Liposome Consolidated with Cyclodextrin Provides Prolonged Drug Retention Resulting in Increased Drug Bioavailability in Brain



 ,
, 
Abstract

1. Introduction
2. Results
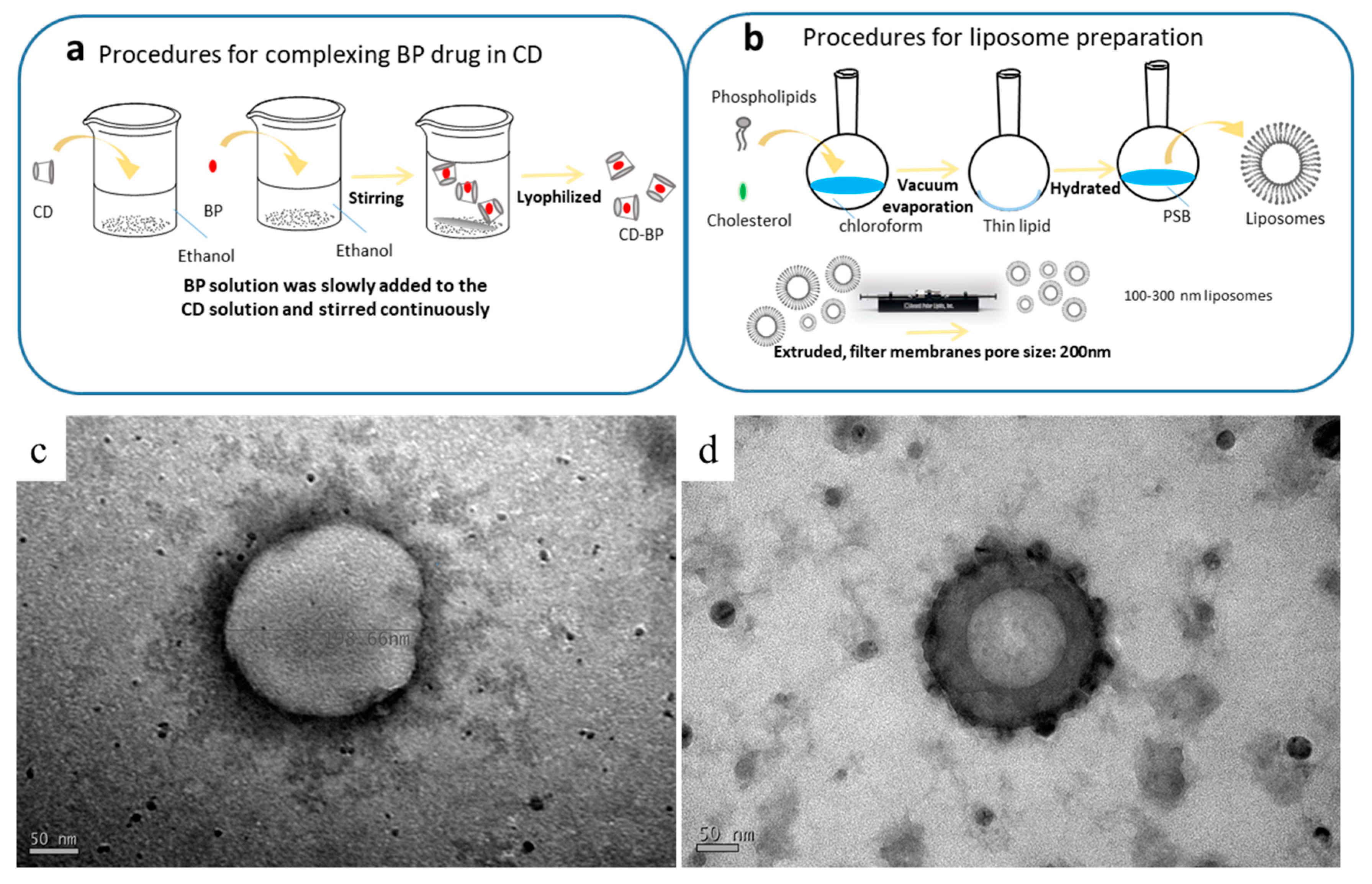
2.1. Characteristics of Drug Formulations
2.2. Appearance of the Liposomes
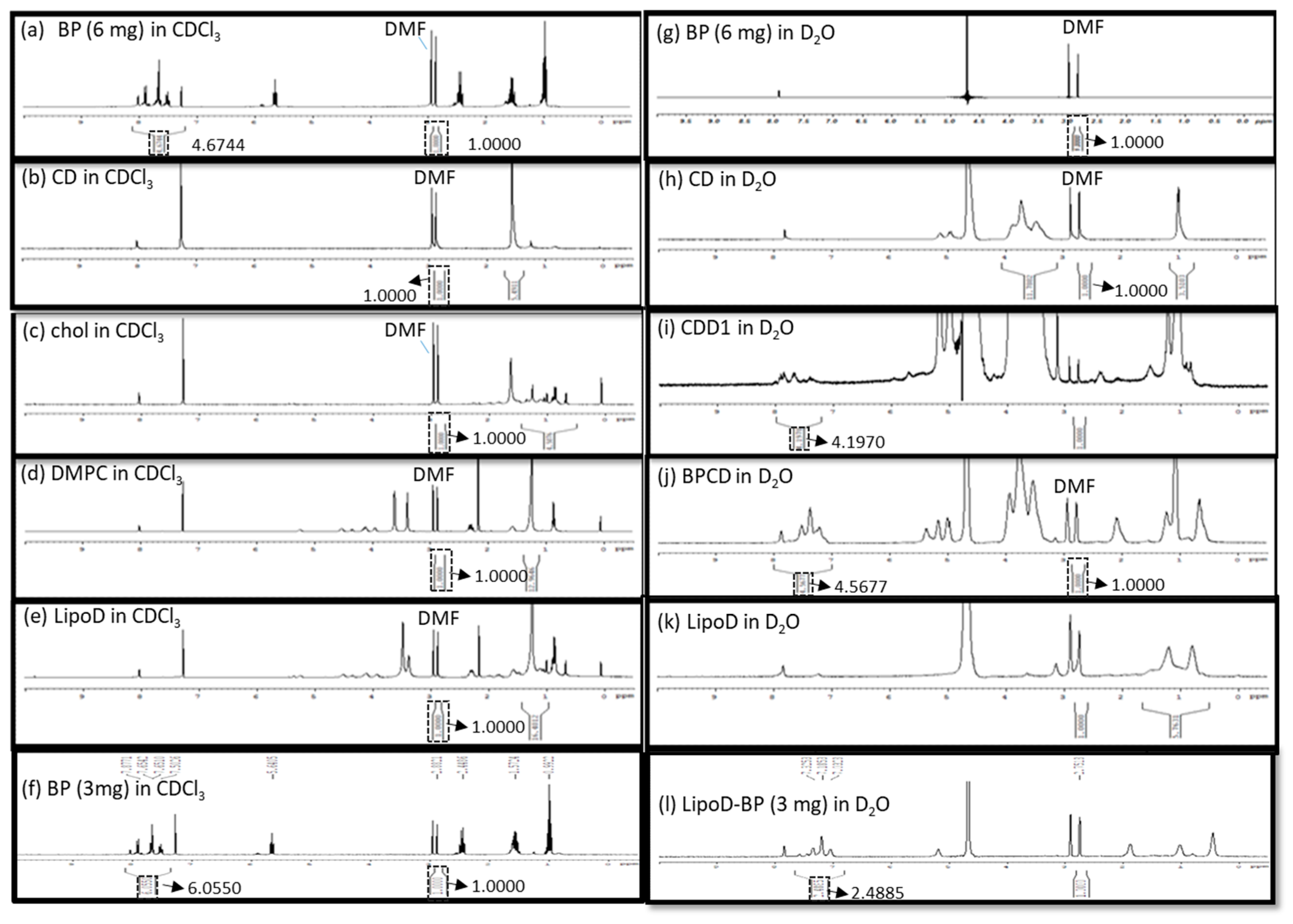
2.3. Characterization of Formulations and the Encapsulation Efficiency
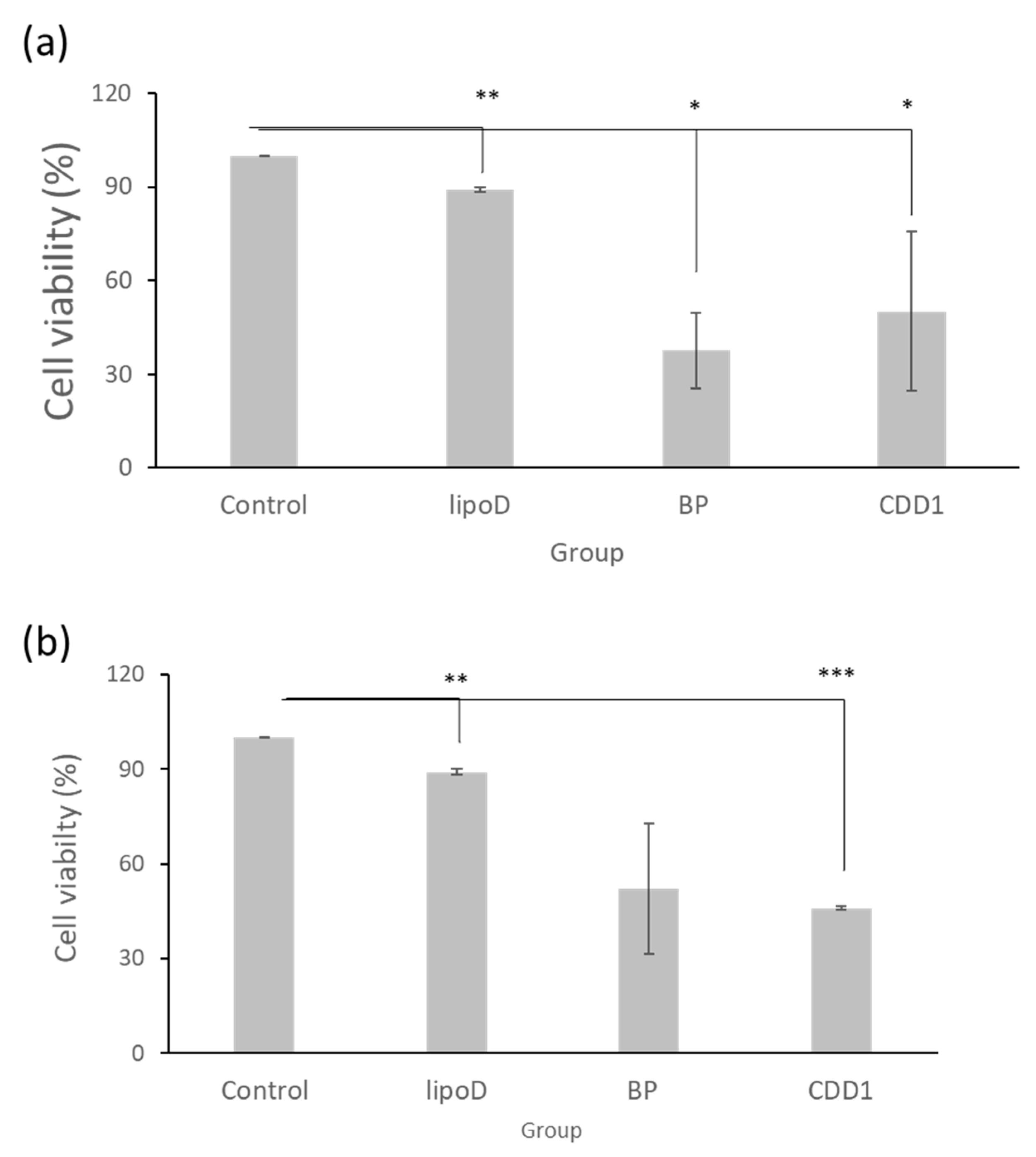
2.4. Cytotoxicity in Drug-Resistant Glioblastoma Multiforme Cells
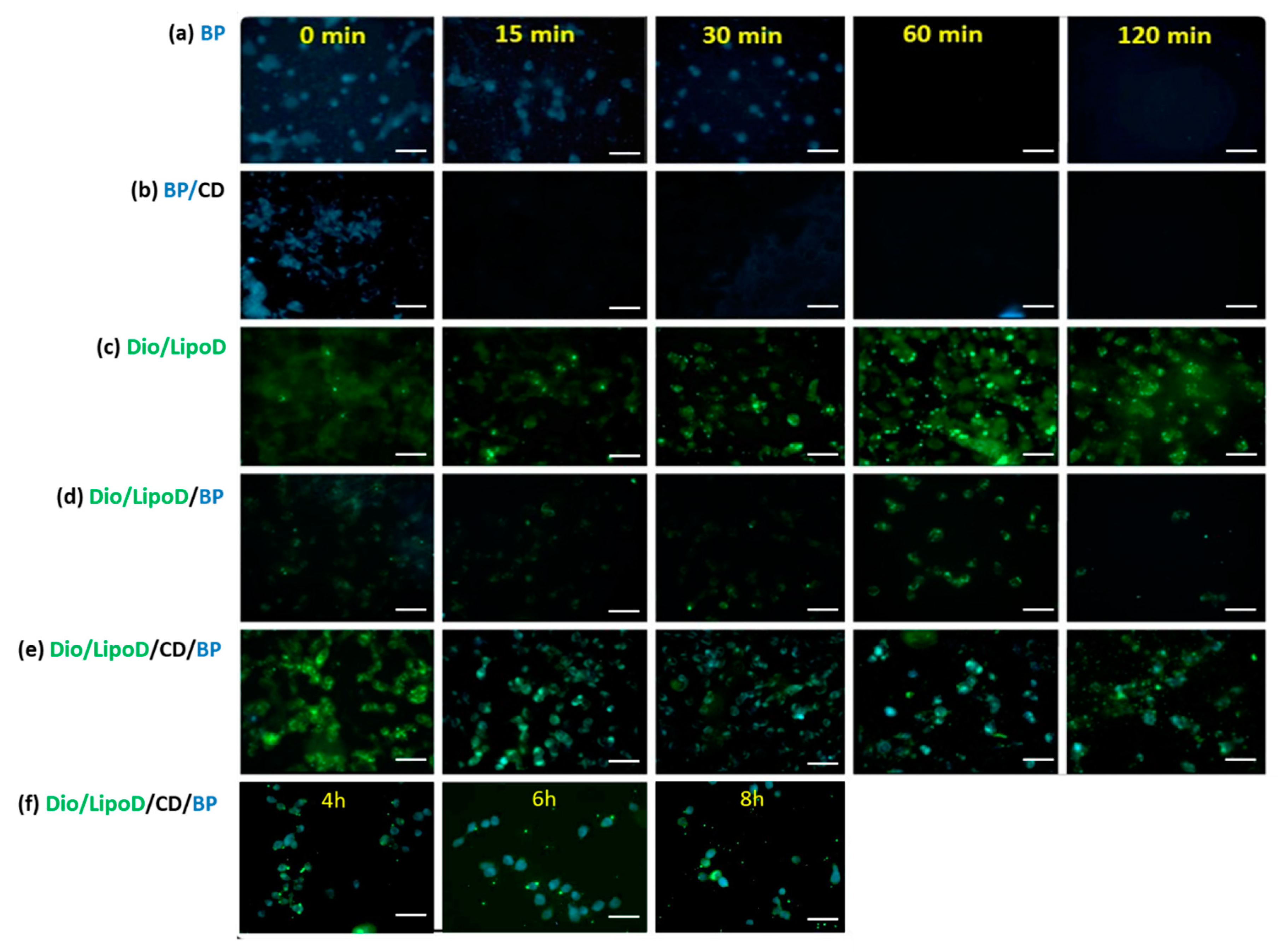
2.5. Dynamic Cellular Uptake of the Formulations
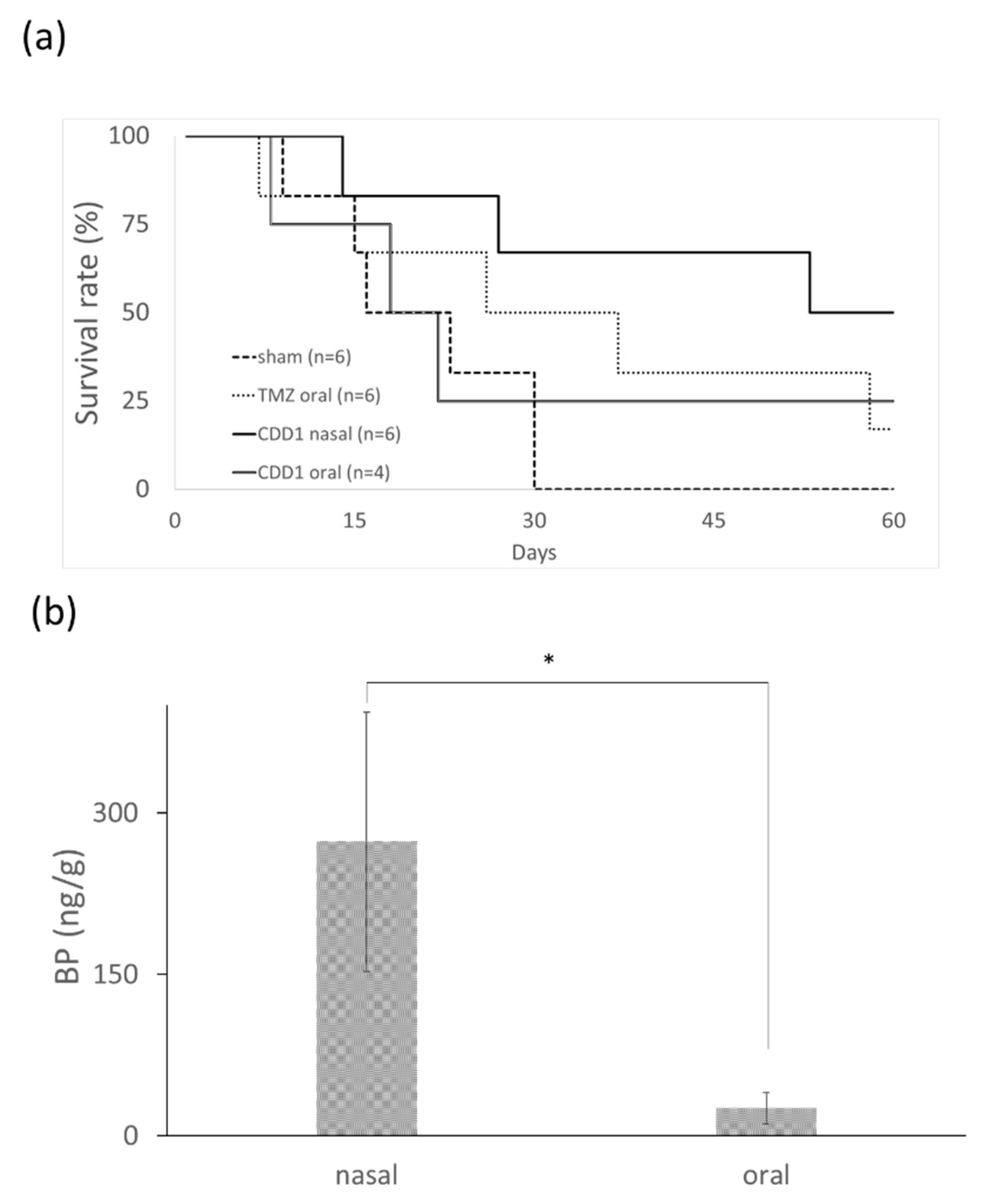
2.6. In Vivo Animal Survival Rate
2.7. In Vivo BP Levels in Brain Tissue
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Encapsulation of BP
4.3. Preparation of Liposomes
4.4. Preparation of BP-Containing Liposomes
4.5. Particle Size and Size Distribution Coefficient Analysis
4.6. EE Analysis by UV–Vis Spectroscopy
4.7. EE Analysis via NMR Spectroscopy
4.8. TEM Analysis
4.9. MTT Assay for Cytotoxicity
4.10. Drug Uptake in Tumor Cells
4.11. Malignant Drug-Resistant Brain Tumor Animal Models and Treatment of Nude Mice
4.12. Biodistribution
4.12.1. Drug Administration
4.12.2. Sample Preparation for LC–MS Analysis
4.12.3. LC–MS Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EE | Encapsulation efficiency |
| ESI | Electrospray ionization |
| MS | Mass spectrometry |
| NLAC | National Laboratory Animal Center |
| NMR | Nuclear magnetic resonance |
| NTA | Nanoparticle tracking analysis |
| SD | Standard deviation |
| TEM | Transmission electron microscopy |
| TERT | Telomerase reverse transcriptase |
