Suppression of Nonsense Mutations by New Emerging Technologies



{kind=link}
{kind=link}
Abstract
1. Introduction
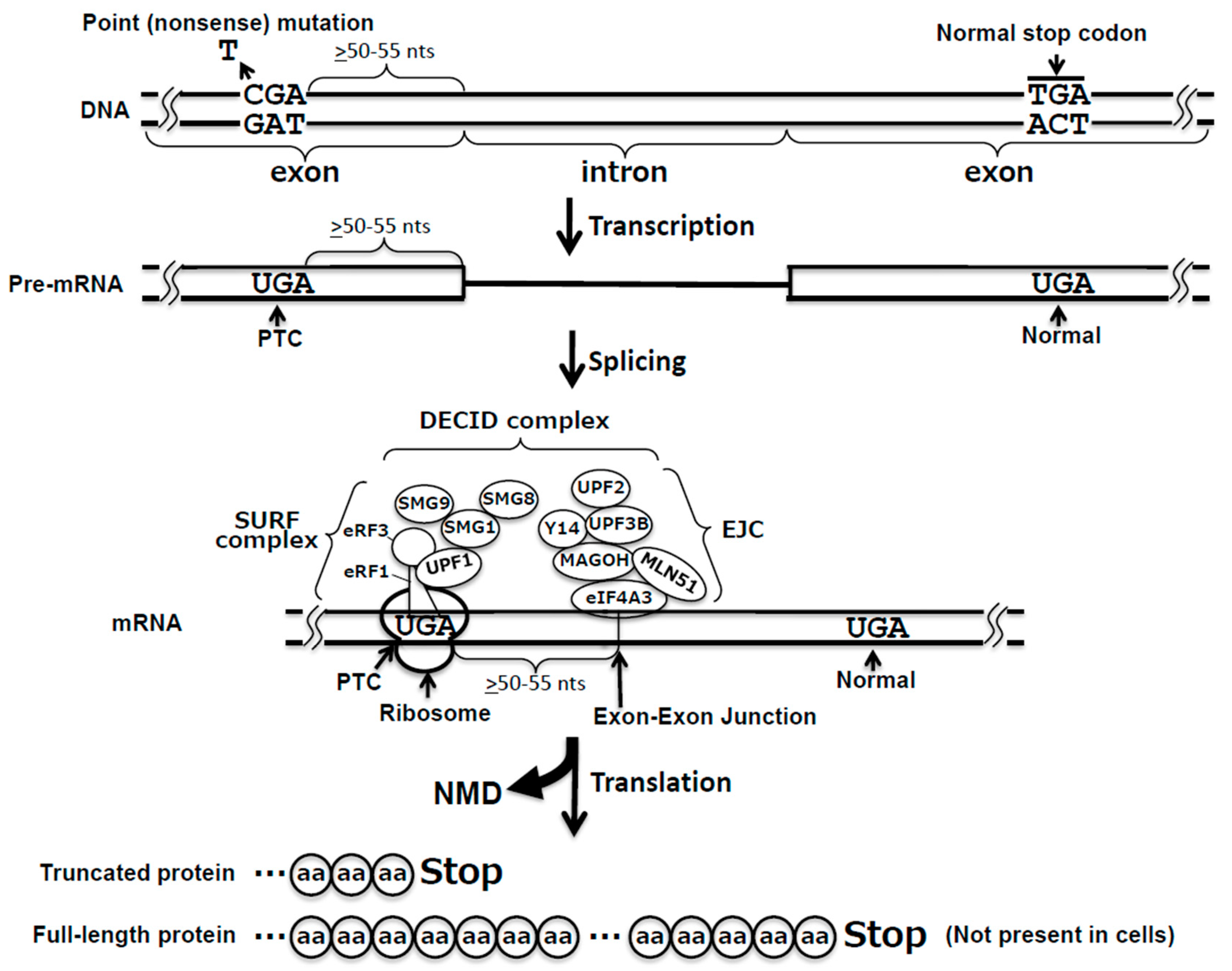
2. Translation Termination: Mechanism of Action
3. Nonsense-Mediated mRNA Decay Pathway
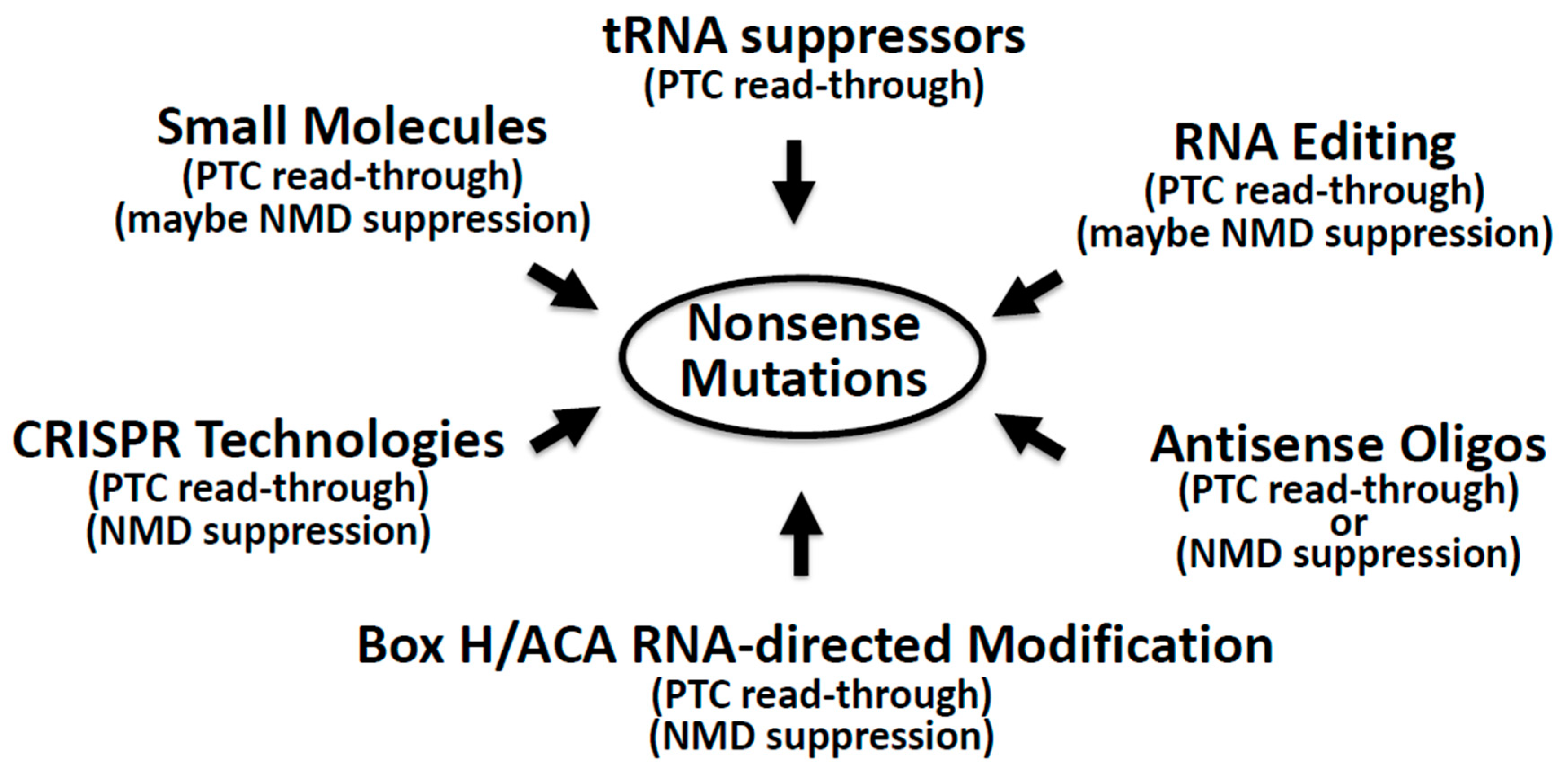
4. Suppression of Nonsense Mutations: Approaches and Challenges
4.1. Nonsense Suppression by Small Molecules
4.2. Nonsense Suppression by Nucleic Acid-Based Approaches
4.2.1. Antisense Oligonucleotides
4.2.2. Suppressor tRNA
4.2.3. ADAR-Catalyzed Editing Directed by Guide RNA
4.2.4. RNA Pseudouridylation Directed by Box H/ACA Guide RNA
4.2.5. CRISPR Technology
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Brenner, S.; Barnett, L.; Katz, E.R.; Crick, F.H.C. UGA: A Third Nonsense Triplet in the Genetic Code. Nature 1967, 213, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Karijolich, J.; Yu, Y.-T. Therapeutic suppression of premature termination codons: Mechanisms and clinical considerations (review). Int. J. Mol. Med. 2014, 34, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.T.; Dietz, H.C. When the message goes awry: Disease-producing mutations that influence mRNA content and performance. Cell 2001, 107, 411–414. [Google Scholar] [CrossRef]
- Atkinson, J.; Martin, R. Mutations to nonsense codons in human genetic disease: Implications for gene therapy by nonsense suppressor tRNAs. Nucleic Acids Res. 1994, 22, 1327–1334. [Google Scholar] [CrossRef]
- Neuhaus, C.; Eisenberger, T.; Decker, C.; Nagl, S.; Blank, C.; Pfister, M.; Kennerknecht, I.; Müller-Hofstede, C.; Charbel Issa, P.; Heller, R.; et al. Next-generation sequencing reveals the mutational landscape of clinically diagnosed Usher syndrome: Copy number variations, phenocopies, a predominant target for translational read-through, and PEX26 mutated in Heimler syndrome. Mol. Genet. Genomic Med. 2017, 5, 531–552. [Google Scholar] [CrossRef]
- Samanta, A.; Stingl, K.; Kohl, S.; Ries, J.; Linnert, J.; Nagel-Wolfrum, K. Ataluren for the Treatment of Usher Syndrome 2A Caused by Nonsense Mutations. Int. J. Mol. Sci. 2019, 20, 6274. [Google Scholar] [CrossRef]
- Sakuru, R.; Bollu, P.C. Hurler Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Fanen, P.; Wohlhuter-Haddad, A.; Hinzpeter, A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int. J. Biochem. Cell Biol. 2014, 52, 94–102. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Bidou, L.; Hatin, I.; Perez, N.; Allamand, V.; Panthier, J.-J.; Rousset, J.-P. Premature stop codons involved in muscular dystrophies show a broad spectrum of readthrough efficiencies in response to gentamicin treatment. Gene Ther. 2004, 11, 619–627. [Google Scholar] [CrossRef]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef]
