Optimized qRT-PCR Approach for the Detection of Intra- and Extra-Cellular SARS-CoV-2 RNAs

 , , , and
, , , and
Abstract
1. Introduction
2. Results
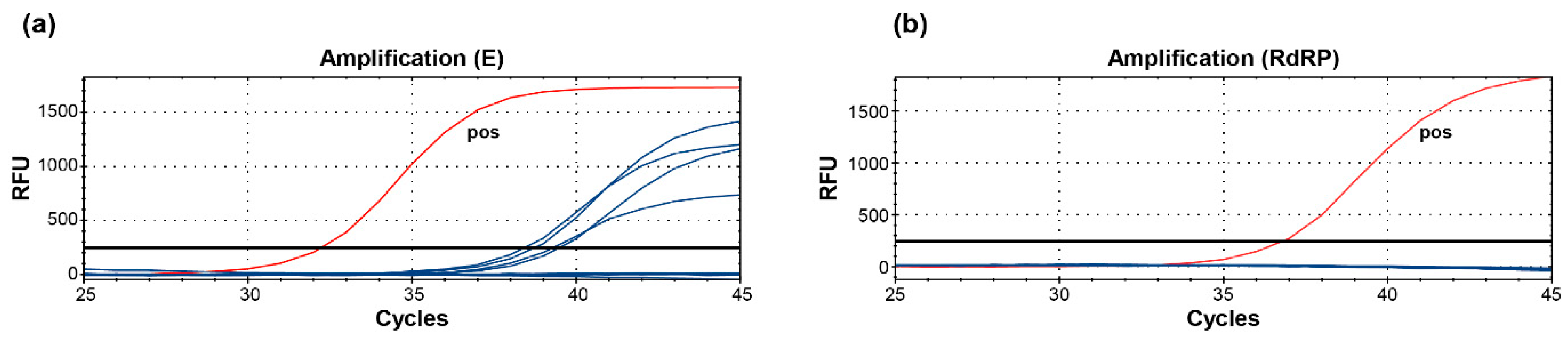
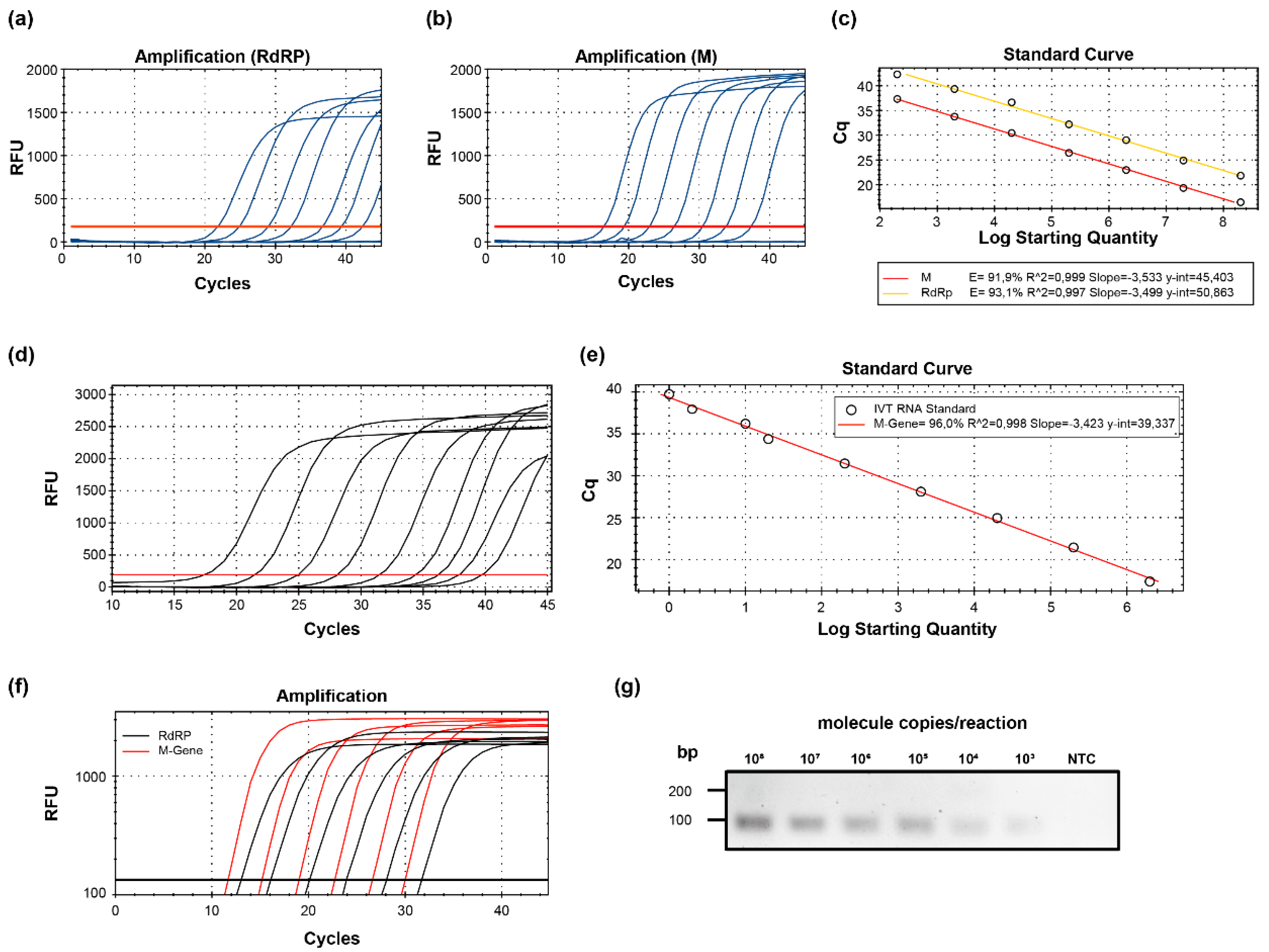
2.1. Evaluation of Specific PCR Approaches for the Detection of SARS-CoV-2 RNA
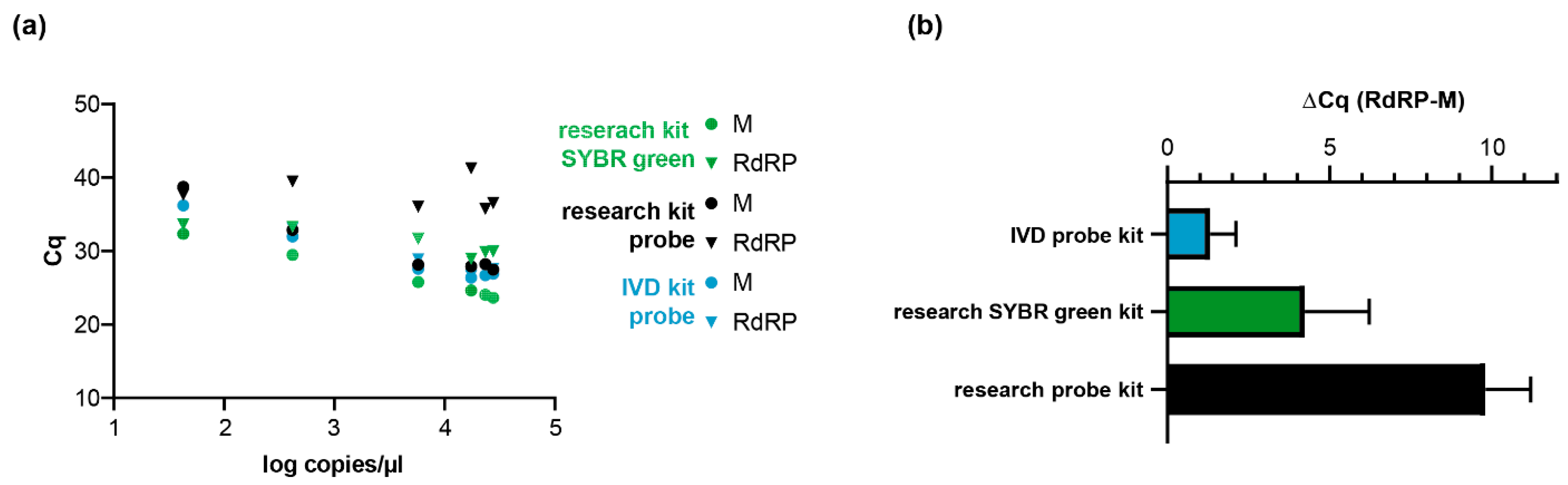
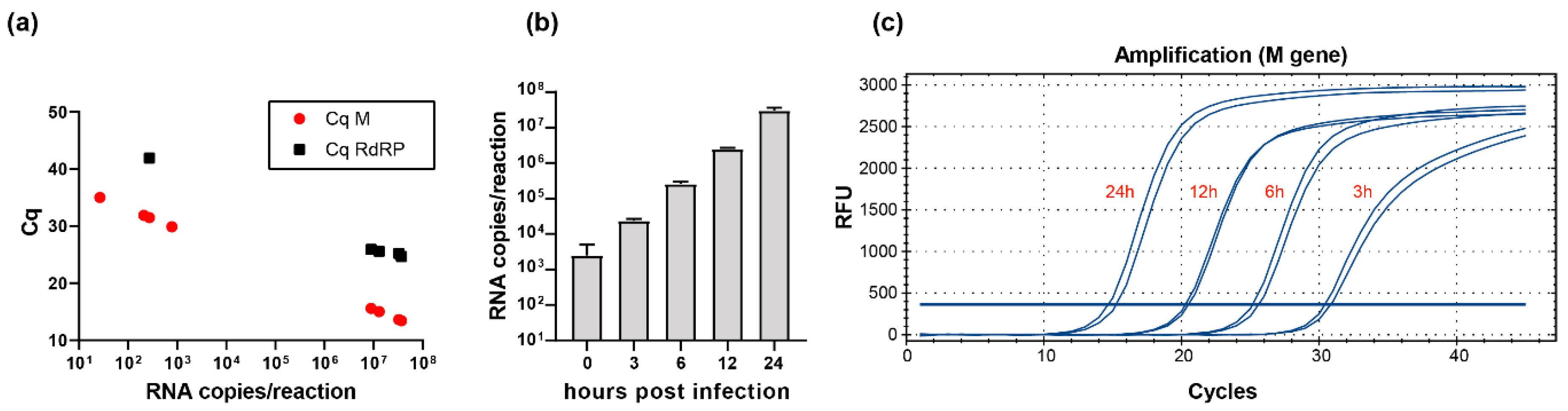
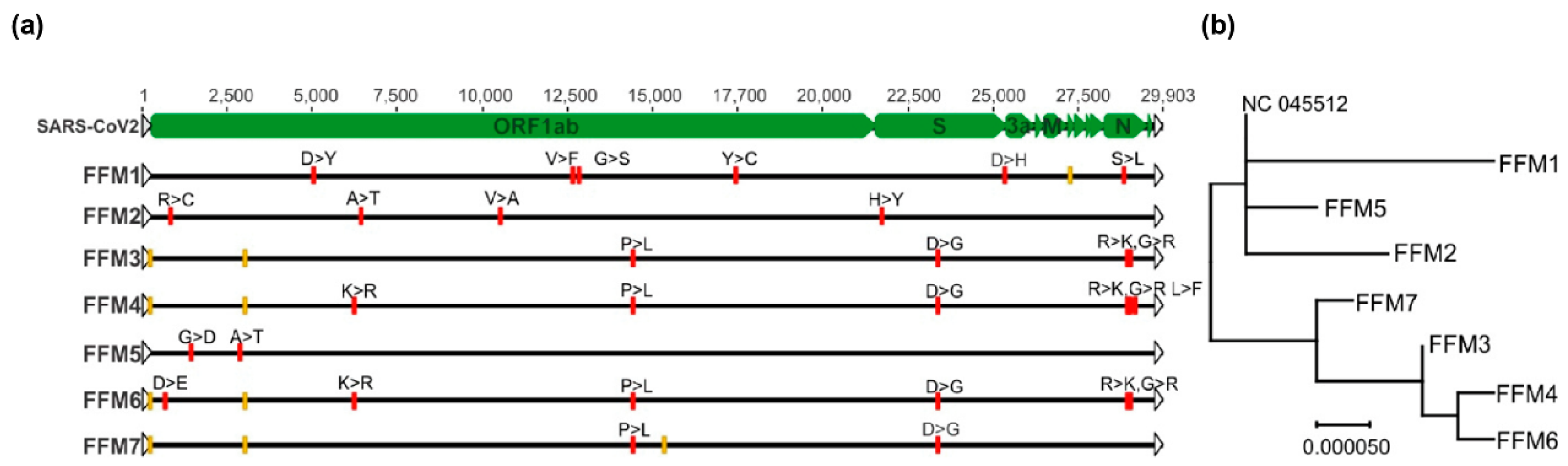
2.2. Detection of SARS-CoV-2 in Clinical and Research Samples Using E-, RdRP-, and M-Gene-Specific Protocols
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Virus Preparation
4.2. Quantification of SARS-CoV-2 RNA
4.3. Primer Design
4.4. Generation of DNA and RNA Standard Curves
4.5. Illumina NGS Sequencing of SARS-CoV-2 Isolates
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| Cq | quantification cycle |
| COVID-19 | coronavirus disease 2019 |
| bp | base pairs |
| RFU | relative fluorescence units |
| IVD | in vitro diagnostic |
References
- WHO. WHO SARS-CoV-2 Situation Report; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. CDC 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel. Revision 2020, 3, 30. [Google