Progressive Skeletal Muscle Atrophy in Muscular Dystrophies: A Role for Toll-Like Receptor-Signaling in Disease Pathogenesis


Abstract
1. Muscular Dystrophies
2. Muscle Atrophy in Health and Disease
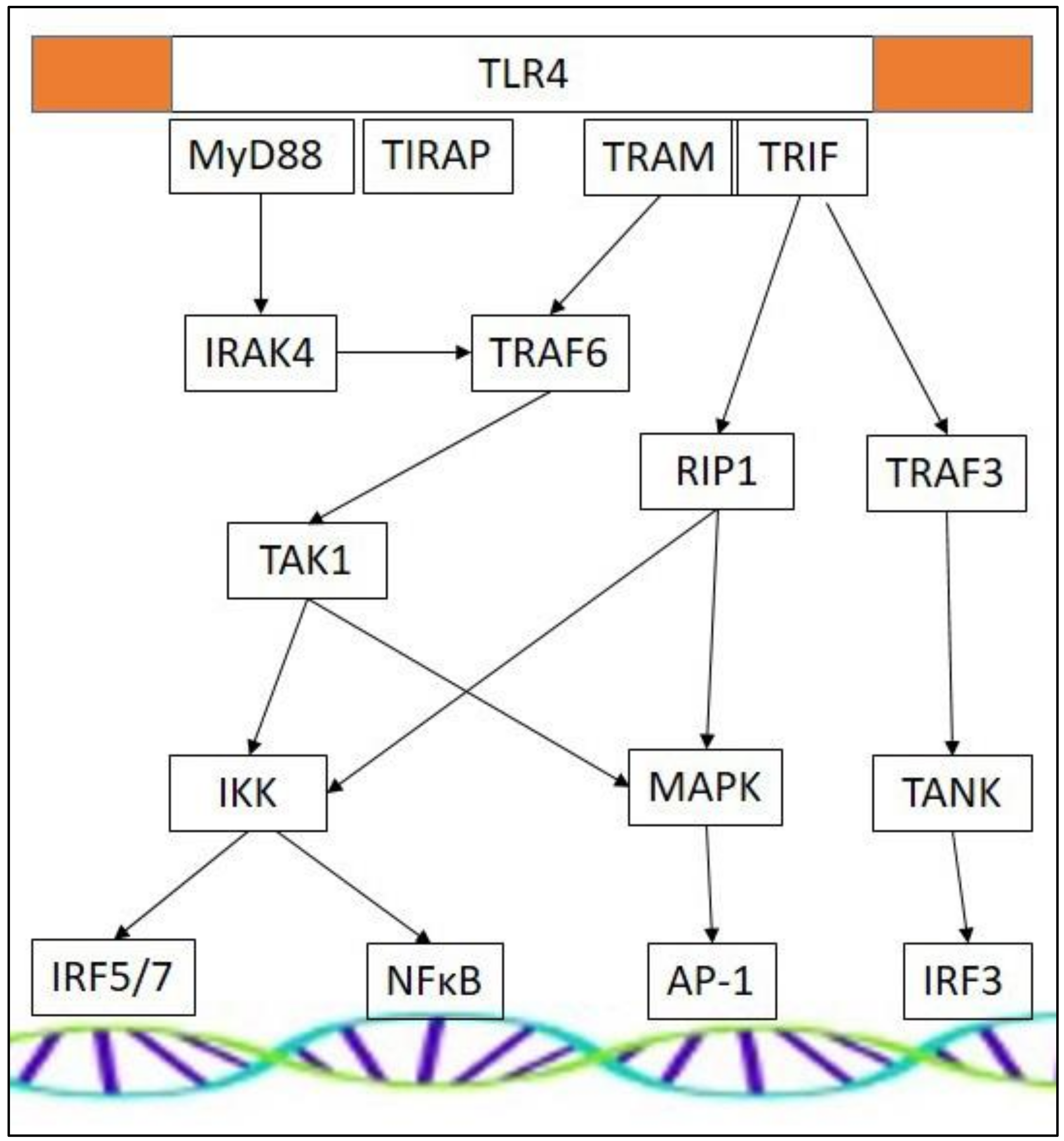
3. Toll-Like Receptor Signaling and Muscular Atrophy
3.1. Cell Surface TLRs
3.2. Intracellular TLRs
3.3. TLR Effectors and Adaptor Molecules
4. Altered TLR Function in Muscular Dystrophies
5. Toll-Like Receptors as a Therapeutic Target for Muscular Dystrophy
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP-1 | Activator protein-1 |
| Bcl-2 | B-cell lymphoma 2 |
| CCL | CC-motif chemokine ligand |
| CSA | Cross sectional area |
| CXCL | CXC-motif chemokine ligand |
| DAMP | Danger-associated molecular pattern |
| DMD | Duchenne muscular dystrophy |
| eIF3f | Eukaryotic translation initiation factor 3f |
| HMGB1 | High mobility group Box 1 |
| HSP | Heat shock protein |
| IFN | Interferon |
| IL | Interleukin |
| IRF | Interferon regulatory factor |
| IκB | Inhibitor κB |
| LGMD | Limb girdle muscular dystrophy |
| LPS | Lipopolysaccharide |
| MHC-I | Major histocompatibility complex I |
| MuRF-1 | Muscle ring finger-1 |
| MyD88 | Myeloid differentiation factor 88 |
| Myf5 | Myogenic factor 5 |
| MyoD | Myogenic determination factor 1 |
| MRF4 | Myogenic regulatory factor 4 |
| NFκB | Nuclear factor κB |
| ROS | Reactive oxygen species |
| SAA1 | Serum amyloid A1 |
| TIR | Toll/IL-1 receptor |
| TIRAP | TIR domain-containing adaptor protein |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| TRAM | TRIF-related adapter molecule |
| TRIF | TIR domain-containing adapter protein inducing interferon-β |
| TRIM32 | E3 ubiquitin ligase tripartite motif 32 |
| UPP | Ubiquitin-proteasome pathway |
References
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Mosquiera, M.; Zeiger, U.; Foerderer, M.; Brinkmeier, H.; Fink, R.H.A. Cardiac and respiratory dysfunction in Duchenne muscular dystrophy and the role of second messengers. Med. Res. Rev. 2013, 5, 1174–1213. [Google Scholar] [CrossRef]
- Mercuri, E.; Boennemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Mouisel, E.; Vignaud, A.; Hourde, C.; Butler-Browne, G.; Ferry, A. Muscle weakness and atrophy are associated with decreased regenerative capacity and changes in MTOR signaling in skeletal muscle of vednerable (18-24-month-old) dystrophic mdx mice. Muscle Nerve 2010, 41, 809–818. [Google Scholar] [CrossRef]
- Madaro, L.; Bouche, M. From innate to adaptive immune response in muscular dystrophies and skeletal muscle regeneration: The role of lymphocytes. BioMed Res. Int. 2014, 2014, 438675. [Google Scholar] [CrossRef] [PubMed]
- Del Rocio Cruz-Guzman, O.; Rodriguez-Cruz, M.; Escobar Cedillo, R.E. Sytemic inflammation in Duchenne muscular dystrophy: Association with muscle function and nutritional status. BioMed Res. Int. 2015, 2015, 891972. [Google Scholar] [CrossRef]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’Amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, B.; Creus, K.K.; Martin, J.J.; De Bleecker, J.L. Upregulation of chemokines and their receptors in Duchenne muscular dystrophy: Potential for attenuation of myofiber necrosis. Muscle Nerve 2012, 46, 917–925. [Google Scholar] [CrossRef]
