Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways

 ,
,
Abstract
1. Introduction
2. Plasma Lipid Profile in IPF Patients
2.1. Fatty Acids and Fatty Acid Elongation in Pulmonary Fibrosis
2.2. Nitrated Fatty Acids in Pulmonary Fibrosis
2.3. Prostaglandins and Leukotrienes in Pulmonary Fibrosis
2.4. PGD2 in Pulmonary Fibrosis
2.5. PGE2 and PGE2 Signaling in the Pathogenesis of Pulmonary Fibrosis
2.5.1. PGE2 Deficiency in Fibrotic Lungs
2.5.2. Modulation of PGE2 Signaling via EP1-4 Receptors Regulating Apoptosis and Proliferation
2.5.3. Epigenetic Regulation of EP Receptors in Fibrotic Lungs
2.5.4. Inter-relationship between Plasminogen Activation and PGE2 Production in Pulmonary Fibrosis
2.5.5. Crosstalk between PGE2 and TGF-β Signaling in Fibroblast Differentiation
2.6. PGF2α and PGF2α Receptor in Pulmonary Fibrosis
2.7. Leukotrienes and Its Role in Pulmonary Fibrosis
Cellular Senescence and Leukotriene Metabolism in Pulmonary Fibrosis
3. Sphingolipids, Sphingolipid Metabolizing Enzymes, and S1p Receptors in Pulmonary Fibrosis
3.1. Sphingomyelin and Sphingomyelinase in Pulmonary Fibrosis
3.2. Ceramide Metabolism and Signaling in Pulmonary Fibrosis
3.3. S1P Signaling Axis in the Pathophysiology of IPF and Animal Models of Pulmonary Fibrosis
3.3.1. S1P Levels Are Altered in IPF and Animal Models of Pulmonary Fibrosis
3.3.2. SPHK1/S1P Signaling Promotes Lung Inflammation and Pulmonary Fibrosis
3.3.3. Dihydro S1P Signaling in Pulmonary Fibrosis
3.3.4. S1P lyase/S1P Signaling in IPF and Pulmonary Fibrosis
3.3.5. Autophagy and S1P lyase/S1P Pathway in IPF and Pulmonary Fibrosis
3.3.6. Serine palmitoyltransferase Modulation of S1P Signaling and Pulmonary Fibrosis
3.3.7. S1P Receptors in Pulmonary Fibrosis
3.3.8. Interaction between TGF-β and S1P Signaling in the Pathogenesis of Pulmonary Fibrosis
3.3.9. SPHK1/S1P Signaling and Mitochondrial ROS in Pulmonary Fibrosis
4. Phospholipids and Phospholipid Metabolizing Enzymes in Pulmonary Fibrosis
4.1. Surfactant Lipids and Surfactant Proteins in IPF and Pulmonary Fibrosis
4.2. Phospholipase D/Phosphatidic acid Signaling Axis in Development of Pulmonary Fibrosis
4.3. Diacylglycerol Kinase in Radiation-Induced Fibrosis
5. Lysophospholipids and Lysophospholipids Metabolizing Enzymes in Pulmonary Fibrosis
5.1. Lysocardiolipin Acyltransferase in Cardiolipin Remodeling
Lysocardiolipin Acyltransferase and Pulmonary Fibrosis
5.2. Autotaxin (Lysophospholipase D), LPA and LPARs
5.2.1. LPA Production in Cells by Autotaxin, Phospholipase D, and Acylglycerol Kinase
5.2.2. LPA Signals via LPARs
5.2.3. PPARγ is an Intracellular Receptor of LPA
5.2.4. ATX/LPA Signaling Axis in Pulmonary Fibrosis
5.2.5. LPA Signaling via LPA Receptors in the Development of IPF and Pulmonary Fibrosis
5.3. Lipid Peroxidation and Oxidized Phospholipids in Pulmonary Fibrosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HPETE | 5-hydroperoxy eicosatetraenoic acid |
| 5-LO | 5-lipoxygenase |
| Abcg1 | ATP-binding cassette sub-family G member 1 |
| AEC | Alveolar epithelial cells |
| ALCAT1 | Acyl-CoA lysocardiolipin acyltransferase |
| AM | Alveolar macrophages |
| ApoM | Apolipoprotein M |
| ASMase | Acid sphingomyelinase |
| ATX | Autotaxin |
| BAL | Bronchoalveolar lavage |
| BPD | Bronchopulmonary dysplasia |
| C1P | Ceramide-1-phosphate |
| CL | Cardiolipin |
| CRTH2 | Chemoattractant receptor homologous with T-helper cell type 2 |
| COPD | Chronic obstructive pulmonary disease |
| COX | Cyclooxygenases |
| CTGF | Connective tissue growth factor |
| cysLT | Cysteinyl leukotrienes |
| DAG | Diacylglycerol |
| DAGK | Diacylglycerol kinase |
| DM PGE2 | 16,16-dimethy PGE2 |
| DP1 | D-prostanoid receptor |
| EBC | Exhaled breath condensate |
| EGR1 | Early growth response 1 |
| EndMT | Endothelial-mesenchymal transdifferentiation |
| ENPP2 | Ectonucleotide pyrophaphatase-phosphodiesterase 2 |
| EP2, EP4 | PGE2 receptors |
| ER | Endoplasmic reticulum |
| GPCRs | G-protein coupled receptors |
| GSH | Glutathione |
| GPx | Glutathione peroxidase |
| H-PGDS | Hematopoietic PGD synthase |
| HLF | Human lung fibroblast |
| IMM | Inner mitochondrial membrane |
| IPF | Idiopathic pulmonary fibrosis |
| LNO2 | 12-nitrolinoleic acid |
| 5-LO | 5-lipoxygenase |
| LPA | Lysophosphatidic acid |
| LPAR | Lysophosphatidic acid receptor |
| LPC | Lysophosphatidylcholine |
| LPE | Lysophosphatidylethanolamine |
| LPP | Lipid phosphate phosphatases |
| LPS | Lipopolysachharide |
| LT | Leukotriene |
| LYCAT | Lysocardiolipin acyltransferase |
| Mfn1 | Mitofusin 1 |
| Mfn2 | Mitofusin 2 |
| MLCL | Monolyso CL |
| mtROS | Mitochondrial ROS |
| NFA | Nitrated fatty acids |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| OA-NO2 | 10-nitro-oleic acid |
| Ox-PC | Oxidized-PC |
| PA | Phosphatidic acid |
| PBMC | Peripheral blood mononuclear cells (PBMCs) |
| PC | Phosphatidylcholine |
| PPARγ | Peroxisome-activated receptor γ |
| PG | Prostaglandin |
| PGD2 | Prostaglandin D2 |
| PGE2 | Prostaglandin E2 |
| PGI2 | Prostacyclin |
| PGF2α | Prostaglandin F2α |
| PGET | PGE2 transporter |
| PF | Pulmonary fibrosis |
| PKC | Protein kinase C |
| PLD | Phospholipase D |
| PTEN | Phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| S1P | Sphingosine-1-phosphate |
| Sgpl1 | S1P lyase |
| SM | Sphingomyelin |
| SMAD2/3/4 | Mothers Against Decapentaplegic Homolog 2/3/4 |
| SMase | Sphingomyelinase |
| SPHK | Sphingosine kinase |
| SPNS2 | Spinster homolog 2 |
| SPP | S1P phosphatase |
| SPT | Serine palmitoyltransferase |
| TRAF2 | TNF receptor-associated factor 2 |
References
- Abrass, C.K. Cellular lipid metabolism and the role of lipids in progressive renal disease. Am. J. Nephrol. 2004, 24, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Role of Lipids in Brain Injury and Diseases. Future Lipidol. 2007, 2, 403–422. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.R.; Hooper, A.J.; Hegele, R.A. Lipids and cardiovascular disease. Pathology 2019, 51, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Sulciner, M.L.; Gartung, A.; Gilligan, M.M.; Serhan, C.N.; Panigrahy, D. Targeting lipid mediators in cancer biology. Cancer Metastasis Rev. 2018, 37, 557–572. [Google Scholar] [CrossRef]
- Higenbottam, T. Lung lipids and disease. Respiration 1989, 55, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhao, X.; Cheng, C.; Li, N.; Liu, Y.; Cao, Y. The implications of signaling lipids in cancer metastasis. Exp. Mol. Med. 2018, 50, 127. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Loomis-King, H.; Flaherty, K.R.; Moore, B.B. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr. Opin. Pharmacol. 2013, 13, 377–385. [Google Scholar] [CrossRef]
- Selman, M.; King, T.E.; Pardo, A.; American Thoracic, S.; European Respiratory, S.; American College of Chest, P. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- Sontake, V.; Gajjala, P.R.; Kasam, R.K.; Madala, S.K. New therapeutics based on emerging concepts in pulmonary fibrosis. Expert Opin. Ther. Targets 2019, 23, 69–81. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Du Bois, R.M. Strategies for treating idiopathic pulmonary fibrosis. Nat. Rev. Drug Discov. 2010, 9, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Noble, P.W. Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am. J. Physiol. Cell Physiol. 2014, 306, C987–C996. [Google Scholar] [CrossRef]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 911–921. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-beta Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef]
- Daniels, C.E.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Murphy, S.J.; Limper, A.H.; Leof, E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Investig. 2004, 114, 1308–1316. [Google Scholar] [CrossRef]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.S.; Tager, A.M.; Borok, Z.; Moore, B.B.; Schwartz, D.A.; Anstrom, K.J.; Bar-Joseph, Z.; Bitterman, P.; Blackburn, M.R.; Bradford, W.; et al. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am. J. Respir. Crit. Care Med. 2014, 189, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Wen, Z.; Wang, R.; Luo, W.; Du, Y.; Wang, W.; Chen, X. Identification of the lipid biomarkers from plasma in idiopathic pulmonary fibrosis by Lipidomics. BMC Pulm. Med. 2017, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.G.; Villalba, J.A.; Liang, X.; Xiong, K.; Tsoyi, K.; Ith, B.; Ayaub, E.A.; Tatituri, R.V.; Byers, D.E.; Hsu, F.F.; et al. Palmitic Acid-Rich High-Fat Diet Exacerbates Experimental Pulmonary Fibrosis by Modulating Endoplasmic Reticulum Stress. Am. J. Respir. Cell Mol. Biol. 2019, 61, 737–746. [Google Scholar] [CrossRef]