- Roy, B.; Friesen, W.J.; Tomizawa, Y.; Leszyk, J.D.; Zhuo, J.; Johnson, B.; Dakka, J.; Trotta, C.R.; Xue, X.; Mutyam, V.; et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. USA 2016, 113, 12508–12513. [Google Scholar] [CrossRef] [PubMed]
- Frolova, L.Y.; Merkulova, T.I.; Kisselev, L.L. Translation termination in eukaryotes: Polypeptide release factor eRF1 is composed of functionally and structurally distinct domains. RNA 2000, 6, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Freistroffer, D.V.; Pavlov, M.Y.; MacDougall, J.; Buckingham, R.H.; Ehrenberg, M. Release factor RF3 in E.coli accelerates the dissociation of release factors RF1 and RF2 from the ribosome in a GTP-dependent manner. EMBO J. 1997, 16, 4126–4133. [Google Scholar] [CrossRef] [PubMed]
- López-Perrote, A.; Castaño, R.; Melero, R.; Zamarro, T.; Kurosawa, H.; Ohnishi, T.; Uchiyama, A.; Aoyagi, K.; Buchwald, G.; Kataoka, N.; et al. Human nonsense-mediated mRNA decay factor UPF2 interacts directly with eRF3 and the SURF complex. Nucleic Acids Res. 2016, 44, 1909–1923. [Google Scholar] [CrossRef]
- Song, H.; Mugnier, P.; Das, A.K.; Webb, H.M.; Evans, D.R.; Tuite, M.F.; Hemmings, B.A.; Barford, D. The Crystal Structure of Human Eukaryotic Release Factor eRF1—Mechanism of Stop Codon Recognition and Peptidyl-tRNA Hydrolysis. Cell 2000, 100, 311–321. [Google Scholar] [CrossRef]
- Kong, C.; Ito, K.; Walsh, M.A.; Wada, M.; Liu, Y.; Kumar, S.; Barford, D.; Nakamura, Y.; Song, H. Crystal Structure and Functional Analysis of the Eukaryotic Class II Release Factor eRF3 from S. pombe. Mol. Cell 2004, 14, 233–245. [Google Scholar] [CrossRef]
- Uchida, N.; Hoshino, S.-I.; Imataka, H.; Sonenberg, N.; Katada, T. A novel role of the mammalian GSPT/eRF3 associating with poly(A)-binding protein in Cap/Poly(A)-dependent translation. J. Biol. Chem. 2002, 277, 50286–50292. [Google Scholar] [CrossRef]
- Kodama, H.; Ito, K.; Nakamura, Y. The role of N-terminal domain of translational release factor eRF3 for the control of functionality and stability in S. cerevisiae. Genes Cells 2007, 12, 639–650. [Google Scholar] [CrossRef]
- Cheng, Z.; Saito, K.; Pisarev, A.V.; Wada, M.; Pisareva, V.P.; Pestova, T.V.; Gajda, M.; Round, A.; Kong, C.; Lim, M.; et al. Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev. 2009, 23, 1106–1118. [Google Scholar] [CrossRef]