- Ajime, T.T.; Serré, J.; Wüst, R.C.I.; Messa, G.A.M.; Poffé, C.; Swaminathan, A.; Maes, K.; Janssens, W.; Troosters, T.; Degens, H.; et al. Two weeks smoking cessation reverses cigarette smoke-induced skeletal muscle atrophy and mitochondrial dysfunction in mice. Nicotine Tob. Res. 2020. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and athrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Models Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Taillandier, D.; Polge, C. Skeletal muscle atrogenes: From rodent models to human pathologies. Biochimie 2019, 166, 251–269. [Google Scholar] [CrossRef]
- Asfour, H.A.; Allouh, M.Z.; Said, R.S. Myogenic regulatory factors: The orchestrators of myogenesis after 30 years of discovery. Exp. Biol. Med. 2018, 243, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Li, L.J. Mitochondrial dysregulation and muscle disuse atrophy. F1000 Res. 2019, 8, e1621. [Google Scholar] [CrossRef]
- De Paepe, B. The changed transcriptome of muscular dystrophy and inflammatory myopathy: Contributions of non-coding RNAs to muscle damage and recovery. OBM Genet. 2019, 3. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, M.; Lian, D.; Li, Y.; Li, Y.; Wang, J.; Deng, S.; Yu, K.; Zhengxing, L. Non-coding RNA regulates the myogenesis of skeletal muscle satellite cells, injury repair and diseases. Cells 2019, 8, 988. [Google Scholar] [CrossRef]
- Brusa, R.; Magri, F.; Bresolin, N.; Comi, G.P.; Corti, S. Noncoding RNAs in Duchenne and Becker muscular dystrophies: Role in pathogenesis and future prognostic and therapeutic perspectives. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Kishi, S.; Aoki, Y.; Komaki, H.; Oya, Y.; Takeda, S.I.; Hashido, K. Three novel serum biomarkers, miR-1, miR-133a, and miR-206 for limb-girdle muscular dystrophy, facioscapulohumeral muscular dystrophy, and Becker muscular dystrophy. Environ. Health Prev. Med. 2014, 19, 452–458. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Incitti, T.; Martone, J.; Cesana, M.; Cazzella, V.; Santini, T.; Sthandier, O.; Bozzoni, I. miR-31 modulates dystrophin expression: New implications for Duchenne muscular dystrophy therapy. EMBO Rep. 2011, 12, 136–141. [Google Scholar] [CrossRef]
- Trifunov, S.; Natera-de Benito, D.; Exposito Escudero, J.M.; Medina, J.; Cuadras, D.; Badosa, C.; Carrera, L.; Nascimento, A.; Jimenez-Mallebrera, C. Longitudinal study of three microRNAs in Duchenne muscular dystrophy and Becker muscular dystrophy. Front. Neurol. 2020, 11, e304. [Google Scholar] [CrossRef]
- Ballarino, M.; Cazzella, V.; D’Andrea, D.; Grassi, L.; Bisceglie, L.; Cipriano, A.; Santini, T.; Pinnarò, C.; Morlando, M.; Tramontano, A.A.; et al. Novel long noncoding RNAs (lncRNAs) in myogenesis: A MiR-31 overlapping lncRNA transcript controls myoblast differentiation. Mol. Cell. Biol. 2015, 35, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’Amico, A.; Bertini, E.; Bozzoni, I. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med. 2011, 3, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Sotgia, F.; Schubert, W.; Park, D.S.; Frank, P.G.; Woodman, S.E.; Insabato, L.; Cammer, M.; Minetti, C.; Lisanti, M.P. Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteins. Am. J. Pathol. 2003, 163, 1663–1675. [Google Scholar] [CrossRef]