- Sunaga, H.; Matsui, H.; Ueno, M.; Maeno, T.; Iso, T.; Syamsunarno, M.R.; Anjo, S.; Matsuzaka, T.; Shimano, H.; Yokoyama, T.; et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat. Commun. 2013, 4, 2563. [Google Scholar] [CrossRef]
- Romero, F.; Hong, X.; Shah, D.; Kallen, C.B.; Rosas, I.; Guo, Z.; Schriner, D.; Barta, J.; Shaghaghi, H.; Hoek, J.B.; et al. Lipid Synthesis Is Required to Resolve Endoplasmic Reticulum Stress and Limit Fibrotic Responses in the Lung. Am. J. Respir. Cell Mol. Biol. 2018, 59, 225–236. [Google Scholar] [CrossRef]
- Baker, P.R.; Schopfer, F.J.; Sweeney, S.; Freeman, B.A. Red cell membrane and plasma linoleic acid nitration products: Synthesis, clinical identification, and quantitation. Proc. Natl. Acad. Sci. USA 2004, 101, 11577–11582. [Google Scholar] [CrossRef]
- Baker, P.R.; Lin, Y.; Schopfer, F.J.; Woodcock, S.R.; Groeger, A.L.; Batthyany, C.; Sweeney, S.; Long, M.H.; Iles, K.E.; Baker, L.M.; et al. Fatty acid transduction of nitric oxide signaling: Multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J. Biol. Chem. 2005, 280, 42464–42475. [Google Scholar] [CrossRef]
- Delmastro-Greenwood, M.; Freeman, B.A.; Wendell, S.G. Redox-dependent anti-inflammatory signaling actions of unsaturated fatty acids. Annu. Rev. Physiol. 2014, 76, 79–105. [Google Scholar] [CrossRef]
- O’Donnell, V.B.; Eiserich, J.P.; Bloodsworth, A.; Chumley, P.H.; Kirk, M.; Barnes, S.; Darley-Usmar, V.M.; Freeman, B.A. Nitration of unsaturated fatty acids by nitric oxide-derived reactive species. Methods Enzymol. 1999, 301, 454–470. [Google Scholar] [CrossRef]
- Michalik, L.; Wahli, W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J. Clin. Investig. 2006, 116, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Burgess, H.A.; Daugherty, L.E.; Thatcher, T.H.; Lakatos, H.F.; Ray, D.M.; Redonnet, M.; Phipps, R.P.; Sime, P.J. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: Implications for therapy of lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L1146–L1153. [Google Scholar] [CrossRef]
- Milam, J.E.; Keshamouni, V.G.; Phan, S.H.; Hu, B.; Gangireddy, S.R.; Hogaboam, C.M.; Standiford, T.J.; Thannickal, V.J.; Reddy, R.C. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L891–L901. [Google Scholar] [CrossRef]
- Reddy, A.T.; Lakshmi, S.P.; Zhang, Y.; Reddy, R.C. Nitrated fatty acids reverse pulmonary fibrosis by dedifferentiating myofibroblasts and promoting collagen uptake by alveolar macrophages. FASEB J. 2014, 28, 5299–5310. [Google Scholar] [CrossRef]
- Olman, M.A. Beyond TGF-beta: A prostaglandin promotes fibrosis. Nat. Med. 2009, 15, 1360–1361. [Google Scholar] [CrossRef] [PubMed]
- Nagase, T.; Uozumi, N.; Ishii, S.; Kita, Y.; Yamamoto, H.; Ohga, E.; Ouchi, Y.; Shimizu, T. A pivotal role of cytosolic phospholipase A(2) in bleomycin-induced pulmonary fibrosis. Nat. Med. 2002, 8, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Ito, A.; Sakurada, K.; Nakamura, J.; Tanaka, K.; Komatsu, M.; Takeda, M.; Saito, K.; Endo, Y.; Kozaki, T.; et al. AK106-001616, a Potent and Selective Inhibitor of Cytosolic Phospholipase A2: In Vivo Efficacy for Inflammation, Neuropathic Pain, and Pulmonary Fibrosis. J. Pharmacol. Exp. Ther. 2019, 369, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Kida, T.; Ayabe, S.; Omori, K.; Nakamura, T.; Maehara, T.; Aritake, K.; Urade, Y.; Murata, T. Prostaglandin D2 Attenuates Bleomycin-Induced Lung Inflammation and Pulmonary Fibrosis. PLoS ONE 2016, 11, e0167729. [Google Scholar] [CrossRef]
- Ando, M.; Murakami, Y.; Kojima, F.; Endo, H.; Kitasato, H.; Hashimoto, A.; Kobayashi, H.; Majima, M.; Inoue, M.; Kondo, H.; et al. Retrovirally introduced prostaglandin D2 synthase suppresses lung injury induced by bleomycin. Am. J. Respir. Cell Mol. Biol. 2003, 28, 582–591. [Google Scholar] [CrossRef]
- Ueda, S.; Fukunaga, K.; Takihara, T.; Shiraishi, Y.; Oguma, T.; Shiomi, T.; Suzuki, Y.; Ishii, M.; Sayama, K.; Kagawa, S.; et al. Deficiency of CRTH2, a Prostaglandin D2 Receptor, Aggravates Bleomycin-induced Pulmonary Inflammation and Fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 60, 289–298. [Google Scholar] [CrossRef]
- Ayabe, S.; Kida, T.; Hori, M.; Ozaki, H.; Murata, T. Prostaglandin D2 inhibits collagen secretion from lung fibroblasts by activating the DP receptor. J. Pharmacol. Sci. 2013, 121, 312–317. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Qu, D.; Yu, J.; Yang, J. The Possible Pathogenesis of Idiopathic Pulmonary Fibrosis considering MUC5B. Biomed Res. Int. 2019, 2019, 9712464. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.C.; Rice, A.B.; Ingram, J.L.; Moomaw, C.R.; Nyska, A.; Bradbury, A.; Sessoms, A.R.; Chulada, P.C.; Morgan, D.L.; Zeldin, D.C.; et al. Susceptibility of cyclooxygenase-2-deficient mice to pulmonary fibrogenesis. Am. J. Pathol. 2002, 161, 459–470. [Google Scholar] [CrossRef]
- Card, J.W.; Voltz, J.W.; Carey, M.A.; Bradbury, J.A.; Degraff, L.M.; Lih, F.B.; Bonner, J.C.; Morgan, D.L.; Flake, G.P.; Zeldin, D.C. Cyclooxygenase-2 deficiency exacerbates bleomycin-induced lung dysfunction but not fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 37, 300–308. [Google Scholar] [CrossRef]
- Dackor, R.T.; Cheng, J.; Voltz, J.W.; Card, J.W.; Ferguson, C.D.; Garrett, R.C.; Bradbury, J.A.; DeGraff, L.M.; Lih, F.B.; Tomer, K.B.; et al. Prostaglandin E(2) protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L645–L655. [Google Scholar] [CrossRef] [PubMed]
- Garbuzenko, O.B.; Ivanova, V.; Kholodovych, V.; Reimer, D.C.; Reuhl, K.R.; Yurkow, E.; Adler, D.; Minko, T. Combinatorial treatment of idiopathic pulmonary fibrosis using nanoparticles with prostaglandin E and siRNA(s). Nanomedicine 2017, 13, 1983–1992. [Google Scholar] [CrossRef]
- Ivanova, V.; Garbuzenko, O.B.; Reuhl, K.R.; Reimer, D.C.; Pozharov, V.P.; Minko, T. Inhalation treatment of pulmonary fibrosis by liposomal prostaglandin E2. Eur. J. Pharm. Biopharm. 2013, 84, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, M.; Serrano-Mollar, A.; Bulbena, O.; Fernandez-Zabalegui, L.; Closa, D.; Marin-Arguedas, A.; Torrego, A.; Mullol, J.; Picado, C.; Xaubet, A. Losartan attenuates bleomycin induced lung fibrosis by increasing prostaglandin E2 synthesis. Thorax 2006, 61, 604–610. [Google Scholar] [CrossRef]
- Lovgren, A.K.; Jania, L.A.; Hartney, J.M.; Parsons, K.K.; Audoly, L.P.; Fitzgerald, G.A.; Tilley, S.L.; Koller, B.H. COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L144–L156. [Google Scholar] [CrossRef]
- Failla, M.; Genovese, T.; Mazzon, E.; Fruciano, M.; Fagone, E.; Gili, E.; Barera, A.; La Rosa, C.; Conte, E.; Crimi, N.; et al. 16,16-Dimethyl prostaglandin E2 efficacy on prevention and protection from bleomycin-induced lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 2009, 41, 50–58. [Google Scholar] [CrossRef]
- Nakanishi, T.; Hasegawa, Y.; Mimura, R.; Wakayama, T.; Uetoko, Y.; Komori, H.; Akanuma, S.; Hosoya, K.; Tamai, I. Prostaglandin Transporter (PGT/SLCO2A1) Protects the Lung from Bleomycin-Induced Fibrosis. PLoS ONE 2015, 10, e0123895. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Gillissen, A.; Buhl, R.; Hoyt, R.F.; Hubbard, R.C.; Ozaki, T.; Rennard, S.I.; Crystal, R.G. Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am. Rev. Respir. Dis. 1991, 144, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Moriguchi, H.; Nakamura, Y.; Kamei, T.; Yasuoka, S.; Ogura, T. Regulatory effect of prostaglandin E2 on fibronectin release from human alveolar macrophages. Am. Rev. Respir. Dis. 1990, 141, 965–969. [Google Scholar] [CrossRef]
- Coward, W.R.; Watts, K.; Feghali-Bostwick, C.A.; Knox, A.; Pang, L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol. Cell. Biol. 2009, 29, 4325–4339. [Google Scholar] [CrossRef]