References
- Anjum, K.; Shagufta, B.I.; Abbas, S.Q.; Patel, S.; Khan, I.; Shah, S.A.A.; Akhter, N.; Hassan, S.S.U. Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: A review. Biomed. Pharmacother. 2017, 92, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.N.; Rewcastle, N.B.; Brasher, P.M.A.; Fulton, D.; MacKinnon, J.A.; Hamilton, M.; Cairncross, J.G.; Forsyth, P. Which glioblastoma multiforme patient will become a long-term survivor? A population-based study. Ann. Neurol. 1999, 46, 183–188. [Google Scholar] [CrossRef]
- Scott, J.N.; Rewcastle, N.B.; Brasher, P.M.; Fulton, D.; Hagen, N.A.; MacKinnon, J.A.; Sutherland, G.; Cairncross, J.G.; Forsyth, P. Long-term glioblastoma multiforme survivors: A population-based study. Can. J. Neurol. Sci. (J. Canadien Sci. Neurol.) 1998, 25, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Janzer, R.C.; Raff, M.C. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Sun, Y.H.; Zhang, Y.Z.; Wang, Z.C.; Sun, M.Z.; Zhao, D.H. [Relationship between the expression of O6-methylguanine-DNA methyltransferase in glioma and the survival time of patients]. Ai Zheng 2004, 23, 1052–1055. [Google Scholar] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Bondy, M.L.; Scheurer, M.E.; Malmer, B.; Barnholtz-Sloan, J.S.; Davis, F.G.; Il’yasova, D.; Kruchko, C.; McCarthy, B.J.; Rajaraman, P.; Schwartzbaum, J.A.; et al. Brain tumor epidemiology: Consensus from the Brain Tumor Epidemiology Consortium. Cancer 2008, 113, 1953–1968. [Google Scholar] [CrossRef]
- Roy, S.; Lahiri, D.; Maji, T.; Biswas, J. Recurrent Glioblastoma: Where we stand. South Asian J. Cancer 2015, 4, 163–173. [Google Scholar] [CrossRef]