- Saenz, A.; Azpitarte, M.; Armananzas, R.; Leturcq, F.; Alzualde, A.; Inza, I.; García-Bragado, F.; De la Herran, G.; Corcuera, J.; Cabello, A.; et al. Gene expression profiling in limb girdle muscular dystrophy 2A. PLoS ONE 2008, 3, e3750. [Google Scholar] [CrossRef]
- Fanin, M.; Nascimbeni, A.C.; Angelini, C. Muscle atrophy in limb girdle muscular dystrophy 2A: A morphometric and molecular study. Neuropathol. Appl. Neurobiol. 2013, 39, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Nascimbeni, A.C.; Angelini, C. Muscle atrophy, ubiquitin-proteasome, and autophagic pathways in dysferlinopathy. Muscle Nerve 2014, 50, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, E.; Kudryashova, D.; Kramerova, I.; Spencer, M.J. Trim 32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 2005, 354, 413–424. [Google Scholar] [CrossRef]
- De Palma, C.; Morisi, F.; Cheli, S.; Pambianco, S.; Cappello, V.; Vezzoli, M.; Revere-Querini, P.; Moggio, M.; Ripolone, M.; Francnolini, M.; et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012, 3, e418. [Google Scholar] [CrossRef]
- Sandri, M.; Coletto, L.; Grumati, P.; Bonaldo, P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci. 2013, 126, 5325–5333. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Spencer, M.J. Calpains and muscular dystrophies. Int. J. Biochem. Cell Biol. 2000, 32, 1–5. [Google Scholar] [CrossRef]
- Sandri, M.; El Meslemani, A.H.; Sandri, C.; Schjerling, P.; Vissing, K.; Andersen, J.L.; Rossini, K.; Carraro, U.; Angelini, C. Caspase 3 expression correlates with skeletal muscle apoptosis in duchenne and facioscapulo human muscular dystrophy. A potential target for pharmacological treatment? J. Neuropathol. Exp. Neurol. 2001, 60, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Cros, D.; Harnden, P.; Pellissier, J.F.; Serratrice, G. Muscle hypertrophy in Duchenne muscular dystrophy. J. Neurol. 1989, 236, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Wokke, B.H.; Van den Bergen, J.C.; Versluis, M.J.; Niks, E.H.; Milles, J.; Webb, A.G.; Van Zwet, E.W.; Aartsma-Rus, A.; Verschuuren, J.J.; Kan, H.E. Quantitative MRI and strenght measurements in the assessment of muscle quality in Duchenne muscular dystrophy. Neuromuscul. Disord. 2014, 24, 409–416. [Google Scholar] [CrossRef]
- Henry, C.C.; Martin, K.S.; Ward, B.B.; Handsfield, G.G.; Pierce, S.M.; Blemker, S.S. Spatial and age-related changes in the microstructure of dystrophic and healthy diaphragms. PLoS ONE 2017, 12, e0183853. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.P.; Hoffman, E.P.; Mittal, P.; Brown, K.J.; Schatzberg, S.J.; Ghimbovschi, S.; Wang, Z.; Kornegay, J.N. Sparing of the dystrophin-deficient cranial Sartorius muscle is associated with classical and novel hypertrophy pathways in GRMD dogs. Am. J. Pathol. 2013, 183, 1411–1424. [Google Scholar] [CrossRef]
- Hahn, A.; Kny, M.; Pablo-Tortola, C.; Todiras, M.; Willenbrock, M.; Schmidt, S.; Schmoeckel, K.; Jorde, I.; Nowak, M.; Jarosch, E.; et al. Serum amyloid A1 mediates myotube atrophy via Toll-like receptors. J. Cachexia Sarcopenia Muscle 2020, 11, 103–119. [Google Scholar] [CrossRef]
- Ono, Y.; Sakamoto, K. Lipopolysaccharide inhibits myogenic differentiation of C2C12 myoblasts through the Toll-like receptor 4-nuclear factor-κB signaling pathway and myoblast-derived tumor necrosis factor-α. PLoS ONE 2017, 12, e0182040. [Google Scholar] [CrossRef]