Scholar]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Eis-Hübinger, A.M.; Hönemann, M.; Wenzel, J.J.; Berger, A.; Widera, M.; Schmidt, B.; Aldabbagh, S.; Marx, B.; Streeck, H.; Ciesek, S.; et al. Ad hoc laboratory-based surveillance of SARS-CoV-2 by real-time RT-PCR using minipools of RNA prepared from routine respiratory samples. J. Clin. Virol. 2020, 127, 104381. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, R. Virus Variation Resource–improved response to emergent viral outbreaks. Nucleic Acids Res. 2016, 45, D482–D490. [Google Scholar] [CrossRef] [PubMed]
- Hoehl, S.; Rabenau, H.; Berger, A.; Kortenbusch, M.; Cinatl, J.; Bojkova, D.; Behrens, P.; Böddinghaus, B.; Götsch, U.; Naujoks, F.; et al. Evidence of SARS-CoV-2 Infection in Returning Travelers from Wuhan, China. N. Engl. J. Med. 2020, 382, 1278–1280. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Klompas, M. Coronavirus Disease 2019 (COVID-19): Protecting Hospitals from the Invisible. Ann. Intern. Med. 2020, 172, 619–620. [Google Scholar] [CrossRef] [PubMed]
- Konrad, R.; Eberle, U.; Dangel, A.; Treis, B.; Berger, A.; Bengs, K.; Fingerle, V.; Liebl, B.; Ackermann, N.; Sing, A. Rapid establishment of laboratory diagnostics for the novel coronavirus SARS-CoV-2 in Bavaria, Germany, February 2020. Eurosurveillance 2020, 25, 2000173. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rothe, C.; Schunk, M.; Sothmann, P.; Bretzel, G.; Froeschl, G.; Wallrauch, C.; Zimmer, T.; Thiel, V.; Janke, C.; Guggemos, W.; et al. Transmission of 2019-nCoV Infection from an Asymptomatic Contact in Germany. N. Engl. J. Med. 2020, 382, 970–971. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligo Name | Oligonucleotide Sequences (5’ to 3’) | Position within the SARS-CoV-2 Genome | Length (nt) | Deg. | Mism. | Ref. |
|---|---|---|---|---|---|---|
| E_Sarbeco_F1 | ACAGGTACGTTAATAGTTAATAGCGT | 26,269–26,294 | 26 | [4] | ||
| E_Sarbeco_R2 | ATATTGCAGCAGTACGCACACA | 26,360–26,381 | 22 | |||
| E_Sarbeco_P1 | 6-Fam ACACTAGCCATCCTTACTGCGCTTCG BBQ1 | 26,332–26,357 | 26 | |||
| RdRP_SARSr-F2 | GTGARATGGTCATGTGTGGCGG | 15,431–15,452 | 22 | 2 | ||
| RdRP_SARSr-R1 | CARATGTTAAASACACTATTAGCATA | 15,505–15,530 | 26 | 4 | 1 1 | |
| RdRP_SARSr-P2 | 6-Fam CAGGTGGAACCTCATCAGGAGATGC BBQ1 | 15,470–15,494 | 25 | |||
| M-475-F | TGTGACATCAAGGACCTGCC | 26,997–27,016 | 20 | |||
| M-574-R | CTGAGTCACCTGCTACACGC | 27,077–27,096 | 20 | |||
| M-507-P | 6-Fam TGTTGCTACATCACGAACGC BHQ1 | 27,029–27,048 | 20 |