- Tsai, N.M.; Chen, Y.L.; Lee, C.C.; Lin, P.C.; Cheng, Y.L.; Chang, W.L.; Lin, S.Z.; Harn, H.J. The natural compound n-butylidenephthalide derived from Angelica sinensis inhibits malignant brain tumor growth in vitro and in vivo. J. Neurochem. 2006, 99, 1251–1262. [Google Scholar] [CrossRef]
- Lin, P.C.; Chen, Y.L.; Chiu, S.C.; Yu, Y.L.; Chen, S.P.; Chien, M.H.; Chen, K.Y.; Chang, W.L.; Lin, S.Z.; Chiou, T.W.; et al. Orphan nuclear receptor, Nurr-77 was a possible target gene of butylidenephthalide chemotherapy on glioblastoma multiform brain tumor. J. Neurochem. 2008, 106, 1017–1026. [Google Scholar] [CrossRef]
- Lin, P.C.; Lin, S.Z.; Chen, Y.L.; Chang, J.S.; Ho, L.I.; Liu, P.Y.; Chang, L.F.; Harn, Y.C.; Chen, S.P.; Sun, L.Y.; et al. Butylidenephthalide suppresses human telomerase reverse transcriptase (TERT) in human glioblastomas. Ann. Surg. Oncol. 2011, 18, 3514–3527. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.H.; Lin, S.Z.; Lin, P.C.; Chiou, T.W.; Harn, Y.W.; Ho, L.I.; Chan, T.M.; Chou, C.W.; Chuang, C.H.; Su, H.L.; et al. Brain tumor senescence might be mediated by downregulation of S-phase kinase-associated protein 2 via butylidenephthalide leading to decreased cell viability. Tumour Biol. J. Int. Soc. Oncodevelopment. Biol. Med. 2014, 35, 4875–4884. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.Y.; Harn, H.J.; Lin, S.Z.; Chen, S.R.; Lin, P.C.; Syu, F.J.; Hsieh, D.K.; Huang, M.H.; Chiou, T.W. (Z)-butylidenephthalide Restores Temozolomide Sensitivity to Temozolomide-resistant Malignant Glioma Cells by Downregulating Expression of the DNA Repair Enzyme MGMT. Eur. J. Cancer 2012, 48, S220–S221. [Google Scholar] [CrossRef]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., 2nd. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Van Woensel, M.; Wauthoz, N.; Rosiere, R.; Mathieu, V.; Kiss, R.; Lefranc, F.; Steelant, B.; Dilissen, E.; Van Gool, S.W.; Mathivet, T.; et al. Development of siRNA-loaded chitosan nanoparticles targeting Galectin-1 for the treatment of glioblastoma multiforme via intranasal administration. J. Control Release 2016, 227, 71–81. [Google Scholar] [CrossRef]
- Merkus, F.W.; Verhoef, J.C.; Marttin, E.; Romeijn, S.G.; van der Kuy, P.H.; Hermens, W.A.; Schipper, N.G. Cyclodextrins in nasal drug delivery. Adv. Drug Deliv. Rev. 1999, 36, 41–57. [Google Scholar] [CrossRef]
- Hsu, C.M.; Tsai, F.J.; Tsai, Y. Inhibitory effect of Angelica sinensis extract in the presence of 2-hydroxypropyl-beta-cyclodextrin. Carbohydr. Polym. 2014, 114, 115–122. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Rai, A.K. Cyclodextrins in delivery systems: Applications. J. Pharm. Bioallied Sci. 2010, 2, 72–79. [Google Scholar] [CrossRef]
- Arima, H.; Yunomae, K.; Miyake, K.; Irie, T.; Hirayama, F.; Uekama, K. Comparative studies of the enhancing effects of cyclodextrins on the solubility and oral bioavailability of tacrolimus in rats. J. Pharm. Sci. 2001, 90, 690–701. [Google Scholar] [CrossRef]
- Arima, H.; Miyaji, T.; Irie, T.; Hirayama, F.; Uekama, K. Enhancing effect of hydroxypropyl-beta-cyclodextrin on cutaneous penetration and activation of ethyl 4-biphenylyl acetate in hairless mouse skin. Eur. J. Pharm. Sci. 1998, 6, 53–59. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Beck, Z.; Matyas, G.R.; Alving, C.R. Detection of liposomal cholesterol and monophosphoryl lipid A by QS-21 saponin and Limulus polyphemus amebocyte lysate. Biochim. Biophys. Acta 2015, 1848, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil(R)--the first FDA-approved nano-drug: Lessons learned. J. Control Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Ashby, L.S.; Smith, K.A.; Stea, B. Gliadel wafer implantation combined with standard radiotherapy and concurrent followed by adjuvant temozolomide for treatment of newly diagnosed high-grade glioma: A systematic literature review. World J. Surg. Oncol. 2016, 14, 225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Q.; Wang, X.; Zhang, W.; Lin, C.; Chen, F.; Yang, X.; Pan, W. Drug-in-cyclodextrin-in-liposomes: A novel drug delivery system for flurbiprofen. Int. J. Pharm. 2015, 492, 40–45. [Google Scholar] [CrossRef]
- Gharib, R.; Greige-Gerges, H.; Fourmentin, S.; Charcosset, C.; Auezova, L. Liposomes incorporating cyclodextrin-drug inclusion complexes: Current state of knowledge. Carbohydr. Polym. 2015, 129, 175–186. [Google Scholar] [CrossRef]
- Ikeda, A.; Iwata, N.; Hino, S.; Mae, T.; Tsuchiya, Y.; Sugikawa, K.; Hirao, T.; Haino, T.; Ohara, K.; Yamaguchi, K. Liposome collapse resulting from an allosteric interaction between 2,6-dimethyl-β-cyclodextrins and lipids. RSC Adv. 2015, 5, 77746–77754. [Google Scholar] [CrossRef]
- Zhao, R.; Tan, T.; Sandström, C. NMR studies on puerarin and its interaction with beta-cyclodextrin. J. Biol. Phys. 2011, 37, 387–400. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Chang, K.-F.; Huang, X.-F.; Hung, C.-L.; Chen, S.-C.; Chao, W.-R.; Liao, K.-W.; Tsai, N.-M. Liposomal n-butylidenephthalide protects the drug from oxidation and enhances its antitumor effects in glioblastoma multiforme. Int. J. Nanomed. 2015, 10, 6009–6020. [Google Scholar] [CrossRef][Green Version]