- Langhans, C.; Weber-Carstens, S.; Schmidt, F.; Hamati, J.; Kny, M.; Zhu, X.; Wollersheim, T.; Koch, S.; Krebs, M.; Schultz, H.; et al. Inflammation-induced acute phase response in skeletal muscle and critical illness myopathy. PLoS ONE 2014, 9, e92048. [Google Scholar] [CrossRef]
- Graber, T.G.; Rawls, B.L.; Tian, B.; Durham, W.J.; Brightwell, C.R.; Brasier, A.R.; Rasmussen, B.B.; Fry, C.S. Repetitive TLR-3 activation in the lung induces skeletal muscle adaptations and cachexia. Exp. Gerontol. 2018, 106, 88–100. [Google Scholar] [CrossRef]
- Li, L.F.; Liu, Y.Y.; Chen, N.H.; Chen, Y.H.; Huang, C.C.; Kao, K.C.; Chang, C.H.; Chuang, L.P.; Chiu, L.C. Attenuation of ventilation-induced diaphragm dysfunction through Toll-like receptor 4 and nuclear factor-κB in a murine endotoxemia model. Lab. Investig. 2018, 98, 1170–1183. [Google Scholar] [CrossRef]
- McKenzie, A.I.; Reidy, P.T.; Nelson, D.S.; Mulvey, J.L.; Yonemura, N.M.; Petrocelli, J.J.; Mamassani, Z.S.; Tippets, T.S.; Summers, S.A.; Funai, K.; et al. Pharmacological inhibition of TLR4 ameliorates muscle and liver ceramide content after disuse in previously physically active mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R503–R511. [Google Scholar] [CrossRef] [PubMed]
- Ulfgren, A.K.; Grundtman, C.; Borg, K.; Alexanderson, H.; Andersson, U.; Harris, H.E.; Lundberg, I.E. Down-regulation of the aberrant expression of the inflammation mediator high mobility group box chromosomal protein 1 in muscle tissue of patients with polymyositis and dermatomyositis treated with corticosteroids. Arthritis Rheum. 2004, 50, 1586–1594. [Google Scholar] [CrossRef]
- Huang, W.; Tang, Y.; Li, L. HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine 2010, 51, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Zhang, X.; Peng, A.; He, M.; Lei, Z.; Wang, Y. TLR4-HMGB1 signaling pathway affects the inflammatory reaction of autoimmune myositis by regulating MHC-I. Int. Immunopharmacol. 2016, 41, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Chai, N.N.; Chen, A.; Jordan, S.; Klein, A. Anti-IL6R attenuates humoral responses to allograft in a mouse model of allosensitization. J. Heart Lung Transplant. 2013, 32, S245. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.D.; Zahoor, M.A.; Abimiku, A.; Osawe, S.; Rosenthal, K.L.; InFANT Study Team. TLR10 Senses HIV-1 Proteins and Significantly Enhances HIV-1 Infection. Front. Immunol. 2019, 10, e482. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.H.; Divangahi, M.; Yahiaoui, L.; Gvozdic, D.; Qureshi, S.; Petrof, B.J. Toll-like receptors differentially regulate CC and CXC chemokines in skeletal muscle via NF-κB and calcineurin. Infect. Immun. 2006, 74, 6829–6838. [Google Scholar] [CrossRef] [PubMed]
- Mojumdar, K.; Giordano, C.; Lemaire, C.; Liang, F.; Divangahi, D.; Qureshi, S.T.; Petrof, B.J. Divergent impact of Toll-like receptor 2 deficiency on repair mechanisms in healthy muscle versus Duchenne muscular dystrophy. J. Pathol. 2016, 239, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, K.R.; Goli, P.; Roy, A.; Sharma, A.K.; Xiong, G.; Gallot, Y.S.; Kumar, A. The toll-like receptor/myd88/xbp1 signaling axis mediates skeletal muscle wasting during cancer cachexia. Mol. Cell. Biol. 2019, 39, e00184-19. [Google Scholar] [CrossRef]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. 