- Taylor, D.; Unbehaun, A.; Li, W.; Das, S.; Lei, J.; Liao, H.Y.; Grassucci, R.A.; Pestova, T.V.; Frank, J. Cryo-EM structure of the mammalian eukaryotic release factor eRF1–eRF3-associated termination complex. Proc. Natl. Acad. Sci. USA 2012, 109, 18413–18418. [Google Scholar] [CrossRef] [PubMed]
- Preis, A.; Heuer, A.; Barrio-Garcia, C.; Hauser, A.; Eyler, D.E.; Berninghausen, O.; Green, R.; Becker, T.; Beckmann, R. Cryoelectron Microscopic Structures of Eukaryotic Translation Termination Complexes Containing eRF1-eRF3 or eRF1-ABCE1. Cell Rep. 2014, 8, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Ito, K. Making sense of mimic in translation termination. Trends Biochem. Sci. 2003, 28, 99–105. [Google Scholar] [CrossRef]
- Hogg, J.R.; Goff, S.P. Upf1 senses 3′UTR length to potentiate mRNA decay. Cell 2010, 143, 379–389. [Google Scholar] [CrossRef]
- Kurosaki, T.; Li, W.; Hoque, M.; Popp, M.W.-L.; Ermolenko, D.N.; Tian, B.; Maquat, L.E. A post-translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev. 2014, 28, 1900–1916. [Google Scholar] [CrossRef]
- Zünd, D.; Gruber, A.R.; Zavolan, M.; Mühlemann, O. Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3′ UTRs. Nat. Struct. Mol. Biol. 2013, 20, 936–943. [Google Scholar] [CrossRef]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Thermann, R.; Neu-Yilik, G.; Deters, A.; Frede, U.; Wehr, K.; Hagemeier, C.; Hentze, M.W.; Kulozik, A.E. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 1998, 17, 3484–3494. [Google Scholar] [CrossRef]
- Le Hir, H.; Izaurralde, E.; Maquat, L.E.; Moore, M.J. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon–exon junctions. EMBO J. 2000, 19, 6860–6869. [Google Scholar] [CrossRef]
- Maquat, L.E.; Li, X. Mammalian heat shock p70 and histone H4 transcripts, which derive from naturally intronless genes, are immune to nonsense-mediated decay. RNA 2001, 7, 445–456. [Google Scholar] [CrossRef]
- Tange, T.Ø.; Shibuya, T.; Jurica, M.S.; Moore, M.J. Biochemical analysis of the EJC reveals two new factors and a stable tetrameric protein core. RNA 2005, 11, 1869–1883. [Google Scholar] [CrossRef]
- Viegas, M.H.; Gehring, N.H.; Breit, S.; Hentze, M.W.; Kulozik, A.E. The abundance of RNPS1, a protein component of the exon junction complex, can determine the variability in efficiency of the Nonsense Mediated Decay pathway. Nucleic Acids Res. 2007, 35, 4542–4551. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Czaplinski, K.; Trifillis, P.; He, F.; Jacobson, A.; Peltz, S.W. Characterization of the biochemical properties of the human Upf1 gene product that is involved in nonsense-mediated mRNA decay. RNA 2000, 6, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Jayachandran, U.; Bonneau, F.; Fiorini, F.; Basquin, C.; Domcke, S.; Le Hir, H.; Conti, E. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell 2011, 41, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Chamieh, H.; Ballut, L.; Bonneau, F.; Le Hir, H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008, 15, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Ohnishi, T.; Kashima, I.; Taya, Y.; Ohno, S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 2001, 15, 2215–2228. [Google Scholar] [CrossRef]
- Yamashita, A.; Izumi, N.; Kashima, I.; Ohnishi, T.; Saari, B.; Katsuhata, Y.; Muramatsu, R.; Morita, T.; Iwamatsu, A.; Hachiya, T.; et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 2009, 23, 1091–1105. [Google Scholar] [CrossRef]
- Deniaud, A.; Karuppasamy, M.; Bock, T.; Masiulis, S.; Huard, K.; Garzoni, F.; Kerschgens, K.; Hentze, M.W.; Kulozik, A.E.; Beck, M.; et al. A network of SMG-8, SMG-9 and SMG-1 C-terminal insertion domain regulates UPF1 substrate recruitment and phosphorylation. Nucleic Acids Res. 2015, 43, 7600–7611. [Google Scholar] [CrossRef]
- Zhu, L.; Li, L.; Qi, Y.; Yu, Z.; Xu, Y. Cryo-EM structure of SMG1-SMG8-SMG9 complex. Cell Res. 2019, 29, 1027–1034. [Google Scholar] [CrossRef]
- Trcek, T.; Sato, H.; Singer, R.H.; Maquat, L.E. Temporal and spatial characterization of nonsense-mediated mRNA decay. Genes Dev. 2013, 27, 541–551. [Google Scholar] [CrossRef]
- Kashima, I.; Yamashita, A.; Izumi, N.; Kataoka, N.; Morishita, R.; Hoshino, S.; Ohno, M.; Dreyfuss, G.; Ohno, S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006, 20, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Eberle, A.B.; Lykke-Andersen, S.; Mühlemann, O.; Jensen, T.H. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol. 2009, 16, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Loh, B.; Jonas, S.; Izaurralde, E. The SMG5–SMG7 heterodimer directly recruits the CCR4–NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev. 2013, 27, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Doma, M.K.; Parker, R. RNA quality control in eukaryotes. Cell 2007, 131, 660–668. [Google Scholar] [CrossRef]
- Schmid, M.; Jensen, T.H. The exosome: A multipurpose RNA-decay machine. Trends Biochem. Sci. 2008, 33, 501–510. [Google Scholar] [CrossRef]
- Peltz, S.W.; Morsy, M.; Welch, E.M.; Jacobson, A. Ataluren as an agent for therapeutic nonsense suppression. Annu. Rev. Med. 2013, 64, 407–425. [Google Scholar] [CrossRef]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef]