- Villano, J.L.; Seery, T.E.; Bressler, L.R. Temozolomide in malignant gliomas: Current use and future targets. Cancer Chemother. Pharmacol. 2009, 64, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Pandy, V. A Simple Method for Animal Dose Calculation in Preclinical Research. EC Pharmacol. Toxicol. 2020, 8, 1. [Google Scholar]
- Harn, H.J.; Lin, S.Z.; Lin, P.C.; Liu, C.Y.; Liu, P.Y.; Chang, L.F.; Yen, S.Y.; Hsieh, D.K.; Liu, F.C.; Tai, D.F.; et al. Local interstitial delivery of z-butylidenephthalide by polymer wafers against malignant human gliomas. Neuro Oncol. 2011, 13, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yue, P.; Tao, T.; Chen, Q.H. Drug brain distribution following intranasal administration of Huperzine A in situ gel in rats. Acta Pharmacol. Sinica 2007, 28, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Rassu, G.; Soddu, E.; Cossu, M.; Brundu, A.; Cerri, G.; Marchetti, N.; Ferraro, L.; Regan, R.F.; Giunchedi, P.; Gavini, E.; et al. Solid microparticles based on chitosan or methyl-β-cyclodextrin: A first formulative approach to increase the nose-to-brain transport of deferoxamine mesylate. J. Controll. Release Off. J. Controll. Release Soc. 2015, 201, 68–77. [Google Scholar] [CrossRef]
- Arima, H.; Hagiwara, Y.; Hirayama, F.; Uekama, K. Enhancement of antitumor effect of doxorubicin by its complexation with gamma-cyclodextrin in pegylated liposomes. J. Drug Target. 2006, 14, 225–232. [Google Scholar] [CrossRef]