- Petkova, D.K.; Clelland, C.A.; Ronan, J.E.; Lewis, S.; Knox, A.J. Reduced expression of cyclooxygenase (COX) in idiopathic pulmonary fibrosis and sarcoidosis. Histopathology 2003, 43, 381–386. [Google Scholar] [CrossRef]
- Maher, T.M.; Evans, I.C.; Bottoms, S.E.; Mercer, P.F.; Thorley, A.J.; Nicholson, A.G.; Laurent, G.J.; Tetley, T.D.; Chambers, R.C.; McAnulty, R.J. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 73–82. [Google Scholar] [CrossRef]
- Ogushi, F.; Endo, T.; Tani, K.; Asada, K.; Kawano, T.; Tada, H.; Maniwa, K.; Sone, S. Decreased prostaglandin E2 synthesis by lung fibroblasts isolated from rats with bleomycin-induced lung fibrosis. Int. J. Exp. Pathol. 1999, 80, 41–49. [Google Scholar] [CrossRef]
- Vancheri, C.; Sortino, M.A.; Tomaselli, V.; Mastruzzo, C.; Condorelli, F.; Bellistri, G.; Pistorio, M.P.; Canonico, P.L.; Crimi, N. Different expression of TNF-alpha receptors and prostaglandin E(2)Production in normal and fibrotic lung fibroblasts: Potential implications for the evolution of the inflammatory process. Am. J. Respir. Cell Mol. Biol. 2000, 22, 628–634. [Google Scholar] [CrossRef]
- Wilborn, J.; Crofford, L.J.; Burdick, M.D.; Kunkel, S.L.; Strieter, R.M.; Peters-Golden, M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J. Clin. Investig. 1995, 95, 1861–1868. [Google Scholar] [CrossRef]
- Moore, B.B.; Peters-Golden, M.; Christensen, P.J.; Lama, V.; Kuziel, W.A.; Paine, R., 3rd; Toews, G.B. Alveolar epithelial cell inhibition of fibroblast proliferation is regulated by MCP-1/CCR2 and mediated by PGE2. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L342–L349. [Google Scholar] [CrossRef]
- Toh, H.; Ichikawa, A.; Narumiya, S. Molecular evolution of receptors for eicosanoids. FEBS Lett. 1995, 361, 17–21. [Google Scholar] [CrossRef]
- Dey, I.; Lejeune, M.; Chadee, K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br. J. Pharmacol. 2006, 149, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.K.; White, E.S.; Wettlaufer, S.H.; Grifka, H.; Hogaboam, C.M.; Thannickal, V.J.; Horowitz, J.C.; Peters-Golden, M. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. 2009, 23, 4317–4326. [Google Scholar] [CrossRef]
- White, E.S.; Atrasz, R.G.; Dickie, E.G.; Aronoff, D.M.; Stambolic, V.; Mak, T.W.; Moore, B.B.; Peters-Golden, M. Prostaglandin E(2) inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am. J. Respir. Cell Mol. Biol. 2005, 32, 135–141. [Google Scholar] [CrossRef]
- Dunkern, T.R.; Feurstein, D.; Rossi, G.A.; Sabatini, F.; Hatzelmann, A. Inhibition of TGF-beta induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur. J. Pharmacol. 2007, 572, 12–22. [Google Scholar] [CrossRef]
- Togo, S.; Liu, X.; Wang, X.; Sugiura, H.; Kamio, K.; Kawasaki, S.; Kobayashi, T.; Ertl, R.F.; Ahn, Y.; Holz, O.; et al. PDE4 inhibitors roflumilast and rolipram augment PGE2 inhibition of TGF-{beta}1-stimulated fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L959–L969. [Google Scholar] [CrossRef]
- Watanabe, T.; Satoh, H.; Togoh, M.; Taniguchi, S.; Hashimoto, Y.; Kurokawa, K. Positive and negative regulation of cell proliferation through prostaglandin receptors in NIH-3T3 cells. J. Cell. Physiol. 1996, 169, 401–409. [Google Scholar] [CrossRef]
- Harding, P.; LaPointe, M.C. Prostaglandin E2 increases cardiac fibroblast proliferation and increases cyclin D expression via EP1 receptor. Leukot. Essent. Fat. Acids 2011, 84, 147–152. [Google Scholar] [CrossRef]
- Huang, S.K.; Wettlaufer, S.H.; Hogaboam, C.M.; Flaherty, K.R.; Martinez, F.J.; Myers, J.L.; Colby, T.V.; Travis, W.D.; Toews, G.B.; Peters-Golden, M. Variable prostaglandin E2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2008, 177, 66–74. [Google Scholar] [CrossRef]
- Moore, B.B.; Ballinger, M.N.; White, E.S.; Green, M.E.; Herrygers, A.B.; Wilke, C.A.; Toews, G.B.; Peters-Golden, M. Bleomycin-induced E prostanoid receptor changes alter fibroblast responses to prostaglandin E2. J. Immunol. 2005, 174, 5644–5649. [Google Scholar] [CrossRef]
- Huang, S.K.; Fisher, A.S.; Scruggs, A.M.; White, E.S.; Hogaboam, C.M.; Richardson, B.C.; Peters-Golden, M. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am. J. Pathol. 2010, 177, 2245–2255. [Google Scholar] [CrossRef]
- Chapman, H.A.; Allen, C.L.; Stone, O.L. Abnormalities in pathways of alveolar fibrin turnover among patients with interstitial lung disease. Am. Rev. Respir. Dis. 1986, 133, 437–443. [Google Scholar] [CrossRef]
- Eitzman, D.T.; McCoy, R.D.; Zheng, X.; Fay, W.P.; Shen, T.; Ginsburg, D.; Simon, R.H. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J. Clin. Investig. 1996, 97, 232–237. [Google Scholar] [CrossRef]
- Bauman, K.A.; Wettlaufer, S.H.; Okunishi, K.; Vannella, K.M.; Stoolman, J.S.; Huang, S.K.; Courey, A.J.; White, E.S.; Hogaboam, C.M.; Simon, R.H.; et al. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Investig. 2010, 120, 1950–1960. [Google Scholar] [CrossRef]
- Allan, E.H.; Martin, T.J. Prostaglandin E2 regulates production of plasminogen activator isoenzymes, urokinase receptor, and plasminogen activator inhibitor-1 in primary cultures of rat calvarial osteoblasts. J. Cell. Physiol. 1995, 165, 521–529. [Google Scholar] [CrossRef]
- Pai, R.; Nakamura, T.; Moon, W.S.; Tarnawski, A.S. Prostaglandins promote colon cancer cell invasion; signaling by cross-talk between two distinct growth factor receptors. FASEB J. 2003, 17, 1640–1647. [Google Scholar] [CrossRef]
- Lim, X.; Bless, D.M.; Munoz-Del-Rio, A.; Welham, N.V. Changes in cytokine signaling and extracellular matrix production induced by inflammatory factors in cultured vocal fold fibroblasts. Ann. Otol. Rhinol. Laryngol. 2008, 117, 227–238. [Google Scholar] [CrossRef]
- Takai, E.; Tsukimoto, M.; Kojima, S. TGF-beta1 downregulates COX-2 expression leading to decrease of PGE2 production in human lung cancer A549 cells, which is involved in fibrotic response to TGF-beta1. PLoS ONE 2013, 8, e76346. [Google Scholar] [CrossRef]
- Thomas, P.E.; Peters-Golden, M.; White, E.S.; Thannickal, V.J.; Moore, B.B. PGE(2) inhibition of TGF-beta1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L417–L428. [Google Scholar] [CrossRef]
- Wettlaufer, S.H.; Scott, J.P.; McEachin, R.C.; Peters-Golden, M.; Huang, S.K. Reversal of the Transcriptome by Prostaglandin E2 during Myofibroblast Dedifferentiation. Am. J. Respir. Cell Mol. Biol. 2016, 54, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Remes Lenicov, F.; Paletta, A.L.; Gonzalez Prinz, M.; Varese, A.; Pavillet, C.E.; Lopez Malizia, A.; Sabatte, J.; Geffner, J.R.; Ceballos, A. Prostaglandin E2 Antagonizes TGF-beta Actions During the Differentiation of Monocytes Into Dendritic Cells. Front. Immunol. 2018, 9, 1441. [Google Scholar] [CrossRef]
- Mukherjee, S.; Sheng, W.; Michkov, A.; Sriarm, K.; Sun, R.; Dvorkin-Gheva, A.; Insel, P.A.; Janssen, L.J. Prostaglandin E2 inhibits profibrotic function of human pulmonary fibroblasts by disrupting Ca(2+) signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L810–L821. [Google Scholar] [CrossRef]
- Oga, T.; Matsuoka, T.; Yao, C.; Nonomura, K.; Kitaoka, S.; Sakata, D.; Kita, Y.; Tanizawa, K.; Taguchi, Y.; Chin, K.; et al. Prostaglandin F(2alpha) receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-beta. Nat. Med. 2009, 15, 1426–1430. [Google Scholar] [CrossRef] [PubMed]
- Aihara, K.; Handa, T.; Oga, T.; Watanabe, K.; Tanizawa, K.; Ikezoe, K.; Taguchi, Y.; Sato, H.; Chin, K.; Nagai, S.; et al. Clinical relevance of plasma prostaglandin F2alpha metabolite concentrations in patients with idiopathic pulmonary fibrosis. PLoS ONE 2013, 8, e66017. [Google Scholar] [CrossRef] [PubMed]
- Radmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Haeggstrom, J.Z. Leukotriene biosynthetic enzymes as therapeutic targets. J. Clin. Investig. 2018, 128, 2680–2690. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, A.J.; Hay, H.; Cromwell, O.; Collins, J.V.; Kay, A.B. Leukotrienes, LTC4 and LTB4, in bronchoalveolar lavage in bronchial asthma and other respiratory diseases. J. Allergy Clin. Immunol. 1989, 84, 19–26. [Google Scholar] [CrossRef]
- Wilborn, J.; Bailie, M.; Coffey, M.; Burdick, M.; Strieter, R.; Peters-Golden, M. Constitutive activation of 5-lipoxygenase in the lungs of patients with idiopathic pulmonary fibrosis. J. Clin. Investig. 1996, 97, 1827–1836. [Google Scholar] [CrossRef]