- Chowdhury, H.M.; Siddiqui, M.A.; Kanneganti, S.; Sharmin, N.; Chowdhury, M.W.; Nasim, M.T. Aminoglycoside-mediated promotion of translation readthrough occurs through a non-stochastic mechanism that competes with translation termination. Hum. Mol. Genet. 2018, 27, 373–384. [Google Scholar] [CrossRef]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef]
- Wangen, J.R.; Green, R. Stop codon context influences genome-wide stimulation of termination codon readthrough by aminoglycosides. Elife 2020, 9, e52611. [Google Scholar] [CrossRef] [PubMed]
- Brar, G.A.; Weissman, J.S. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Frew, J.; Baradaran-Heravi, A.; Balgi, A.D.; Wu, X.; Yan, T.D.; Arns, S.; Shidmoossavee, F.S.; Tan, J.; Jaquith, J.B.; Jansen-West, K.R.; et al. Premature termination codon readthrough upregulates progranulin expression and improves lysosomal function in preclinical models of GRN deficiency. Mol. Neurodegener. 2020, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Mutyam, V.; Tang, L.; Biswas, S.; Du, M.; Jackson, L.A.; Dai, Y.; Belakhov, V.; Shalev, M.; Chen, F.; et al. Synthetic Aminoglycosides Efficiently Suppress Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations and Are Enhanced by Ivacaftor. Am. J. Respir. Cell Mol. Biol. 2013, 50, 805–816. [Google Scholar] [CrossRef]
- Leubitz, A.; Frydman-Marom, A.; Sharpe, N.; Duzer, J.; Van Campbell, K.C.M.; Vanhoutte, F. Safety, Tolerability, and Pharmacokinetics of Single Ascending Doses of ELX-02, a Potential Treatment for Genetic Disorders Caused by Nonsense Mutations, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2019, 8, 984–994. [Google Scholar] [CrossRef]
- Lentini, L.; Melfi, R.; Di Leonardo, A.; Spinello, A.; Barone, G.; Pace, A.; Palumbo Piccionello, A.; Pibiri, I. Toward a rationale for the PTC124 (Ataluren) promoted readthrough of premature stop codons: A computational approach and GFP-reporter cell-based assay. Mol. Pharm. 2014, 11, 653–664. [Google Scholar] [CrossRef]
- Pibiri, I.; Lentini, L.; Melfi, R.; Tutone, M.; Baldassano, S.; Ricco Galluzzo, P.; Di Leonardo, A.; Pace, A. Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur. J. Med. Chem. 2018, 159, 126–142. [Google Scholar] [CrossRef]
- Tutone, M.; Pibiri, I.; Lentini, L.; Pace, A.; Almerico, A.M. Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An in Silico Compared Study. ACS Med. Chem. Lett. 2019, 10, 522–527. [Google Scholar] [CrossRef]
- Campofelice, A.; Lentini, L.; Di Leonardo, A.; Melfi, R.; Tutone, M.; Pace, A.; Pibiri, I. Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs). Int. J. Mol. Sci. 2019, 20, 3329. [Google Scholar] [CrossRef]