| Patient | Virus Isolate | Material | Age/Gender | Cluster of Infection | Symptoms | Accession |
|---|---|---|---|---|---|---|
| Pat.1 | FFM1 | throat swab | 44/female | Hubei, China | dry cough, sore throat | https://www.ncbi.nlm.nih.gov/nuccore/MT358638 |
| Pat.2 | FFM2 | throat swab | 58/male | Hubei, China | asymptomatic | https://www.ncbi.nlm.nih.gov/nuccore/MT539726 |
| Pat.12 | FFM3 | nasopharyngeal swab | 30/male | Italy, Austria | diarrhea, rhinitis | https://www.ncbi.nlm.nih.gov/nuccore/MT358639 |
| Pat.11 | FFM4 | throat swab | 32/male | Italy | cough, muscle ache, fever | https://www.ncbi.nlm.nih.gov/nuccore/MT358640 |
| Pat.15 | FFM5 | respiratory swab | 42/female | Germany | unknown | https://www.ncbi.nlm.nih.gov/nuccore/MT358641 |
| Pat.14 | FFM6 | nasopharyngeal swab | 27/male | Italy | cough, rhinitis, headache, muscle ache, abdominal pain | https://www.ncbi.nlm.nih.gov/nuccore/MT358642 |
| Pat.7 | FFM7 | throat swab | 61/male | Israel | sore throat | https://www.ncbi.nlm.nih.gov/nuccore/MT358643 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toptan, T.; Hoehl, S.; Westhaus, S.; Bojkova, D.; Berger, A.; Rotter, B.; Hoffmeier, K.; Cinatl, J., Jr.; Ciesek, S.; Widera, M. Optimized