- Shimbori, C.; Shiota, N.; Okunishi, H. Involvement of leukotrienes in the pathogenesis of silica-induced pulmonary fibrosis in mice. Exp. Lung Res. 2010, 36, 292–301. [Google Scholar] [CrossRef]
- Peters-Golden, M.; Bailie, M.; Marshall, T.; Wilke, C.; Phan, S.H.; Toews, G.B.; Moore, B.B. Protection from pulmonary fibrosis in leukotriene-deficient mice. Am. J. Respir. Crit. Care Med. 2002, 165, 229–235. [Google Scholar] [CrossRef]
- Beller, T.C.; Friend, D.S.; Maekawa, A.; Lam, B.K.; Austen, K.F.; Kanaoka, Y. Cysteinyl leukotriene 1 receptor controls the severity of chronic pulmonary inflammation and fibrosis. Proc. Natl. Acad. Sci. USA 2004, 101, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Yokomizo, T.; Nakamura, M.; Shimizu, T. Leukotriene receptors as potential therapeutic targets. J. Clin. Investig. 2018, 128, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Zdanov, S.; Bernard, D.; Debacq-Chainiaux, F.; Martien, S.; Gosselin, K.; Vercamer, C.; Chelli, F.; Toussaint, O.; Abbadie, C. Normal or stress-induced fibroblast senescence involves COX-2 activity. Exp. Cell Res. 2007, 313, 3046–3056. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Rodilossi, S.; Caprari, P.; Coppola, V.; Procopio, A. 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. EMBO J. 2005, 24, 170–179. [Google Scholar] [CrossRef]
- Wiley, C.D.; Brumwell, A.N.; Davis, S.S.; Jackson, J.R.; Valdovinos, A.; Calhoun, C.; Alimirah, F.; Castellanos, C.A.; Ruan, R.; Wei, Y.; et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Lagares, D.; Santos, A.; Grasberger, P.E.; Liu, F.; Probst, C.K.; Rahimi, R.A.; Sakai, N.; Kuehl, T.; Ryan, J.; Bhola, P.; et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Haynes, C.A.; Allegood, J.C.; Park, H.; Sullards, M.C. Sphingolipidomics: Methods for the comprehensive analysis of sphingolipids. J. Chromatogr. B 2009, 877, 2696–2708. [Google Scholar] [CrossRef]
- Goins, L.; Spassieva, S. Sphingoid bases and their involvement in neurodegenerative diseases. Adv. Biol. Regul. 2018, 70, 65–73. [Google Scholar] [CrossRef]
- Bieberich, E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem. Phys. Lipids 2018, 216, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Pavoine, C.; Pecker, F. Sphingomyelinases: Their regulation and roles in cardiovascular pathophysiology. Cardiovasc. Res. 2009, 82, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Noe, J.; Petrusca, D.; Rush, N.; Deng, P.; VanDemark, M.; Berdyshev, E.; Gu, Y.; Smith, P.; Schweitzer, K.; Pilewsky, J.; et al. CFTR regulation of intracellular pH and ceramides is required for lung endothelial cell apoptosis. Am. J. Respir. Cell Mol. Biol. 2009, 41, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gomez-Munoz, A. Ceramide and ceramide 1-phosphate in health and disease. Lipids Health Dis. 2010, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Testai, F.D.; Xu, H.L.; Kilkus, J.; Suryadevara, V.; Gorshkova, I.; Berdyshev, E.; Pelligrino, D.A.; Dawson, G. Changes in the metabolism of sphingolipids after subarachnoid hemorrhage. J. Neurosci. Res. 2015, 93, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Shea, B.S.; Tager, A.M. Sphingolipid regulation of tissue fibrosis. Open Rheumatol. J. 2012, 6, 123–129. [Google Scholar] [CrossRef]
- Suryadevara, V.; Fu, P.; Ebenezer, D.L.; Berdyshev, E.; Bronova, I.A.; Huang, L.S.; Harijith, A.; Natarajan, V. Sphingolipids in Ventilator Induced Lung Injury: Role of Sphingosine-1-Phosphate Lyase. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.; Brindley, D.N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid Res. 2015, 56, 2048–2060. [Google Scholar] [CrossRef]
- Wakashima, T.; Abe, K.; Kihara, A. Dual functions of the trans-2-enoyl-CoA reductase TER in the sphingosine 1-phosphate metabolic pathway and in fatty acid elongation. J. Biol. Chem. 2014, 289, 24736–24748. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, D.L.; Fu, P.; Suryadevara, V.; Zhao, Y.; Natarajan, V. Epigenetic regulation of pro-inflammatory cytokine secretion by sphingosine 1-phosphate (S1P) in acute lung injury: Role of S1P lyase. Adv. Biol. Regul. 2017, 63, 156–166. [Google Scholar] [CrossRef]
- Ichikawa, S.; Sakiyama, H.; Suzuki, G.; Hidari, K.I.; Hirabayashi, Y. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 12654. [Google Scholar] [CrossRef]
- D’Angelo, G.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and functions. FEBS J. 2013, 280, 6338–6353. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Kono, K.; Liu, H.; Shimizugawa, T.; Minekura, H.; Spiegel, S.; Kohama, T. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J. Biol. Chem. 2002, 277, 23294–23300. [Google Scholar] [CrossRef] [PubMed]
- Bornancin, F. Ceramide kinase: The first decade. Cell. Signal. 2011, 23, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Hoeferlin, L.A.; Wijesinghe, D.S.; Chalfant, C.E. The role of ceramide-1-phosphate in biological functions. Handb. Exp. Pharmacol. 2013, 153–166. [Google Scholar] [CrossRef]
- Berger, A.; Rosenthal, D.; Spiegel, S. Sphingosylphosphocholine, a signaling molecule which accumulates in Niemann-Pick disease type A, stimulates DNA-binding activity of the transcription activator protein AP-1. Proc. Natl. Acad. Sci. USA 1995, 92, 5885–5889. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Canals, D.; Hannun, Y.A. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell. Signal. 2009, 21, 836–846. [Google Scholar] [CrossRef]
- Beckmann, N.; Becker, K.A.; Kadow, S.; Schumacher, F.; Kramer, M.; Kuhn, C.; Schulz-Schaeffer, W.J.; Edwards, M.J.; Kleuser, B.; Gulbins, E.; et al. Acid Sphingomyelinase Deficiency Ameliorates Farber Disease. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Teichgraber, V.; Ulrich, M.; Endlich, N.; Riethmuller, J.; Wilker, B.; De Oliveira-Munding, C.C.; van Heeckeren, A.M.; Barr, M.L.; von Kurthy, G.; Schmid, K.W.; et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 2008, 14, 382–391. [Google Scholar] [CrossRef]
- Becker, K.A.; Riethmuller, J.; Luth, A.; Doring, G.; Kleuser, B.; Gulbins, E. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 716–724. [Google Scholar] [CrossRef]
- Riethmuller, J.; Anthonysamy, J.; Serra, E.; Schwab, M.; Doring, G.; Gulbins, E. Therapeutic efficacy and safety of amitriptyline in patients with cystic fibrosis. Cell. Physiol. Biochem. 2009, 24, 65–72. [Google Scholar] [CrossRef]
- Dhami, R.; He, X.; Schuchman, E.H. Acid sphingomyelinase deficiency attenuates bleomycin-induced lung inflammation and fibrosis in mice. Cell. Physiol. Biochem. 2010, 26, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Simonaro, C.M.; Park, J.H.; Eliyahu, E.; Shtraizent, N.; McGovern, M.M.; Schuchman, E.H. Imprinting at the SMPD1 locus: Implications for acid sphingomyelinase-deficient Niemann-Pick disease. Am. J. Hum. Genet. 2006, 78, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.V.; Gorshkova, I.; Skobeleva, A.; Bittman, R.; Lu, X.; Dudek, S.M.; Mirzapoiazova, T.; Garcia, J.G.; Natarajan, V. FTY720 inhibits ceramide synthases and up-regulates dihydrosphingosine 1-phosphate formation in human lung endothelial cells. J. Biol. Chem. 2009, 284, 5467–5477. [Google Scholar] [CrossRef] [PubMed]
- Scholte, B.J.; Horati, H.; Veltman, M.; Vreeken, R.J.; Garratt, L.W.; Tiddens, H.; Janssens, H.M.; Stick, S.M. Australian Respiratory Early Surveillance Team for Cystic, F. Oxidative stress and abnormal bioactive lipids in early cystic fibrosis lung disease. J. Cyst. Fibros. 2019, 18, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Tibboel, J.; Reiss, I.; de Jongste, J.C.; Post, M. Ceramides: A potential therapeutic target in pulmonary emphysema. Respir. Res 2013, 14, 96. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Berdyshev, E.; Mathew, B.; Fu, P.; Gorshkova, I.A.; He, D.; Ma, W.; Noth, I.; Ma, S.F.; Pendyala, S.; et al. Targeting sphingosine kinase 1 attenuates bleomycin-induced pulmonary fibrosis. FASEB J. 2013, 27, 1749–1760. [Google Scholar] [CrossRef]