- Benhabiles, H.; Gonzalez-Hilarion, S.; Amand, S.; Bailly, C.; Prévotat, A.; Reix, P.; Hubert, D.; Adriaenssens, E.; Rebuffat, S.; Tulasne, D.; et al. Optimized approach for the identification of highly efficient correctors of nonsense mutations in human diseases. PLoS ONE 2017, 12, e0187930. [Google Scholar] [CrossRef]
- Trzaska, C.; Amand, S.; Bailly, C.; Leroy, C.; Marchand, V.; Duvernois-Berthet, E.; Saliou, J.-M.; Benhabiles, H.; Werkmeister, E.; Chassat, T.; et al. 2,6-Diaminopurine as a highly potent corrector of UGA nonsense mutations. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wilton, S.D.; Lloyd, F.; Carville, K.; Fletcher, S.; Honeyman, K.; Agrawal, S.; Kole, R. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 1999, 9, 330–338. [Google Scholar] [CrossRef]
- Lu, Q.L.; Morris, G.E.; Wilton, S.D.; Ly, T.; Artem’yeva, O.V.; Strong, P.; Partridge, T.A. Massive Idiosyncratic Exon Skipping Corrects the Nonsense Mutation in Dystrophic Mouse Muscle and Produces Functional Revertant Fibers by Clonal Expansion. J. Cell Biol. 2000, 148, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Wilton, S.D.; Dye, D.E.; Laing, N.G. Dystrophin gene transcripts skipping the mdx mutation. Muscle Nerve 1997, 20, 728–734. [Google Scholar] [CrossRef]
- Barny, I.; Perrault, I.; Michel, C.; Goudin, N.; Defoort-Dhellemmes, S.; Ghazi, I.; Kaplan, J.; Rozet, J.-M.; Gerard, X. AON-Mediated Exon Skipping to Bypass Protein Truncation in Retinal Dystrophies Due to the Recurrent CEP290 c.4723A > T Mutation. Fact or Fiction? Genes 2019, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Molinari, E.; Ramsbottom, S.A.; Srivastava, S.; Booth, P.; Alkanderi, S.; McLafferty, S.M.; Devlin, L.A.; White, K.; Gunay-Aygun, M.; Miles, C.G.; et al. Targeted exon skipping rescues ciliary protein composition defects in Joubert syndrome patient fibroblasts. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Huang, L.; Low, A.; Damle, S.S.; Keenan, M.M.; Kuntz, S.; Murray, S.F.; Monia, B.P.; Guo, S. Antisense suppression of the nonsense mediated decay factor Upf3b as a potential treatment for diseases caused by nonsense mutations. Genome Biol. 2018, 19, 4. [Google Scholar] [CrossRef]
- Keenan, M.M.; Huang, L.; Jordan, N.J.; Wong, E.; Cheng, Y.; Valley, H.C.; Mahiou, J.; Liang, F.; Bihler, H.; Mense, M.; et al. Nonsense-mediated RNA Decay Pathway Inhibition Restores Expression and Function of W1282X CFTR. Am. J. Respir. Cell Mol. Biol. 2019, 61, 290–300. [Google Scholar] [CrossRef]
- Huang, L.; Aghajan, M.; Quesenberry, T.; Low, A.; Murray, S.F.; Monia, B.P.; Guo, S. Targeting Translation Termination Machinery with Antisense Oligonucleotides for Diseases Caused by Nonsense Mutations. Nucleic Acid Ther. 2019, 29, 175–186. [Google Scholar] [CrossRef]