qRT-PCR Approach for the Detection of Intra- and Extra-Cellular SARS-CoV-2 RNAs. Int. J. Mol. Sci. 2020, 21, 4396. https://doi.org/10.3390/ijms21124396
Toptan T, Hoehl S, Westhaus S, Bojkova D, Berger A, Rotter B, Hoffmeier K, Cinatl J Jr., Ciesek S, Widera M. Optimized qRT-PCR Approach for the Detection of Intra- and Extra-Cellular SARS-CoV-2 RNAs. International Journal of Molecular Sciences. 2020; 21(12):4396. https://doi.org/10.3390/ijms21124396
Chicago/Turabian StyleToptan, Tuna, Sebastian Hoehl, Sandra Westhaus, Denisa Bojkova, Annemarie Berger, Björn Rotter, Klaus Hoffmeier, Jindrich Cinatl, Jr., Sandra Ciesek, and Marek Widera. 2020. "Optimized qRT-PCR Approach for the Detection of Intra- and Extra-Cellular SARS-CoV-2 RNAs" International Journal of Molecular Sciences 21, no. 12: 4396. https://doi.org/10.3390/ijms21124396
APA StyleToptan, T., Hoehl, S., Westhaus, S., Bojkova, D., Berger, A., Rotter, B., Hoffmeier, K., Cinatl, J., Jr., Ciesek, S., & Widera, M. (2020). Optimized qRT-PCR Approach for the Detection of Intra- and Extra-Cellular SARS-CoV-2 RNAs. International Journal of Molecular Sciences, 21(12), 4396. https://doi.org/10.3390/ijms21124396