2010, 298, C542–C549. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.; Zhang, G.; Abdel Fattah, E.A.; Eissa, N.T.; Lui, Y.P. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J. 2011, 25, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Dehoux, M.J.; Van Beneden, R.P.; Fernandez-Celemin, L.; Lause, P.L.; Thissen, J.P. Induction of MafBx and Murf ubiquitin ligase mRNAs in rat skeletal muscle after LPS injection. FEBS Lett. 2003, 544, 214–217. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.P. Curcumin prevents lipopolysaccharide-induced atrogin-1/MAFbx upregulation and muscle mass loss. J. Cell. Biochem. 2007, 100, 960–969. [Google Scholar] [CrossRef]
- Wan, J.; Chen, D.; Yu, B.; Luo, Y.; Mao, X.; Zheng, P.; Yu, J.; Luo, J.; He, J. Leucine protects against skeletal muscle atrophy in lipopolysaccharide-challenged rats. J. Med. Food 2017, 20, 93–101. [Google Scholar] [CrossRef]
- Kawanishi, N.; Nozaki, R.; Naito, H.; Machida, S. TLR4-defective (C3H/HeJ) mice are not protected from cast immobilization-induced muscle atrophy. Physiol. Rep. 2017, 5, e13255. [Google Scholar] [CrossRef]
- Sin, T.K.; Zhang, G.; Zhang, Z.; Gao, S.; Li, M.; Li, Y.P. Cancer takes a toll on skeletal muscle by releasing heat shock proteins—An emerging mechanism of cancer-induced cachexia. Cancers 2019, 11, 1272. [Google Scholar] [CrossRef]
- Ewald, S.E.; Lee, B.L.; Lau, L.; Wickliffe, K.E.; Shi, G.P.; Chapman, H.A.; Barton, G.M. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 2008, 456, 658–662. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Geto, Z.; Molla, M.D.; Challa, F.; Belay, Y.; Getahun, T. Mitochondrial dynamic dysfunction as a main triggering factor for inflammation associated chronic non-communicable diseases. J. Inflamm. Res. 2020, 13, 97–107. [Google Scholar] [CrossRef]
- Duregotti, E.; Negro, S.; Scorzeto, M.; Zornetta, I.; Dickinson, B.C.; Chang, C.J.; Montecucco, C.; Rigoni, M. Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc. Natl. Acad. Sci. USA 2015, 112, e497–e505. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Rodriguez-Nuevo, A.; Dias-Ramos, A.; Noguera, E.; Diaz-Saez, F.; Duran, X.; Munoz, J.P.; Romero, M.; Plana, N.; Sebastian, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nguyen, P.T.; Wang, X.; Zhao, Y.; Meacham, C.E.; Zou, Z.; Bordieanu, B.; Johanns, M.; Vertommen, D.; Wijshake, T.; et al. TLR9 and beclin 1 crosstalk regulates muscle AMPK activation in exercise. Nature 2020, 578, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar] [PubMed]
- Raby, A.C.; Colmont, C.S.; Kift-Morgan, A.; Köhl, J.; Eberl, M.; Fraser, D.; Tpley, N.; Labeta, M.O. Toll-like receptors 2 and 4 are potential therapeutic targets in peritoneal dialysis-associated fibrosis. J. Am. Soc. Nephrol. 2017, 28, 461–478. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathways. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Verstak, B.; Stack, J.; Ve, T.; Mangan, M.; Hjerrild, K.; Jeon, J.; Stahl, R.; Latz, E.; Gay, N.; Kobe, B.; et al. The TLR signaling adaptor TRAM interacts with TRAF6 to mediate activation of the inflammatory response by TLR4. J. Leukoc. Biol. 2014, 96, 427–436. [Google Scholar] [CrossRef]