- Nomakuchi, T.T.; Rigo, F.; Aznarez, I.; Krainer, A.R. Antisense oligonucleotide–directed inhibition of nonsense-mediated mRNA decay. Nat. Biotechnol. 2016, 34, 164–166. [Google Scholar] [CrossRef]
- Lueck, J.D.; Yoon, J.S.; Perales-Puchalt, A.; Mackey, A.L.; Infield, D.T.; Behlke, M.A.; Pope, M.R.; Weiner, D.B.; Skach, W.R.; McCray, P.B.; et al. Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hagemeijer, M.C.; Siegwart, D.J.; Strug, L.J.; Cebotaru, L.; Torres, M.J.; Sofoluwe, A.; Beekman, J.M. Translational research to enable personalized treatment of cystic fibrosis. J. Cyst. Fibros. 2018, 17, S46–S51. [Google Scholar] [CrossRef] [PubMed]
- Woolf, T.M.; Chase, J.M.; Stinchcomb, D.T. Toward the therapeutic editing of mutated RNA sequences. Proc. Natl. Acad. Sci. USA 1995, 92, 8298–8302. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Gonzalez, M.F.; Vallecillo-Viejo, I.; Yudowski, G.A.; Rosenthal, J.J.C. Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl. Acad. Sci. USA 2013, 110, 18285–18290. [Google Scholar] [CrossRef] [PubMed]
- Wettengel, J.; Reautschnig, P.; Geisler, S.; Kahle, P.J.; Stafforst, T. Harnessing human ADAR2 for RNA repair—Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 2017, 45, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- Merkle, T.; Merz, S.; Reautschnig, P.; Blaha, A.; Li, Q.; Vogel, P.; Wettengel, J.; Li, J.B.; Stafforst, T. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019, 37, 133–138. [Google Scholar] [CrossRef]
- Katrekar, D.; Chen, G.; Meluzzi, D.; Ganesh, A.; Worlikar, A.; Shih, Y.-R.; Varghese, S.; Mali, P. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 2019, 16, 239–242. [Google Scholar] [CrossRef]
- Desterro, J.M.P.; Keegan, L.P.; Lafarga, M.; Berciano, M.T.; O’Connell, M.; Carmo-Fonseca, M. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 2003, 116, 1805–1818. [Google Scholar] [CrossRef]
- Jayan, G.C.; Casey, J.L. Increased RNA Editing and Inhibition of Hepatitis Delta Virus Replication by High-Level Expression of ADAR1 and ADAR2. J. Virol. 2002, 76, 3819–3827. [Google Scholar] [CrossRef]
- Monteleone, L.R.; Matthews, M.M.; Palumbo, C.M.; Thomas, J.M.; Zheng, Y.; Chiang, Y.; Fisher, A.J.; Beal, P.A. A Bump-Hole Approach for Directed RNA Editing. Cell Chem. Biol. 2019, 26, 269–277.e5. [Google Scholar] [CrossRef]