- Naiim, M.; Boualem, A.; Ferre, C.; Jabloun, M.; Jalocha, A.; Ravier, P. Multiangle dynamic light scattering for the improvement of multimodal particle size distribution measurements. Soft Matter 2015, 11, 28–32. [Google Scholar] [CrossRef]
- Owen, J.; Stride, E. Technique for the Characterization of Phospholipid Microbubbles Coatings by Transmission Electron Microscopy. Ultrasound Med. Biol. 2015, 41, 3253–3258. [Google Scholar] [CrossRef]
- Yefimova, S.L.; Kurilchenko, I.Y.; Tkacheva, T.N.; Rozhkov, V.A.; Sorokin, A.V.; Lukianova, N.Y.; Bezdenezhnykh, N.A.; Malyukin, Y.V.; Chekhun, V.F. Comparative study of dye-loaded liposome accumulation in sensitive and resistant human breast cancer cells. Exp. Oncol. 2012, 34, 101–106. [Google Scholar]
- Baumann, B.C.; Benci, J.L.; Santoiemma, P.P.; Chandrasekaran, S.; Hollander, A.B.; Kao, G.D.; Dorsey, J.F. An Integrated Method for Reproducible and Accurate Image-Guided Stereotactic Cranial Irradiation of Brain Tumors Using the Small Animal Radiation Research Platform. Transl. Oncol. 2012, 5, 230–237. [Google Scholar] [CrossRef][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | DMPC mg (%) | CL mg (%) | BP mg (%) | CD mg (%) |
|---|---|---|---|---|
| LipoD | 55 (78.57%) | 15 (21.43%) | 0 (0%) | 0 (0%) |
| CDD1 | 55 (9.82%) | 15 (2.68%) | 140 (25%) | 350 (62%) |
| CDD2 | 55 (17.46%) | 15 (4.76%) | 70 (22%) | 175 (55.56%) |
| CDD3 | 55 (28.57%) | 15 (7.79%) | 35 (18.18%) | 87.5 (45.45%) |
| Formulation (BP%) | Z-Average (nm) | PDI | %EE (±SD) |
|---|---|---|---|
| LipoD (0%) | 211.2 ± 0.60 | 0.08 ± 0.03 | 0 |
| CDD3 (18%) | 253.0 ± 6.05 | 0.33 ± 0.01 | 58.2 ± 2.3 |
