Novel Dominant KCNQ2 Exon 7 Partial In-Frame Duplication in a Complex Epileptic and Neurodevelopmental Delay Syndrome

 , ,
, ,  , , , and
, , , and 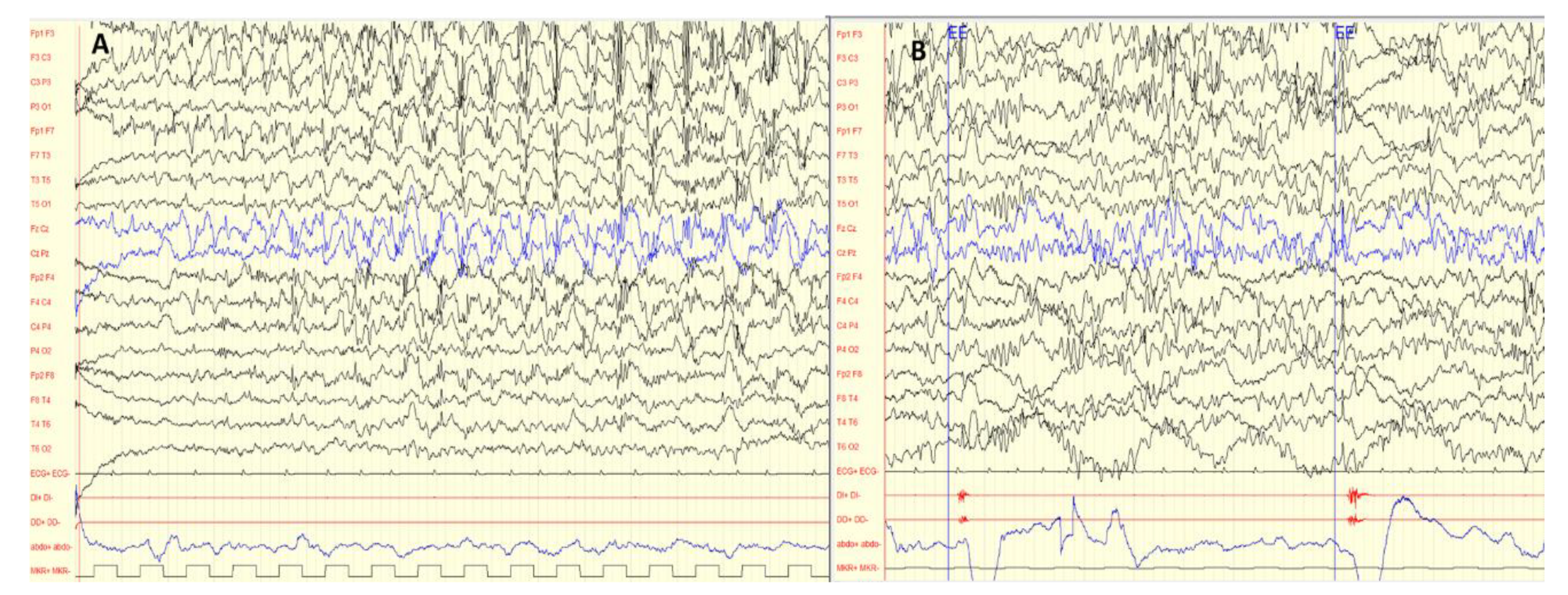{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Epileptic, Cerebral Palsy, and Neuromotor Developmental Delay Phenotype of Patient
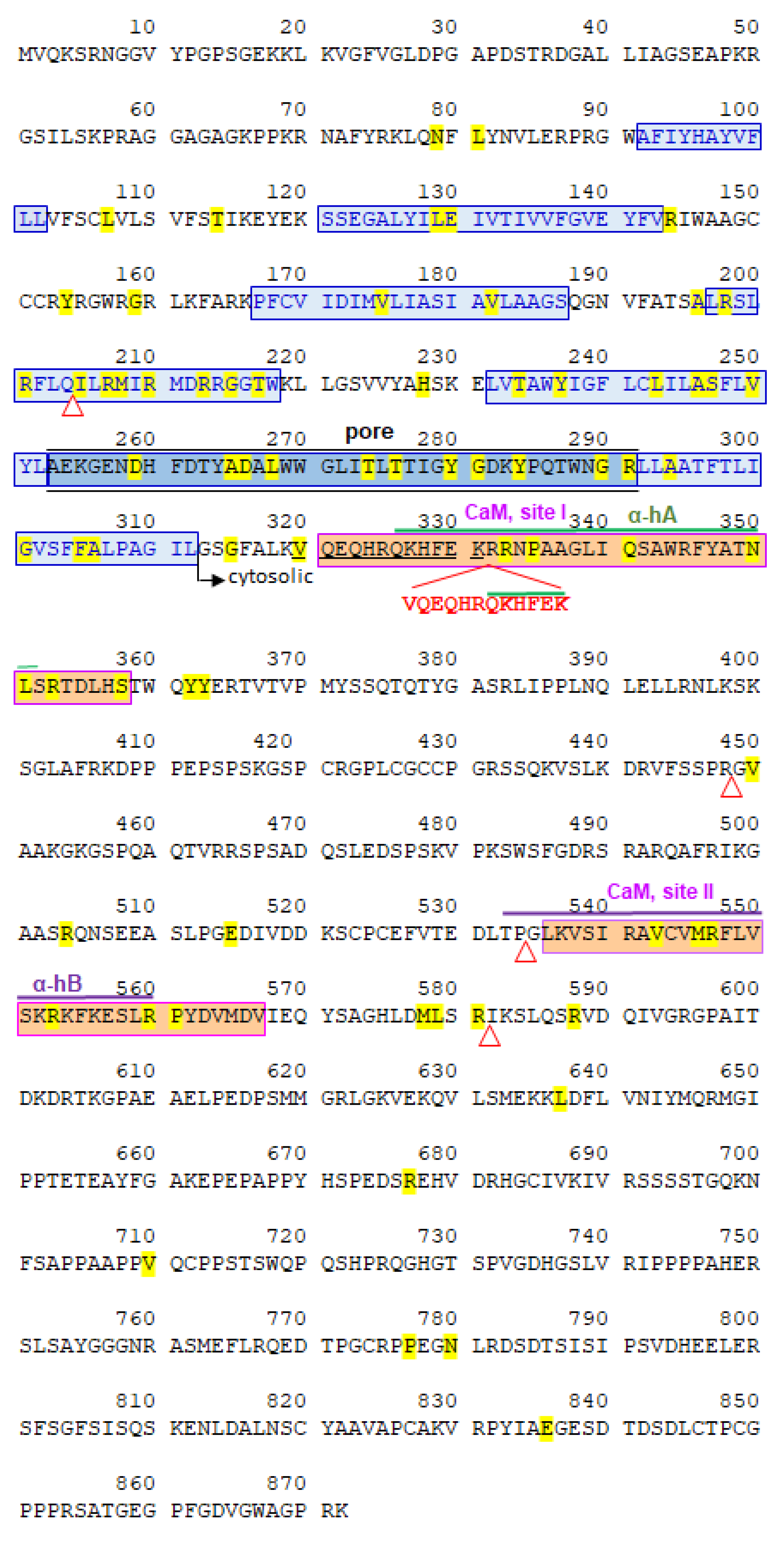
2.2. Partial de Novo KCNQ2 Exon 7 Duplication
2.3. Dynamic Molecular Modelling of KCNQ2 Partial Duplication
3. Discussion
4. Methods
4.1. Standard Protocol Approvals, Registrations, and Patient Consents
4.2. Whole-Exome Sequencing
4.3. Analysis of Gene Alterations and Clinical Phenotype
4.4. Molecular Dynamics Simulation and Structural Modeling of KCNQ2
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data availability statement
References
- Wang, J.; Ou, S.W.; Wang, Y.J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels (Austin) 2017, 11, 534–554. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.S.; Walsh, C.A. Ion Channel Functions in Early Brain Development. Trends Neurosci. 2020, 43, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Battefeld, A.; Tran, B.T.; Gavrilis, J.; Cooper, E.C.; Kole, M.H. Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J. Neurosci. 2014, 34, 3719–3732. [Google Scholar] [CrossRef] [PubMed]
- Misonou, H. Precise localizations of voltage-gated sodium and potassium channels in neurons. Dev. Neurobiol. 2018, 78, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef]
- Cooper, E.C.; Harrington, E.; Jan, Y.N.; Jan, L.Y. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci. 2001, 21, 9529–9540. [Google Scholar] [CrossRef]
- Lombardo, J.; Sun, J.; Harrington, M.A. Rapid activity-dependent modulation of the intrinsic excitability through up-regulation of KCNQ/Kv7 channel function in neonatal spinal motoneurons. PLoS ONE 2018, 13, e0193948. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ramirez, M.B.; Laville, A.; Tapia, D.; Duhne, M.; Lara-Gonzalez, E.; Bargas, J.; Galarraga, E. KV7 Channels Regulate Firing during Synaptic Integration in GABAergic Striatal Neurons. Neural. Plast. 2015, 2015, 472676. [Google Scholar] [CrossRef]
- Lee, I.C.; Yang, J.J.; Li, S.Y. KCNQ2-Associated Epilepsy: A Review of Variable Phenotypes and Neurodevelopmental Outcomes. Neuropsychiatry 2018, 8, 318–323. [Google Scholar] [CrossRef]
- Mefford, H.C.; Muhle, H.; Ostertag, P.; von Spiczak, S.; Buysse, K.; Baker, C.; Franke, A.; Malafosse, A.; Genton, P.; Thomas, P.; et al. Genome-wide copy number variation in epilepsy: Novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010, 6, e1000962. [Google Scholar] [CrossRef] [PubMed]