- Mathew, B.; Jacobson, J.R.; Berdyshev, E.; Huang, Y.; Sun, X.; Zhao, Y.; Gerhold, L.M.; Siegler, J.; Evenoski, C.; Wang, T.; et al. Role of sphingolipids in murine radiation-induced lung injury: Protection by sphingosine 1-phosphate analogs. FASEB J. 2011, 25, 3388–3400. [Google Scholar] [CrossRef]
- Christofidou-Solomidou, M.; Pietrofesa, R.A.; Arguiri, E.; Schweitzer, K.S.; Berdyshev, E.V.; McCarthy, M.; Corbitt, A.; Alwood, J.S.; Yu, Y.; Globus, R.K.; et al. Space radiation-associated lung injury in a murine model. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L416–L428. [Google Scholar] [CrossRef]
- Zhao, Y.D.; Yin, L.; Archer, S.; Lu, C.; Zhao, G.; Yao, Y.; Wu, L.; Hsin, M.; Waddell, T.K.; Keshavjee, S.; et al. Metabolic heterogeneity of idiopathic pulmonary fibrosis: A metabolomic study. BMJ Open Respir. Res. 2017, 4, e000183. [Google Scholar] [CrossRef]
- Okajima, F. Plasma lipoproteins behave as carriers of extracellular sphingosine 1-phosphate: Is this an atherogenic mediator or an anti-atherogenic mediator? Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2002, 1582, 132–137. [Google Scholar] [CrossRef]
- Xu, N.; Dahlback, B. A novel human apolipoprotein (apoM). J. Biol. Chem. 1999, 274, 31286–31290. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnstrom, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, D.L.; Berdyshev, E.V.; Bronova, I.A.; Liu, Y.; Tiruppathi, C.; Komarova, Y.; Benevolenskaya, E.V.; Suryadevara, V.; Ha, A.W.; Harijith, A.; et al. Pseudomonas aeruginosa stimulates nuclear sphingosine-1-phosphate generation and epigenetic regulation of lung inflammatory injury. Thorax 2019, 74, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, V.; Dudek, S.M.; Jacobson, J.R.; Moreno-Vinasco, L.; Huang, L.S.; Abassi, T.; Mathew, B.; Zhao, Y.; Wang, L.; Bittman, R.; et al. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Ebenezer, D.L.; Ha, A.W.; Suryadevara, V.; Harijith, A.; Natarajan, V. Nuclear lipid mediators: Role of nuclear sphingolipids and sphingosine-1-phosphate signaling in epigenetic regulation of inflammation and gene expression. J. Cell. Biochem. 2018, 119, 6337–6353. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.D. Fifty years of lyase and a moment of truth: Sphingosine phosphate lyase from discovery to disease. J. Lipid Res. 2019, 60, 456–463. [Google Scholar] [CrossRef]
- Ebenezer, D.L.; Fu, P.; Natarajan, V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol. Ther. 2016, 168, 143–157. [Google Scholar] [CrossRef]
- Ksiazek, M.; Chacinska, M.; Chabowski, A.; Baranowski, M. Sources, metabolism, and regulation of circulating sphingosine-1-phosphate. J. Lipid Res. 2015, 56, 1271–1281. [Google Scholar] [CrossRef]
- Nagahashi, M.; Takabe, K.; Terracina, K.P.; Soma, D.; Hirose, Y.; Kobayashi, T.; Matsuda, Y.; Wakai, T. Sphingosine-1-phosphate transporters as targets for cancer therapy. Biomed Res. Int. 2014, 2014, 651727. [Google Scholar] [CrossRef]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef]
- Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; et al. BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. 2011, 31, 6850–6857. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Etemadi, N.; Chopin, M.; Anderton, H.; Tanzer, M.C.; Rickard, J.A.; Abeysekera, W.; Hall, C.; Spall, S.K.; Wang, B.; Xiong, Y.; et al. TRAF2 regulates TNF and NF-kappaB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gorshkova, I.A.; Berdyshev, E.; He, D.; Fu, P.; Ma, W.; Su, Y.; Usatyuk, P.V.; Pendyala, S.; Oskouian, B.; et al. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am. J. Respir. Cell Mol. Biol. 2011, 45, 426–435. [Google Scholar] [CrossRef]
- McVerry, B.J.; Peng, X.; Hassoun, P.M.; Sammani, S.; Simon, B.A.; Garcia, J.G. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am. J. Respir. Crit. Care Med. 2004, 170, 987–993. [Google Scholar] [CrossRef]
- Peng, X.; Hassoun, P.M.; Sammani, S.; McVerry, B.J.; Burne, M.J.; Rabb, H.; Pearse, D.; Tuder, R.M.; Garcia, J.G. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am. J. Respir. Crit. Care Med. 2004, 169, 1245–1251. [Google Scholar] [CrossRef]
- Harijith, A.; Pendyala, S.; Reddy, N.M.; Bai, T.; Usatyuk, P.V.; Berdyshev, E.; Gorshkova, I.; Huang, L.S.; Mohan, V.; Garzon, S.; et al. Sphingosine kinase 1 deficiency confers protection against hyperoxia-induced bronchopulmonary dysplasia in a murine model: Role of S1P signaling and Nox proteins. Am. J. Pathol. 2013, 183, 1169–1182. [Google Scholar] [CrossRef]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef]
- Ammit, A.J.; Hastie, A.T.; Edsall, L.C.; Hoffman, R.K.; Amrani, Y.; Krymskaya, V.P.; Kane, S.A.; Peters, S.P.; Penn, R.B.; Spiegel, S.; et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J. 2001, 15, 1212–1214. [Google Scholar] [CrossRef]
- Huang, L.S.; Natarajan, V. Sphingolipids in pulmonary fibrosis. Adv. Biol. Regul. 2015, 57, 55–63. [Google Scholar] [CrossRef]
- Milara, J.; Navarro, R.; Juan, G.; Peiro, T.; Serrano, A.; Ramon, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dudek, S.M. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc. Res. 2009, 77, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.Q.; Wong, W.S.; Leung, B.P. Sphingosine kinase and sphingosine 1-phosphate in asthma. Biosci. Rep. 2011, 31, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Sudhadevi, T.; Ha, A.W.; Ebenezer, D.L.; Fu, P.; Putherickal, V.; Natarajan, V.; Harijith, A. Advancements in understanding the role of lysophospholipids and their receptors in lung disorders including bronchopulmonary dysplasia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158685. [Google Scholar] [CrossRef]
- Gorshkova, I.; Zhou, T.; Mathew, B.; Jacobson, J.R.; Takekoshi, D.; Bhattacharya, P.; Smith, B.; Aydogan, B.; Weichselbaum, R.R.; Natarajan, V.; et al. Inhibition of serine palmitoyltransferase delays the onset of radiation-induced pulmonary fibrosis through the negative regulation of sphingosine kinase-1 expression. J. Lipid Res. 2012, 53, 1553–1568. [Google Scholar] [CrossRef]
- Morishima, Y.; Nomura, A.; Uchida, Y.; Noguchi, Y.; Sakamoto, T.; Ishii, Y.; Goto, Y.; Masuyama, K.; Zhang, M.J.; Hirano, K.; et al. Triggering the induction of myofibroblast and fibrogenesis by airway epithelial shedding. Am. J. Respir. Cell Mol. Biol. 2001, 24, 1–11. [Google Scholar] [CrossRef]
- Leach, H.G.; Chrobak, I.; Han, R.; Trojanowska, M. Endothelial cells recruit macrophages and contribute to a fibrotic milieu in bleomycin lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1093–1101. [Google Scholar] [CrossRef]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Kim, S.-J.; Cheresh, P.; Huang, L.; Watanabe, S.; Joshi, N.; Williams, K.; Piseaux-Allion, R.; Chi, M.; Yeldandi, A.; Lam, A. The Sphingosine Kinase 1 Inhibitor, PF543, Mitigates Asbestos-Induced Pulmonary Fibrosis and Lung mtDNA Damage in Mice in D107. Mitochondria and er Stress in Homeostasis and Repair. Presented at American Thoracis Society 2019 International Conference, Dallas, TX, USA, 17–22 May 2019; p. A7220. [Google Scholar]
- Bu, S.; Kapanadze, B.; Hsu, T.; Trojanowska, M. Opposite effects of dihydrosphingosine 1-phosphate and sphingosine 1-phosphate on transforming growth factor-beta/Smad signaling are mediated through the PTEN/PPM1A-dependent pathway. J. Biol. Chem. 2008, 283, 19593–19602. [Google Scholar] [CrossRef]
- Bu, S.; Yamanaka, M.; Pei, H.; Bielawska, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.; Trojanowska, M. Dihydrosphingosine 1-phosphate stimulates MMP1 gene expression via activation of ERK1/2-Ets1 pathway in human fibroblasts. FASEB J. 2006, 20, 184–186. [Google Scholar] [CrossRef]
- Yamanaka, M.; Shegogue, D.; Pei, H.; Bu, S.; Bielawska, A.; Bielawski, J.; Pettus, B.; Hannun, Y.A.; Obeid, L.; Trojanowska, M. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J. Biol. Chem. 2004, 279, 53994–54001. [Google Scholar] [CrossRef]