| CDD2 (22%) | 331.4 ± 4.74 | 0.47 ± 0.06 | 89.9 ± 1.0 |
| CDD1 (25%) | 360.9 ± 23.06 | 0.53 ± 0.07 | 98.4 ± 3.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, E.-Y.; Chen, Y.-S.; Li, Y.-S.; Chen, S.-R.; Lee, C.-H.; Huang, M.-H.; Chuang, H.-M.; Harn, H.-J.; Yang, H.-H.; Lin, S.-Z.; et al. Liposome Consolidated with Cyclodextrin Provides Prolonged Drug Retention Resulting in Increased Drug Bioavailability in Brain. Int. J. Mol. Sci. 2020, 21, 4408. https://doi.org/10.3390/ijms21124408
Lin E-Y, Chen Y-S, Li Y-S, Chen S-R, Lee C-H, Huang M-H, Chuang H-M, Harn H-J, Yang H-H, Lin S-Z, et al. Liposome Consolidated with Cyclodextrin Provides Prolonged Drug Retention Resulting in Increased Drug Bioavailability in Brain. International Journal of Molecular Sciences. 2020; 21(12):4408. https://doi.org/10.3390/ijms21124408
Chicago/Turabian StyleLin, En-Yi, Yu-Shuan Chen, Yuan-Sheng Li, Syuan-Rong Chen, Chia-Hung Lee, Mao-Hsuan Huang, Hong-Meng Chuang, Horng-Jyh Harn, Hsueh-Hui Yang, Shinn-Zong Lin, and et al. 2020. "Liposome Consolidated with Cyclodextrin Provides Prolonged Drug Retention Resulting in Increased Drug Bioavailability in Brain" International Journal of Molecular Sciences 21, no. 12: 4408. https://doi.org/10.3390/ijms21124408
APA StyleLin, E.-Y., Chen, Y.-S., Li, Y.-S., Chen, S.-R., Lee, C.-H., Huang, M.-H., Chuang, H.-M., Harn, H.-J., Yang, H.-H., Lin, S.-Z., Tai, D.-F., & Chiou, T.-W. (2020). Liposome Consolidated with Cyclodextrin Provides Prolonged Drug Retention Resulting in Increased Drug Bioavailability in Brain. International Journal of Molecular Sciences, 21(12), 4408. https://doi.org/10.3390/ijms21124408