- Vallecillo-Viejo, I.C.; Liscovitch-Brauer, N.; Montiel-Gonzalez, M.F.; Eisenberg, E.; Rosenthal, J.J.C. Abundant off-target edits from site-directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol. 2018, 15, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Yi, Z.; Zhu, S.; Wang, C.; Cao, Z.; Zhou, Z.; Yuan, P.; Yu, Y.; Tian, F.; Liu, Z.; et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019, 37, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.-H.E.; Bahn, J.H.; Yang, Y.; Lin, X.; Tran, S.; Yang, E.-W.; Quinones-Valdez, G.; Xiao, X. RNA editing in nascent RNA affects pre-mRNA splicing. Genome Res. 2018, 28, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Riedmann, E.M.; Schopoff, S.; Hartner, J.C.; Jantsch, M.F. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA 2008, 14, 1110–1118. [Google Scholar] [CrossRef]
- Karijolich, J.; Yu, Y.-T. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 2011, 474, 395–398. [Google Scholar] [CrossRef]
- Meier, U.T. RNA modification in Cajal bodies. RNA Biol. 2017, 14, 693–700. [Google Scholar] [CrossRef]
- Fernández, I.S.; Ng, C.L.; Kelley, A.C.; Wu, G.; Yu, Y.-T.; Ramakrishnan, V. Unusual base pairing during the decoding of a stop codon by the ribosome. Nature 2013, 500, 107–110. [Google Scholar] [CrossRef]
- De Zoysa, M.D.; Wu, G.; Katz, R.; Yu, Y.-T. Guide-substrate base-pairing requirement for box H/ACA RNA-guided RNA pseudouridylation. RNA 2018, 24, 1106–1117. [Google Scholar] [CrossRef]
- Kelly, E.K.; Czekay, D.P.; Kothe, U. Base-pairing interactions between substrate RNA and H/ACA guide RNA modulate the kinetics of pseudouridylation, but not the affinity of substrate binding by H/ACA small nucleolar ribonucleoproteins. RNA 2019, 25, 1393–1404. [Google Scholar] [CrossRef]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Lee, C.; Hyun Jo, D.; Hwang, G.-H.; Yu, J.; Kim, J.H.; Park, S.; Kim, J.-S.; Kim, J.H.; Bae, S. CRISPR-Pass: Gene Rescue of Nonsense Mutations Using Adenine Base Editors. Mol. Ther. 2019, 27, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morais, P.; Adachi, H.; Yu, Y.-T. Suppression of Nonsense Mutations by New Emerging Technologies. Int. J. Mol. Sci. 2020, 21, 4394. https://doi.org/10.3390/ijms21124394
Morais P, Adachi H, Yu Y-T. Suppression of Nonsense Mutations by New Emerging Technologies. International Journal of Molecular Sciences. 2020; 21(12):4394. https://doi.org/10.3390/ijms21124394
Chicago/Turabian StyleMorais, Pedro, Hironori Adachi, and Yi-Tao Yu. 2020. "Suppression of Nonsense Mutations by New Emerging Technologies" International Journal of Molecular Sciences 21, no. 12: 4394. https://doi.org/10.3390/ijms21124394
APA StyleMorais, P., Adachi, H., & Yu, Y.-T. (2020). Suppression of Nonsense Mutations by New Emerging Technologies. International Journal of Molecular Sciences, 21(12), 4394. https://doi.org/10.3390/ijms21124394