Igarashi, T.; Goussev, S.; Werb, Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J. Neurosci. 2002, 22, 7526–7535. [Google Scholar] [CrossRef]
- Lv, L.; Sun, Y.; Han, X.; Xu, C.-C.; Tang, Y.-P.; Dong, Q. Valproic acid improves outcome after rodent spinal cord injury: Potential roles of histone deacetylase inhibition. Brain Res. 2011, 1396, 60–68. [Google Scholar] [CrossRef]
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, 1619, 1–11. [Google Scholar] [CrossRef]
- Nusinzon, I.; Horvath, C.M. Positive and Negative Regulation of the Innate Antiviral Response and Beta Interferon Gene Expression by Deacetylation. Mol. Cell. Biol. 2006, 26, 3106–3113. [Google Scholar] [CrossRef]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. BioMed Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef]
- Leoni, F.; Fossati, G.; Lewis, E.C.; Lee, J.-K.; Porro, G.; Pagani, P.; Modena, D.; Moras, M.L.; Pozzi, P.; Reznikov, L.L.; et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol. Med. 2005, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Leoni, F.; Zaliani, A.; Bertolini, G.; Porro, G.; Pagani, P.; Pozzi, P.; Donà, G.; Fossati, G.; Sozzani, S.; Azam, T.; et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. USA 2002, 99, 2995–3000. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Han, C.Y.; Reid, C.; Kim, S.O. HDAC8-mediated epigenetic reprogramming plays a key role in resistance to anthrax lethal toxin-induced pyroptosis in macrophages. J. Immunol. 2014, 193, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Reid, C.; Meshkibaf, S.; Kim, S.O. Inhibition of Interleukin 1beta (IL-1beta) Expression by Anthrax Lethal Toxin (LeTx) Is Reversed by Histone Deacetylase 8 (HDAC8) Inhibition in Murine Macrophages. J. Biol. Chem. 2016, 291, 8745–8755. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Kee, H.J.; Kurz, T.; Hansen, F.K.; Ryu, Y.; Kim, G.R.; Lin, M.Q.; Jin, L.; Piao, Z.H.; Jeong, M.H. Class I HDACs specifically regulate E-cadherin expression in human renal epithelial cells. J. Cell. Mol. Med. 2016, 20, 2289–2298. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, H.S.; Choi, H.Y.; Oh, T.H.; Ju, B.G.; Yune, T.Y. Valproic acid attenuates blood-spinal cord barrier disruption by inhibiting matrix metalloprotease-9 activity and improves functional recovery after spinal cord injury. J. Neurochem. 2012, 121, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Abdanipour, A.; Schluesener, H.J.; Tiraihi, T. Effects of valproic acid, a histone deacetylase inhibitor, on improvement of locomotor function in rat spinal cord injury based on epigenetic science. Iran. Biomed. J. 2012, 16, 90–100. [Google Scholar]
- Steward, O.; Zheng, B.; Tessier-Lavigne, M. False resurrections: Distinguishing regenerated from spared axons in the injured central nervous system. J. Comp. Neurol. 2003, 459, 1–8. [Google Scholar] [CrossRef]
- Dooley, D.; Lemmens, E.; Vangansewinkel, T.; Le Blon, D.; Hoornaert, C.; Ponsaerts, P.; Hendrix, S. Cell-Based Delivery of Interleukin-13 Directs Alternative Activation of Macrophages Resulting in Improved Functional Outcome after Spinal Cord Injury. Stem Cell Rep. 2016, 7, 1099–1115. [Google Scholar] [CrossRef]
- Nelissen, S.; Vangansewinkel, T.; Geurts, N.; Geboes, L.; Lemmens, E.; Vidal, P.M.; Lemmens, S.; Willems, L.; Boato, F.; Dooley, D.; et al. Mast cells protect from post-traumatic spinal cord damage in mice by degrading inflammation-associated cytokines via mouse mast cell protease 4. Neurobiol. Dis. 2014, 62, 260–272. [Google Scholar] [CrossRef]
- Slaets, H.; Nelissen, S.; Janssens, K.; Vidal, P.M.; Lemmens, E.; Stinissen, P.; Hendrix, S.; Hellings, N. Oncostatin M reduces lesion size and promotes functional recovery and neurite outgrowth after spinal cord injury. Mol. Neurobiol. 2014, 50, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Vangansewinkel, T.; Geurts, N.; Quanten, K.; Nelissen, S.; Lemmens, S.; Geboes, L.; Dooley, D.; Vidal, P.M.; Pejler, G.; Hendrix, S. Mast cells promote scar remodeling and functional recovery after spinal cord injury via mouse mast cell protease 6. FASEB J. 2016, 30, 2040–2057. [Google Scholar] [CrossRef] [PubMed]
- Vidal, P.M.; Lemmens, E.; Avila, A.; Vangansewinkel, T.; Chalaris, A.; Rose-John, S.; Hendrix, S. ADAM17 is a survival factor for microglial cells in vitro and in vivo after spinal cord injury in mice. Cell Death Dis. 2013, 4, e954. [Google Scholar] [CrossRef] [PubMed]
- Mietto, B.S.; Mostacada, K.; Martinez, A.M. Neurotrauma and inflammation: CNS and PNS responses. Mediat. Inflamm. 2015, 2015, 251204. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chang, M.; Hansen, C.N.; Basso, D.M.; Noble-Haeusslein, L.J. Role of matrix metalloproteinases and therapeutic benefits of their inhibition in spinal cord injury. Neurotherapeutics 2011, 8, 206–220. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrix, S.; Sanchez, S.; Ventriglia, E.; Lemmens, S. HDAC8 Inhibition Reduces Lesional Iba-1+ Cell Infiltration after Spinal Cord Injury without Effects on Functional Recovery. Int. J. Mol. Sci. 2020, 21, 4539. https://doi.org/10.3390/ijms21124539
Hendrix S, Sanchez S, Ventriglia E, Lemmens S. HDAC8 Inhibition Reduces Lesional Iba-1+ Cell Infiltration after Spinal Cord Injury without Effects on Functional Recovery. International Journal of Molecular Sciences. 2020; 21(12):4539. https://doi.org/10.3390/ijms21124539
Chicago/Turabian StyleHendrix, Sven, Selien Sanchez, Elissia Ventriglia, and Stefanie Lemmens. 2020. "HDAC8 Inhibition Reduces Lesional Iba-1+ Cell Infiltration after Spinal Cord Injury without Effects on Functional Recovery" International Journal of Molecular Sciences 21, no. 12: 4539. https://doi.org/10.3390/ijms21124539
APA StyleHendrix, S., Sanchez, S., Ventriglia, E., & Lemmens, S. (2020). HDAC8 Inhibition Reduces Lesional Iba-1+ Cell Infiltration after Spinal Cord Injury without Effects on Functional Recovery. International Journal of Molecular Sciences, 21(12), 4539. https://doi.org/10.3390/ijms21124539