- Bu, S.; Asano, Y.; Bujor, A.; Highland, K.; Hant, F.; Trojanowska, M. Dihydrosphingosine 1-phosphate has a potent antifibrotic effect in scleroderma fibroblasts via normalization of phosphatase and tensin homolog levels. Arthritis Rheum. 2010, 62, 2117–2126. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Berdyshev, E.V.; Tran, J.T.; Xie, L.; Chen, J.; Ebenezer, D.L.; Mathew, B.; Gorshkova, I.; Zhang, W.; Reddy, S.P.; et al. Sphingosine-1-phosphate lyase is an endogenous suppressor of pulmonary fibrosis: Role of S1P signalling and autophagy. Thorax 2015, 70, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Chang, C.L.; Tang, C.H.; Lin, Y.C.; Ju, T.K.; Huang, W.P.; Lee, H. Extrinsic sphingosine 1-phosphate activates S1P5 and induces autophagy through generating endoplasmic reticulum stress in human prostate cancer PC-3 cells. Cell. Signal. 2014, 26, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Huang, W.P.; Lee, H. Roles of sphingosine 1-phosphate on tumorigenesis. World J. Biol. Chem. 2011, 2, 25–34. [Google Scholar] [CrossRef]
- Lepine, S.; Allegood, J.C.; Park, M.; Dent, P.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 2011, 18, 350–361. [Google Scholar] [CrossRef]
- Sheng, R.; Zhang, T.T.; Felice, V.D.; Qin, T.; Qin, Z.H.; Smith, C.D.; Sapp, E.; Difiglia, M.; Waeber, C. Preconditioning stimuli induce autophagy via sphingosine kinase 2 in mouse cortical neurons. J. Biol. Chem. 2014, 289, 20845–20857. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.L.; Ho, M.C.; Lee, P.H.; Hsu, C.Y.; Huang, W.P.; Lee, H. S1P(5) is required for sphingosine 1-phosphate-induced autophagy in human prostate cancer PC-3 cells. Am. J. Physiol. Cell Physiol. 2009, 297, C451–G458. [Google Scholar] [CrossRef]
- Slattum, G.; Gu, Y.; Sabbadini, R.; Rosenblatt, J. Autophagy in oncogenic K-Ras promotes basal extrusion of epithelial cells by degrading S1P. Curr. Biol. 2014, 24, 19–28. [Google Scholar] [CrossRef]
- Taniguchi, M.; Kitatani, K.; Kondo, T.; Hashimoto-Nishimura, M.; Asano, S.; Hayashi, A.; Mitsutake, S.; Igarashi, Y.; Umehara, H.; Takeya, H.; et al. Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: Reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J. Biol. Chem. 2012, 287, 39898–39910. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Li, J.; Liu, D.; Conforti, F.; Brereton, C.J.; Yao, L.; Zhou, Y.; Alzetani, A.; Chee, S.J.; Marshall, B.G.; et al. Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in pulmonary fibrosis. Cell Death Dis. 2019, 10, 591. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Ye, Z.W.; Zhang, J.; Hammad, S.M.; Townsend, D.M.; Rockey, D.C.; Kim, S.H. 3-ketodihydrosphingosine reductase mutation induces steatosis and hepatic injury in zebrafish. Sci. Rep. 2019, 9, 1138. [Google Scholar] [CrossRef]
- Weiss, B.; Stoffel, W. Human and murine serine-palmitoyl-CoA transferase--cloning, expression and characterization of the key enzyme in sphingolipid synthesis. Eur. J. Biochem. 1997, 249, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Yamaji-Hasegawa, A.; Takahashi, A.; Tetsuka, Y.; Senoh, Y.; Kobayashi, T. Fungal metabolite sulfamisterin suppresses sphingolipid synthesis through inhibition of serine palmitoyltransferase. Biochemistry 2005, 44, 268–277. [Google Scholar] [CrossRef]
- Wadsworth, J.M.; Clarke, D.J.; McMahon, S.A.; Lowther, J.P.; Beattie, A.E.; Langridge-Smith, P.R.; Broughton, H.B.; Dunn, T.M.; Naismith, J.H.; Campopiano, D.J. The chemical basis of serine palmitoyltransferase inhibition by myriocin. J. Am. Chem. Soc. 2013, 135, 14276–14285. [Google Scholar] [CrossRef]
- Watson, M.L.; Coghlan, M.; Hundal, H.S. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem. J. 2009, 417, 791–801. [Google Scholar] [CrossRef]
- Shea, B.S.; Brooks, S.F.; Fontaine, B.A.; Chun, J.; Luster, A.D.; Tager, A.M. Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am. J. Respir. Cell Mol. Biol. 2010, 43, 662–673. [Google Scholar] [CrossRef]
- Shea, B.S.; Probst, C.K.; Brazee, P.L.; Rotile, N.J.; Blasi, F.; Weinreb, P.H.; Black, K.E.; Sosnovik, D.E.; Van Cott, E.M.; Violette, S.M.; et al. Uncoupling of the profibrotic and hemostatic effects of thrombin in lung fibrosis. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Dudek, S.M.; Camp, S.M.; Chiang, E.T.; Singleton, P.A.; Usatyuk, P.V.; Zhao, Y.; Natarajan, V.; Garcia, J.G. Pulmonary endothelial cell barrier enhancement by FTY720 does not require the S1P1 receptor. Cell. Signal. 2007, 19, 1754–1764. [Google Scholar] [CrossRef]
- Wang, L.; Sammani, S.; Moreno-Vinasco, L.; Letsiou, E.; Wang, T.; Camp, S.M.; Bittman, R.; Garcia, J.G.; Dudek, S.M. FTY720 (s)-phosphonate preserves sphingosine 1-phosphate receptor 1 expression and exhibits superior barrier protection to FTY720 in acute lung injury. Crit. Care Med. 2014, 42, e189–e199. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Okamoto, Y.; Asano, Y.; Ishimaru, K.; Aki, S.; Yoshioka, K.; Takuwa, N.; Wada, T.; Inagaki, Y.; Takahashi, C.; et al. Sphingosine-1-phosphate receptor-2 facilitates pulmonary fibrosis through potentiating IL-13 pathway in macrophages. PLoS ONE 2018, 13, e0197604. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Im, D.S. Deficiency of Sphingosine-1-Phosphate Receptor 2 (S1P2) Attenuates Bleomycin-Induced Pulmonary Fibrosis. Biomol. Ther. 2019, 27, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kohno, M.; Kadoya, M.; Nagahara, H.; Fujii, W.; Seno, T.; Yamamoto, A.; Oda, R.; Fujiwara, H.; Kubo, T.; et al. Knock out of S1P3 receptor signaling attenuates inflammation and fibrosis in bleomycin-induced lung injury mice model. PLoS ONE 2014, 9, e106792. [Google Scholar] [CrossRef]
- Sobel, K.; Menyhart, K.; Killer, N.; Renault, B.; Bauer, Y.; Studer, R.; Steiner, B.; Bolli, M.H.; Nayler, O.; Gatfield, J. Sphingosine 1-phosphate (S1P) receptor agonists mediate pro-fibrotic responses in normal human lung fibroblasts via S1P2 and S1P3 receptors and Smad-independent signaling. J. Biol. Chem. 2013, 288, 14839–14851. [Google Scholar] [CrossRef]
- Kono, Y.; Nishiuma, T.; Nishimura, Y.; Kotani, Y.; Okada, T.; Nakamura, S.; Yokoyama, M. Sphingosine kinase 1 regulates differentiation of human and mouse lung fibroblasts mediated by TGF-beta1. Am. J. Respir. Cell Mol. Biol. 2007, 37, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Antel, J.; Comi, G.; Montalban, X.; O’Connor, P.; Polman, C.H.; Haas, T.; Korn, A.A.; Karlsson, G.; Radue, E.W.; et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 355, 1124–1140. [Google Scholar] [CrossRef]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef]
- Hoyt, D.G.; Lazo, J.S. Alterations in pulmonary mRNA encoding procollagens, fibronectin and transforming growth factor-beta precede bleomycin-induced pulmonary fibrosis in mice. J. Pharmacol. Exp. Ther. 1988, 246, 765–771. [Google Scholar] [PubMed]
- Yi, E.S.; Bedoya, A.; Lee, H.; Chin, E.; Saunders, W.; Kim, S.J.; Danielpour, D.; Remick, D.G.; Yin, S.; Ulich, T.R. Radiation-induced lung injury in vivo: Expression of transforming growth factor-beta precedes fibrosis. Inflammation 1996, 20, 339–352. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhuang, F.; Liu, Y.H.; Xu, B.; Del Moral, P.; Deng, W.; Chai, Y.; Kolb, M.; Gauldie, J.; Warburton, D.; et al. TGF-beta receptor II in epithelia versus mesenchyme plays distinct roles in the developing lung. Eur. Respir. J. 2008, 32, 285–295. [Google Scholar] [CrossRef]
- Li, M.; Krishnaveni, M.S.; Li, C.; Zhou, B.; Xing, Y.; Banfalvi, A.; Li, A.; Lombardi, V.; Akbari, O.; Borok, Z.; et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Investig. 2011, 121, 277–287. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, W.; Wang, Y.L.; Chen, H.; Bringas, P., Jr.; Datto, M.B.; Frederick, J.P.; Wang, X.F.; Warburton, D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L585–L593. [Google Scholar] [CrossRef] [PubMed]
- Gyorfi, A.H.; Matei, A.E.; Distler, J.H.W. Targeting TGF-beta signaling for the treatment of fibrosis. Matrix Biol. 2018, 68–69, 8–27. [Google Scholar] [CrossRef]
- Cencetti, F.; Bernacchioni, C.; Nincheri, P.; Donati, C.; Bruni, P. Transforming growth factor-beta1 induces transdifferentiation of myoblasts into myofibroblasts via up-regulation of sphingosine kinase-1/S1P3 axis. Mol. Biol. Cell 2010, 21, 1111–1124. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef]