- Fahey, M.C.; Maclennan, A.H.; Kretzschmar, D.; Gecz, J.; Kruer, M.C. The genetic basis of cerebral palsy. Dev. Med. Child. Neurol. 2017, 59, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Patel, A. Chromosomal Microarrays: Understanding Genetics of Neurodevelopmental Disorders and Congenital Anomalies. J. Pediatric Genet. 2017, 6, 42–50. [Google Scholar] [CrossRef]
- Lindy, A.S.; Stosser, M.B.; Butler, E.; Downtain-Pickersgill, C.; Shanmugham, A.; Retterer, K.; Brandt, T.; Richard, G.; McKnight, D.A. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 2018, 59, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Borlot, F.; Regan, B.M.; Bassett, A.S.; Stavropoulos, D.J.; Andrade, D.M. Prevalence of Pathogenic Copy Number Variation in Adults With Pediatric-Onset Epilepsy and Intellectual Disability. JAMA Neurol. 2017, 74, 1301–1311. [Google Scholar] [CrossRef]
- McMichael, G.; Bainbridge, M.N.; Haan, E.; Corbett, M.; Gardner, A.; Thompson, S.; van Bon, B.W.; van Eyk, C.L.; Broadbent, J.; Reynolds, C.; et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiatry 2015, 20, 176–182. [Google Scholar] [CrossRef]
- Emrick, L.T.; DiCarlo, S.M. The Expanding Role of Genetics in Cerebral Palsy. Phys. Med. Rehabil. Clin. N. Am. 2020, 31, 15–24. [Google Scholar] [CrossRef]
- Risch, N.; Spiker, D.; Lotspeich, L.; Nouri, N.; Hinds, D.; Hallmayer, J.; Kalaydjieva, L.; McCague, P.; Dimiceli, S.; Pitts, T.; et al. A genomic screen of autism: Evidence for a multilocus etiology. Am. J. Hum. Genet. 1999, 65, 493–507. [Google Scholar] [CrossRef]
- Liu, X.; Shimada, T.; Otowa, T.; Wu, Y.Y.; Kawamura, Y.; Tochigi, M.; Iwata, Y.; Umekage, T.; Toyota, T.; Maekawa, M.; et al. Genome-wide Association Study of Autism Spectrum Disorder in the East Asian Populations. Autism. Res. 2015, 9, 340–349. [Google Scholar] [CrossRef]
- Yus-Najera, E.; Santana-Castro, I.; Villarroel, A. The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J. Biol. Chem. 2002, 277, 28545–28553. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Plaschkes, I.; Oz-Levi, D.; Alkelai, A.; Olender, T.; Zimmerman, S.; Twik, M.; Belinky, F.; Fishilevich, S.; Nudel, R.; et al. VarElect: The phenotype-based variation prioritizer of the GeneCards Suite. BMC Genom. 2016, 17 (Suppl. 2), 444. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Paciorkowski, A.R.; Darras, B.T. Making sense of genetic heterogeneity: Emergence of pathways in developmental brain disorders. Neurology 2013. [Google Scholar] [CrossRef]