- Trinchieri, G.; Sher, A. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 2007, 7, 179–190. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Sanjo, H.; Uematsu, S.; Kaisho, T.; Hoshino, K.; Takeuchi, O.; Kobayashi, M.; Fujita, T.; et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 2002, 420, 324–329. [Google Scholar] [CrossRef]
- Hemmi, H.; Akira, S. TLR signaling and the function of dendritic cells. Chem. Immunol. Allergy 2005, 86, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, e461. [Google Scholar] [CrossRef] [PubMed]
- Cappelletti, C.; Salerno, F.; Canioni, E.; Mora, M.; Mantegazza, R.; Bernasconi, P.; Maggi, L. Up-regulation of Toll-like receptors 7 and 9 and its potential implications in the pathogenic mechanisms of LMNA-related myopathies. Nucleus 2018, 9, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef]
- Giordano, C.; Mojumdar, K.; Liang, F.; Lemaire, C.; Li, T.; Richardson, J.; Divangahi, M.; Qureshi, S.; Petrof, B.J. Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum. Mol. Genet. 2015, 24, 2147–2162. [Google Scholar] [CrossRef]
- Chen, Y.W.; Nagaraju, K.; Bakay, M.; McIntyre, O.; Rawat, R.; Shi, R.; Hoffman, E.P. Early onset of inflammation and later involvement of TGFβ in Duchenne muscular dystrophy. Neurology 2005, 65, 826–834. [Google Scholar] [CrossRef]
- Hathout, Y.; Brody, E.; Clemens, P.R.; Cripe, L.; DeLisle, R.K.; Furlong, P.; Gordish-Dressman, H.; Hache, L.; Henricson, E.; Hoffman, E.P.; et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 7153–7158. [Google Scholar] [CrossRef]
- Davalos, D.; Akassoglou, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2011, 34, 43–62. [Google Scholar] [CrossRef]
- Henriques-Pons, A.; Yu, Q.; Rayavarapu, S.; Cohen, T.V.; Ampong, B.; Cha, H.J.; Jahnke, V.; Van der Meulen, J.; Wang, D.; Jiang, W.; et al. Role of Toll-like receptors in the pathogenesis of dystrophin-deficient skeletal and heart muscle. Hum. Mol. Genet. 2014, 23, 2604–2617. [Google Scholar] [CrossRef]
- Gallot, Y.S.; Straughn, A.R.; Bohnert, K.R.; Xiong, G.; Hindi, S.M.; Kumar, A. MyD88 is required for satellite cell-mediated myofiber regeneration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2018, 27, 3449–3463. [Google Scholar] [CrossRef]
- Uaesoontrachoon, K.; Cha, H.J.; Ampong, B.; Sali, A.; Vandermeulen, J.; Wei, B.; Creeden, B.; Huynh, T.; Quinn, J.; Tatem, K.; et al. The effects of MyD88 deficiency on disease phenotype in dysferlin-deficient A/J mice: Role of endogeneous TLR ligands. J. Pathol. 2013, 231, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Calore, F.; Londhe, P.; Fadda, P.; Nigita, G.; Casadei, L.; Marceca, G.P.; Fassan, M.; Lovat, F.; Gasparini, P.; Rizzotto, L.; et al. The TLR7/8/9 Antagonist IMO-8503 inhibits cancer-induced cachexia. Cancer Res. 2018, 78, 6680–6690. [Google Scholar] [CrossRef]
- Kandimalla, E.R.; Bhagat, L.; Wang, D.; Yu, D.; Sullivan, T.; La Monica, N.; Agrawal, S. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res. 2013, 41, 3947–3961. [Google Scholar] [CrossRef][Green Version]
- Terrill, J.R.; Radley-Crabb, H.G.; Grounds, M.D.; Arthur, P.G. N-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul. Disord. 2012, 22, 427–434. [Google Scholar] [CrossRef]
- Head, S.I. Antioxidant therapy in a mouse model of Duchenne muscular dystrophy: Some promising results but with a weighty caveat. J. Physiol. 2017, 595, e7015. [Google Scholar] [CrossRef]