- Kurundkar, A.; Thannickal, V.J. Redox mechanisms in age-related lung fibrosis. Redox Biol. 2016, 9, 67–76. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J. Transl. Med. 2017, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.C.; et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011, 15, 607–619. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Turn, C.S.; Lockey, R.F.; Kolliputi, N. Putting the brakes on age-related idiopathic pulmonary fibrosis: Can Nox4 inhibitors suppress IPF? Exp. Gerontol. 2015, 63, 81–82. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Lee, J.H.; Hwang, S.C.; Choi, K.S.; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Fu, P.; Usatyuk, P.V.; Jacobson, J.; Cress, A.E.; Garcia, J.G.; Salgia, R.; Natarajan, V. Role played by paxillin and paxillin tyrosine phosphorylation in hepatocyte growth factor/sphingosine-1-phosphate-mediated reactive oxygen species generation, lamellipodia formation, and endothelial barrier function. Pulm. Circ. 2015, 5, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Harijith, A.; Pendyala, S.; Ebenezer, D.L.; Ha, A.W.; Fu, P.; Wang, Y.T.; Ma, K.; Toth, P.T.; Berdyshev, E.V.; Kanteti, P.; et al. Hyperoxia-induced p47phox activation and ROS generation is mediated through S1P transporter Spns2, and S1P/S1P1&2 signaling axis in lung endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L337–L351. [Google Scholar] [CrossRef]
- Fu, P.; Shaaya, M.; Harijith, A.; Jacobson, J.R.; Karginov, A.; Natarajan, V. Sphingolipids Signaling in Lamellipodia Formation and Enhancement of Endothelial Barrier Function. Curr. Top. Membr. 2018, 82, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Lehmann, W.; Martin, T.; Delaunay, C.; Hueber, A.; Barys, L.; Niu, H.; Billy, E.; Wartmann, M.; Ito, M.; et al. The tyrosine phosphatase PTPN14 is a negative regulator of YAP activity. PLoS ONE 2013, 8, e61916. [Google Scholar] [CrossRef] [PubMed]
- Boggiano, J.C.; Fehon, R.G. Growth control by committee: Intercellular junctions, cell polarity, and the cytoskeleton regulate Hippo signaling. Dev. Cell 2012, 22, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef]
- Ninou, I.; Kaffe, E.; Muller, S.; Budd, D.C.; Stevenson, C.S.; Ullmer, C.; Aidinis, V. Pharmacologic targeting of the ATX/LPA axis attenuates bleomycin-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2018, 52, 32–40. [Google Scholar] [CrossRef]
- Robinson, P.C.; Watters, L.C.; King, T.E.; Mason, R.J. Idiopathic pulmonary fibrosis. Abnormalities in bronchoalveolar lavage fluid phospholipids. Am. Rev. Respir. Dis. 1988, 137, 585–591. [Google Scholar] [CrossRef]
- Han, S.; Mallampalli, R.K. The Role of Surfactant in Lung Disease and Host Defense against Pulmonary Infections. Ann. Am. Thorac. Soc. 2015, 12, 765–774. [Google Scholar] [CrossRef]
- Low, R.B. Bronchoalveolar lavage lipids in idiopathic pulmonary fibrosis. Chest 1989, 95, 3–5. [Google Scholar] [CrossRef]
- Gunther, A.; Schmidt, R.; Nix, F.; Yabut-Perez, M.; Guth, C.; Rosseau, S.; Siebert, C.; Grimminger, F.; Morr, H.; Velcovsky, H.G.; et al. Surfactant abnormalities in idiopathic pulmonary fibrosis, hypersensitivity pneumonitis and sarcoidosis. Eur. Respir. J. 1999, 14, 565–573. [Google Scholar] [CrossRef]
- Vazquez-de-Lara, L.G.; Tlatelpa-Romero, B.; Romero, Y.; Fernandez-Tamayo, N.; Vazquez-de-Lara, F.; M Justo-Janeiro, J.; Garcia-Carrasco, M.; de-la-Rosa Paredes, R.; Cisneros-Lira, J.G.; Mendoza-Milla, C.; et al. Phosphatidylethanolamine Induces an Antifibrotic Phenotype in Normal Human Lung Fibroblasts and Ameliorates Bleomycin-Induced Lung Fibrosis in Mice. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- El Nady, M.; Kaddah, S.; El Hinnawy, Y.; Halim, R.M.; Kandeel, R. Plasma surfactant protein-D as a potential biomarker in idiopathic pulmonary fibrosis. Egypt. J. Bronchol. 2019, 13, 214–218. [Google Scholar] [CrossRef]
- Greene, K.E.; King, T.E., Jr.; Kuroki, Y.; Bucher-Bartelson, B.; Hunninghake, G.W.; Newman, L.S.; Nagae, H.; Mason, R.J. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur. Respir. J. 2002, 19, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, C.; Hirschbach, L.; Nef, H.; Seeger, W.; Guenther, A.; Markart, P. Surfactant protein B proforms as potential new biomarkers for idiopathic pulmonary fibrosis. Eur. Respir. J. 2014, 44, P772. [Google Scholar]
- McCormack, F.X.; King, T.E., Jr.; Bucher, B.L.; Nielsen, L.; Mason, R.J. Surfactant protein A predicts survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1995, 152, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Lawson, W.E.; Polosukhin, V.V.; Stathopoulos, G.T.; Zoia, O.; Han, W.; Lane, K.B.; Li, B.; Donnelly, E.F.; Holburn, G.E.; Lewis, K.G.; et al. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am. J. Pathol. 2005, 167, 1267–1277. [Google Scholar] [CrossRef]
- Chung, K.P.; Hsu, C.L.; Fan, L.C.; Huang, Z.; Bhatia, D.; Chen, Y.J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390. [Google Scholar] [CrossRef]
- Frohman, M.A. The phospholipase D superfamily as therapeutic targets. Trends Pharmacol. Sci. 2015, 36, 137–144. [Google Scholar] [CrossRef]
- Gomez-Cambronero, J. New concepts in phospholipase D signaling in inflammation and cancer. Sci. World J. 2010, 10, 1356–1369. [Google Scholar] [CrossRef]
- Cummings, R.; Parinandi, N.; Wang, L.; Usatyuk, P.; Natarajan, V. Phospholipase D/phosphatidic acid signal transduction: Role and physiological significance in lung. Mol. Cell. Biochem. 2002, 234–235, 99–109. [Google Scholar] [CrossRef]
- Brown, H.A.; Thomas, P.G.; Lindsley, C.W. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat. Rev. Drug Discov. 2017, 16, 351–367. [Google Scholar] [CrossRef]
- Carman, G.M.; Han, G.S. Phosphatidic acid phosphatase, a key enzyme in the regulation of lipid synthesis. J. Biol. Chem. 2009, 284, 2593–2597. [Google Scholar] [CrossRef] [PubMed]
- Siniossoglou, S. Phospholipid metabolism and nuclear function: Roles of the lipin family of phosphatidic acid phosphatases. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2013, 1831, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, H.; Aoki, J.; Hiramatsu, T.; Ishida, M.; Bandoh, K.; Nagai, Y.; Taguchi, R.; Inoue, K.; Arai, H. A novel phosphatidic acid-selective phospholipase A1 that produces lysophosphatidic acid. J. Biol. Chem. 2002, 277, 34254–34263. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Tchoua, U.; Okamoto, M.; Tojo, H. Purification and properties of a phospholipase A2/lipase preferring phosphatidic acid, bis(monoacylglycerol) phosphate, and monoacylglycerol from rat testis. J. Biol. Chem. 2002, 277, 43674–43681. [Google Scholar] [CrossRef]
- Gomez-Cambronero, J.; Kantonen, S. A river runs through it: How autophagy, senescence, and phagocytosis could be linked to phospholipase D by Wnt signaling. J. Leukoc. Biol. 2014, 96, 779–784. [Google Scholar] [CrossRef]
- Henkels, K.M.; Miller, T.E.; Ganesan, R.; Wilkins, B.A.; Fite, K.; Gomez-Cambronero, J. A Phosphatidic Acid (PA) conveyor system of continuous intracellular transport from cell membrane to nucleus maintains EGF receptor homeostasis. Oncotarget 2016, 7, 47002–47017. [Google Scholar] [CrossRef]
- Patel, R.B.; Kotha, S.R.; Sherwani, S.I.; Sliman, S.M.; Gurney, T.O.; Loar, B.; Butler, S.O.; Morris, A.J.; Marsh, C.B.; Parinandi, N.L. Pulmonary fibrosis inducer, bleomycin, causes redox-sensitive activation of phospholipase D and cytotoxicity through formation of bioactive lipid signal mediator, phosphatidic acid, in lung microvascular endothelial cells. Int. J. Toxicol. 2011, 30, 69–90. [Google Scholar] [CrossRef]
- Suryadevara, V.; Huang, L.; Kim, S.J.; Cheresh, P.; Shaaya, M.; Bandela, M.; Fu, P.; Feghali-Bostwick, C.; Di Paolo, G.; Kamp, D.W.; et al. Role of phospholipase D in bleomycin-induced mitochondrial reactive oxygen species generation, mitochondrial DNA damage, and pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L175–L187. [Google Scholar] [CrossRef]
- Usatyuk, P.V.; Gorshkova, I.A.; He, D.; Zhao, Y.; Kalari, S.K.; Garcia, J.G.; Natarajan, V. Phospholipase D-mediated activation of IQGAP1 through Rac1 regulates hyperoxia-induced p47phox translocation and reactive oxygen species generation in lung endothelial cells. J. Biol. Chem. 2009, 284, 15339–15352. [Google Scholar] [CrossRef]