- Hernandez, C.C.; Zaika, O.; Tolstykh, G.P.; Shapiro, M.S. Regulation of neural KCNQ channels: Signalling pathways, structural motifs and functional implications. J. Physiol. 2008, 586, 1811–1821. [Google Scholar] [CrossRef]
- Alaimo, A.; Alberdi, A.; Gomis-Perez, C.; Fernandez-Orth, J.; Bernardo-Seisdedos, G.; Malo, C.; Millet, O.; Areso, P.; Villarroel, A. Pivoting between calmodulin lobes triggered by calcium in the Kv7.2/calmodulin complex. PLoS ONE 2014, 9, e86711. [Google Scholar] [CrossRef]
- Bernardo-Seisdedos, G.; Nunez, E.; Gomis-Perez, C.; Malo, C.; Villarroel, A.; Millet, O. Structural basis and energy landscape for the Ca(2+) gating and calmodulation of the Kv7.2 K(+) channel. Proc. Natl. Acad. Sci. USA 2018, 115, 2395–2400. [Google Scholar] [CrossRef]
- Abidi, A.; Devaux, J.J.; Molinari, F.; Alcaraz, G.; Michon, F.X.; Sutera-Sardo, J.; Becq, H.; Lacoste, C.; Altuzarra, C.; Afenjar, A.; et al. A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol. Dis. 2015, 80, 80–92. [Google Scholar] [CrossRef]
- Kim, E.C.; Zhang, J.; Pang, W.; Wang, S.; Lee, K.Y.; Cavaretta, J.P.; Walters, J.; Procko, E.; Tsai, N.P.; Chung, H.J. Reduced axonal surface expression and phosphoinositide sensitivity in Kv7 channels disrupts their function to inhibit neuronal excitability in Kcnq2 epileptic encephalopathy. Neurobiol. Dis. 2018, 118, 76–93. [Google Scholar] [CrossRef]
- Devaux, J.; Abidi, A.; Roubertie, A.; Molinari, F.; Becq, H.; Lacoste, C.; Villard, L.; Milh, M.; Aniksztejn, L. A Kv7.2 mutation associated with early onset epileptic encephalopathy with suppression-burst enhances Kv7/M channel activity. Epilepsia 2016, 57, e87–e93. [Google Scholar] [CrossRef]
- Spagnoli, C.; Salerno, G.G.; Iodice, A.; Frattini, D.; Pisani, F.; Fusco, C. KCNQ2 encephalopathy: A case due to a de novo deletion. Brain Dev. 2018, 40, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Otto, J.F.; Yang, Y.; Frankel, W.N.; White, H.S.; Wilcox, K.S. A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J. Neurosci. 2006, 26, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- King, C.H.; Lancaster, E.; Salomon, D.; Peles, E.; Scherer, S.S. Kv7.2 regulates the function of peripheral sensory neurons. J. Comp. Neurol. 2014, 522, 3262–3280. [Google Scholar] [CrossRef] [PubMed]