- Burns, D.P.; Drummond, S.E.; Bolger, D.; Coiscaud, A.; Murphy, K.H.; Edge, D.; O’Halloran, K.D. N-acetylcysteine decreases fibrosis and increases force-generating capacity of mdx diaphragm. Antioxidants 2019, 8, 581. [Google Scholar] [CrossRef]
- Pinniger, G.J.; Terrill, J.R.; Assan, E.B.; Grounds, M.D.; Arthur, P.G. Pre-clinical evaluation of N-acetylcysteine reveals side effects in the mdx mouse model of Duchenne muscular dystrophy. J. Physiol. 2017, 595, 7093–7107. [Google Scholar] [CrossRef]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol. 2008, 101, 14–19. [Google Scholar] [CrossRef]
- Giri, S.S.; Sen, S.S.; Jun, J.W.; Sukumaran, V.; Park, S.C. Protective effects of leucine against lipopolysaccharide-induced inflammatory response in Labeo rohita fingerlings. Fish Shellfish Immunol. 2016, 52, 239–247. [Google Scholar] [CrossRef]
- Mendell, J.R.; Griggs, R.C.; Moxley, R.T., III; Fenichel, G.M.; Brooke, M.H.; Miller, J.P.; Province, M.A.; Dodson, W.E. Clinical investigation in Duchenne muscular dystrophy: IV. Double-blind controlled trial of leucine. Muscle Nerve 1984, 7, 535–541. [Google Scholar] [CrossRef]
- Kessel, A.; Toubi, E.; Pavlotzky, E.; Mogilner, J.; Coran, A.G.; Lurie, M.; Karry, R.; Sukhotnik, I. Treatment with glutamine is associated with down-regulation of Toll-like receptor-4 and myeloid differentiation factor 88 expression and decrease in intestinal mucosal injury caused by lipopolysaccharide endotoxaemia in a rat. Clin. Exp. Immunol. 2008, 151, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Mok, E.; Letellier, G.; Cuisset, J.M.; Denjean, A.; Gottrand, F.; Alberti, C.; Hankard, R. Lack of functional benefit with glutamine versus placebo in Duchenne muscular dystrophy: A randomized crossover trial. PLoS ONE 2009, 4, e5448. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Wang, W.; Tamaki, Z.; Shi, B.; Yeldandi, A.; Tsukimi, Y.; Yamasaki, M.; Varga, J. Pharmacological inhibition of Toll-like receptor-4 signaling by TAK242 prevents and induces regression of experimental organ fibrosis. Front. Immunol. 2018, 9, e2434. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Carlson, C.G.; Dole, E.; Stefanski, C.; Bayless, D. The effect of specific IKKβ inhibitors on the cytosolic expression of IκB-α and the nuclear expression of p65 in dystrophic (MDX) muscle. Am. J. Transl. Res. 2015, 7, 670–682. [Google Scholar]
- Charan, R.A.; Niizawa, G.; Nakai, H.; Clemens, P.R. Adeno-associated virus serotype 8 (AAV8) delivery of recombinant A20 to skeletal muscle reduces pathological activation of nuclear factor (NF)-kappaB in muscle of mdx mice. Mol. Med. 2013, 18, 1527–1535. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Line/Animal Strain | Treatment | Atrophic Effect | Reference |
|---|---|---|---|
| C2C12 myotubes | SAA1 | Myotube atrophy and increased IL-6 expression. | [36] |
| C2C12 myotubes | SAA1 + anti-TLR2 anti-TLR4 | Attenuated myotube atrophy and IL-6 expression. | [36] |
| C2C12 myotubes | Pam3Csk4 | Myotube atrophy. | [36] |
| C2C12 myotubes | LPS | Muscle cell atrophy. | [37] |
| C2C12 myotubes | LPS + TAK242 | Attenuated muscle cell atrophy. | [37] |
| C2C12 myotubes | LPS | Myotube atrophy and increased SAA1 mRNA and protein. | [38] |
| C57BL/6J mice | Cecal ligation and puncture sepsis model | Increased expression of TLR2 and TLR4 in tibialis anterior and gastrocnemius/plantaris. | [36] |
| C57BL/6J mice | Poly(I:C) | Atrophy of gastrocnemius, tibilalis anterior, soleus, and plantaris, but not in extensor digitorum longus. Lower mean muscle fiber cross-sectional areas in gastrocnemius, plantaris, and diaphragm. | [39] |
| C57BL/6J mice | Mechanical ventilation + LPS | Diaphragmic atrophy. | [40] |
| C57BL/6J Tlr4Lps-del TLR4-deficient mice | Mechanical ventilation + LPS | Normal diaphragmic muscle fiber diameter. | [40] |
| C57BL/6J mice | Hindlimb suspension + TAK242 | Smaller gastrocnemius, but muscle fiber size unaltered. | [41] |
| Gene Deficiency | Target | Regulation in Dystrophy | Reference |
|---|---|---|---|
| DMD | TLR2/4 | Expression of TLR2 and TLR4 is significantly increased in DMD muscle. | [79] |
| TLR4 expression is increased in mdx diaphragm. | [75] | ||
| HMGB1 is overexpressed in the muscle fiber cytoplasm of DMD tissues. | [75] | ||
| Soluble HSP70 is increased in serum of DMD patients. | [77] | ||
| TLR7/8/9 | TLR7 is expressed in inflammatory cells and blood vessels in muscle from DMD patients. | [76] | |
| Strong TLR7 staining is present in muscle fibers and inflammatory cells of DMD tissue. | [79] | ||
| The vast majority of muscle cells isolated from mdx express TLR9. | [79] | ||
| adaptors | MyD88 is upregulated in skeletal muscle of DMD patients. | [76] | |
| MyD88 protein levels are strongly increased in mdx muscle. | [80] | ||
| MyD88 staining is increased in satellite cells of mdx mice. | [80] | ||
| DYSF | TLR2/4 | Primary muscle cells prepared from SJL/J mice express TLR2 and TLR4. | [74] |
| TLR7/8/9 | TLR7 expression is significantly upregulated in muscle from A/J mice. | [81] | |
| TLR8 gene expression is not significantly altered in A/J mice. | [81] | ||
| LMNA | TLR2/4 | TLR4 is strongly expressed on the sarcolemma of a subset of muscle fibers and capillaries in patient biopsies. | [73] |
| TLR7/8/9 | TLR7 is expressed in the endomysial space, on blood vessels, a minority of inflammatory cells, and occasionally at the sarcolemma of degenerated muscle fibers in patient biopsies. | [73] | |
| In patient muscle, TLR9 is expressed in the endomysial space, at the muscle fiber membrane, on capillaries, and rare inflammatory cells. | [73] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Paepe, B. Progressive Skeletal Muscle Atrophy in Muscular Dystrophies: A Role for Toll-Like Receptor-Signaling in Disease Pathogenesis. Int. J. Mol. Sci. 2020, 21, 4440. https://doi.org/10.3390/ijms21124440
De Paepe B. Progressive Skeletal Muscle Atrophy in Muscular Dystrophies: A Role for Toll-Like Receptor-Signaling in Disease Pathogenesis. International Journal of Molecular Sciences. 2020; 21(12):4440. https://doi.org/10.3390/ijms21124440
Chicago/Turabian StyleDe Paepe, Boel. 2020. "Progressive Skeletal Muscle Atrophy in Muscular Dystrophies: A Role for Toll-Like Receptor-Signaling in Disease Pathogenesis" International Journal of Molecular Sciences 21, no. 12: 4440. https://doi.org/10.3390/ijms21124440
APA StyleDe Paepe, B. (2020). Progressive Skeletal Muscle Atrophy in Muscular Dystrophies: A Role for Toll-Like Receptor-Signaling in Disease Pathogenesis. International Journal of Molecular Sciences, 21(12), 4440. https://doi.org/10.3390/ijms21124440