- Niday, Z.; Hawkins, V.E.; Soh, H.; Mulkey, D.K.; Tzingounis, A.V. Epilepsy-Associated KCNQ2 Channels Regulate Multiple Intrinsic Properties of Layer 2/3 Pyramidal Neurons. J. Neurosci. 2017, 37, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Hewson, S.; Puka, K.; Mercimek-Mahmutoglu, S. Variable expressivity of a likely pathogenic variant in KCNQ2 in a three-generation pedigree presenting with intellectual disability with childhood onset seizures. Am. J. Med. Genet. A 2017, 173, 2226–2230. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Chang, T.M.; Liang, J.S.; Li, S.Y. KCNQ2 mutations in childhood nonlesional epilepsy: Variable phenotypes and a novel mutation in a case series. Mol. Genet. Genom. Med. 2019, 7, e00816. [Google Scholar] [CrossRef]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The Genetics of Epilepsy. Annu Rev. Genom. Hum. Genet. 2020, 20. [Google Scholar] [CrossRef]
- Marcos-Alcalde, I.; Mendieta-Moreno, J.I.; Puisac, B.; Gil-Rodriguez, M.C.; Hernandez-Marcos, M.; Soler-Polo, D.; Ramos, F.J.; Ortega, J.; Pie, J.; Mendieta, J.; et al. Two-step ATP-driven opening of cohesin head. Sci. Rep. 2017, 7, 3266. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–38. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazo, P.A.; García, J.L.; Gómez-Puertas, P.; Marcos-Alcalde, Í.; Arjona, C.; Villarroel, A.; González-Sarmiento, R.; Fons, C. Novel Dominant KCNQ2 Exon 7 Partial In-Frame Duplication in a Complex Epileptic and Neurodevelopmental Delay Syndrome. Int. J. Mol. Sci. 2020, 21, 4447. https://doi.org/10.3390/ijms21124447
Lazo PA, García JL, Gómez-Puertas P, Marcos-Alcalde Í, Arjona C, Villarroel A, González-Sarmiento R, Fons C. Novel Dominant KCNQ2 Exon 7 Partial In-Frame Duplication in a Complex Epileptic and Neurodevelopmental Delay Syndrome. International Journal of Molecular Sciences. 2020; 21(12):4447. https://doi.org/10.3390/ijms21124447
Chicago/Turabian StyleLazo, Pedro A., Juan L. García, Paulino Gómez-Puertas, Íñigo Marcos-Alcalde, Cesar Arjona, Alvaro Villarroel, Rogelio González-Sarmiento, and Carmen Fons. 2020. "Novel Dominant KCNQ2 Exon 7 Partial In-Frame Duplication in a Complex Epileptic and Neurodevelopmental Delay Syndrome" International Journal of Molecular Sciences 21, no. 12: 4447. https://doi.org/10.3390/ijms21124447
APA StyleLazo, P. A., García, J. L., Gómez-Puertas, P., Marcos-Alcalde, Í., Arjona, C., Villarroel, A., González-Sarmiento, R., & Fons, C. (2020). Novel Dominant KCNQ2 Exon 7 Partial In-Frame Duplication in a Complex Epileptic and Neurodevelopmental Delay Syndrome. International Journal of Molecular Sciences, 21(12), 4447. https://doi.org/10.3390/ijms21124447