- Foster, D.A. Regulation of mTOR by phosphatidic acid? Cancer Res. 2007, 67, 1–4. [Google Scholar] [CrossRef]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Kumar, R.K.; Iyer, A.; Boswell, S.; Gerarduzzi, C.; Dadhania, V.P.; Herbert, Z.; Joshi, N.; Luyendyk, J.P.; Humphreys, B.D.; et al. Targeting Phospholipase D4 Attenuates Kidney Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 3579–3589. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.S.; Schmezer, P.; Popanda, O. Diacylglycerol Kinase Alpha in Radiation-Induced Fibrosis: Potential as a Predictive Marker or Therapeutic Target. Front. Oncol. 2020, 10, 737. [Google Scholar] [CrossRef] [PubMed]
- Weigel, C.; Veldwijk, M.R.; Oakes, C.C.; Seibold, P.; Slynko, A.; Liesenfeld, D.B.; Rabionet, M.; Hanke, S.A.; Wenz, F.; Sperk, E.; et al. Epigenetic regulation of diacylglycerol kinase alpha promotes radiation-induced fibrosis. Nat. Commun. 2016, 7, 10893. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Liu, K.; Sasaki, S.; Kunii, N.; Sakai, H.; Mizuno, H.; Saga, H.; Sakane, F. Evaluations of the selectivities of the diacylglycerol kinase inhibitors R59022 and R59949 among diacylglycerol kinase isozymes using a new non-radioactive assay method. Pharmacology 2013, 92, 99–107. [Google Scholar] [CrossRef]
- Kulkarni, Y.M.; Dutta, S.; Iyer, A.K.; Venkatadri, R.; Kaushik, V.; Ramesh, V.; Wright, C.A.; Semmes, O.J.; Yakisich, J.S.; Azad, N. A proteomics approach to identifying key protein targets involved in VEGF inhibitor mediated attenuation of bleomycin-induced pulmonary fibrosis. Proteomics 2016, 16, 33–46. [Google Scholar] [CrossRef]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2008, 1781, 513–518. [Google Scholar] [CrossRef]
- Rindlisbacher, B.; Schmid, C.; Geiser, T.; Bovet, C.; Funke-Chambour, M. Serum metabolic profiling identified a distinct metabolic signature in patients with idiopathic pulmonary fibrosis—A potential biomarker role for LysoPC. Respir Res 2018, 19, 7. [Google Scholar] [CrossRef]
- Tan, Y.; Jia, D.; Lin, Z.; Guo, B.; He, B.; Lu, C.; Xiao, C.; Liu, Z.; Zhao, N.; Bian, Z.; et al. Potential Metabolic Biomarkers to Identify Interstitial Lung Abnormalities. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Montesi, S.B.; Mathai, S.K.; Brenner, L.N.; Gorshkova, I.A.; Berdyshev, E.V.; Tager, A.M.; Shea, B.S. Docosatetraenoyl LPA is elevated in exhaled breath condensate in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2014, 14, 5. [Google Scholar] [CrossRef]
- Park, G.Y.; Lee, Y.G.; Berdyshev, E.; Nyenhuis, S.; Du, J.; Fu, P.; Gorshkova, I.A.; Li, Y.; Chung, S.; Karpurapu, M.; et al. Autotaxin production of lysophosphatidic acid mediates allergic asthmatic inflammation. Am. J. Respir. Crit. Care Med. 2013, 188, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Turkenburg, M.; Poll-The, B.T.; Karall, D.; Perez-Cerda, C.; Morrone, A.; Malvagia, S.; Wanders, R.J.; Kulik, W.; Vaz, F.M. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Mathew, B.; Li, H.; Zhao, Y.; Ma, S.F.; Noth, I.; Reddy, S.P.; Harijith, A.; Usatyuk, P.V.; Berdyshev, E.V.; et al. The mitochondrial cardiolipin remodeling enzyme lysocardiolipin acyltransferase is a novel target in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 189, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Saric, A.; Andreau, K.; Armand, A.S.; Moller, I.M.; Petit, P.X. Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front. Genet. 2015, 6, 359. [Google Scholar] [CrossRef]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8. [Google Scholar] [CrossRef]
- Baile, M.G.; Sathappa, M.; Lu, Y.W.; Pryce, E.; Whited, K.; McCaffery, J.M.; Han, X.; Alder, N.N.; Claypool, S.M. Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J. Biol. Chem. 2014, 289, 1768–1778. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Bakovic, M. Formation and regulation of mitochondrial membranes. Int. J. Cell Biol. 2014, 2014, 709828. [Google Scholar] [CrossRef]
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 2022–2031. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1837, 408–417. [Google Scholar] [CrossRef]
- Borisenko, G.G. Mitochondrial phospholipid cardiolipin and its triggering functions in the cells. Lipid Technol. 2016, 28, 40–43. [Google Scholar] [CrossRef]
- Ray, N.B.; Durairaj, L.; Chen, B.B.; McVerry, B.J.; Ryan, A.J.; Donahoe, M.; Waltenbaugh, A.K.; O’Donnell, C.P.; Henderson, F.C.; Etscheidt, C.A.; et al. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat. Med. 2010, 16, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Raja, V.; Greenberg, M.L. The functions of cardiolipin in cellular metabolism-potential modifiers of the Barth syndrome phenotype. Chem. Phys. Lipids 2014, 179, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kiebish, M.A.; Yang, K.; Liu, X.; Mancuso, D.J.; Guan, S.; Zhao, Z.; Sims, H.F.; Cerqua, R.; Cade, W.T.; Han, X.; et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013, 54, 1312–1325. [Google Scholar] [CrossRef]
- Huang, L.S.; Jiang, P.; Feghali-Bostwick, C.; Reddy, S.P.; Garcia, J.G.N.; Natarajan, V. Lysocardiolipin acyltransferase regulates TGF-beta mediated lung fibroblast differentiation. Free Radic. Biol. Med. 2017, 112, 162–173. [Google Scholar] [CrossRef]
- Ackerman, S.J.; Park, G.Y.; Christman, J.W.; Nyenhuis, S.; Berdyshev, E.; Natarajan, V. Polyunsaturated lysophosphatidic acid as a potential asthma biomarker. Biomark. Med. 2016, 10, 123–135. [Google Scholar] [CrossRef]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef]
- Federico, L.; Jeong, K.J.; Vellano, C.P.; Mills, G.B. Autotaxin, a lysophospholipase D with pleomorphic effects in oncogenesis and cancer progression. J. Lipid Res. 2016, 57, 25–35. [Google Scholar] [CrossRef]
- Van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Ninou, I.; Magkrioti, C.; Aidinis, V. Autotaxin in Pathophysiology and Pulmonary Fibrosis. Front. Med. 2018, 5, 180. [Google Scholar] [CrossRef]
- Benesch, M.G.; Tang, X.; Venkatraman, G.; Bekele, R.T.; Brindley, D.N. Recent advances in targeting the autotaxin-lysophosphatidate-lipid phosphate phosphatase axis in vivo. J. Biomed. Res. 2016, 30, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar] [PubMed]
- Murata, J.; Lee, H.Y.; Clair, T.; Krutzsch, H.C.; Arestad, A.A.; Sobel, M.E.; Liotta, L.A.; Stracke, M.L. cDNA cloning of the human tumor motility-stimulating protein, autotaxin, reveals a homology with phosphodiesterases. J. Biol. Chem. 1994, 269, 30479–30484. [Google Scholar] [PubMed]
- Stefan, C.; Jansen, S.; Bollen, M. NPP-type ectophosphodiesterases: Unity in diversity. Trends Biochem. Sci. 2005, 30, 542–550. [Google Scholar] [CrossRef]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef]
- Zhao, Y.; Natarajan, V. Lysophosphatidic acid (LPA) and its receptors: Role in airway inflammation and remodeling. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2013, 1831, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Bektas, M.; Payne, S.G.; Liu, H.; Goparaju, S.; Milstien, S.; Spiegel, S. A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J. Cell Biol. 2005, 169, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Kalari, S.; Zhao, Y.; Spannhake, E.W.; Berdyshev, E.V.; Natarajan, V. Role of acylglycerol kinase in LPA-induced IL-8 secretion and transactivation of epidermal growth factor-receptor in human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L328–L336. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Stroud, D.A.; Baker, M.J.; De Souza, D.P.; Frazier, A.E.; Liem, M.; Tull, D.; Mathivanan, S.; McConville, M.J.; Thorburn, D.R.; et al. Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 2017, 67, 457–470.e455. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.E.; Kaplan, M.G.; Barnard, E.A. Identification of 6H1 as a P2Y purinoceptor: P2Y5. Biochem. Biophys. Res. Commun. 1996, 219, 105–110. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Huang, J.; Fajas, L.; Li, A.; Welch, J.; Najib, J.; Witztum, J.L.; Auwerx, J.; Palinski, W.; Glass, C.K. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1998, 95, 7614–7619. [Google Scholar] [CrossRef]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; St Hilaire, A.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, H.F.; Thatcher, T.H.; Kottmann, R.M.; Garcia, T.M.; Phipps, R.P.; Sime, P.J. The Role of PPARs in Lung Fibrosis. PPAR Res. 2007, 2007, 71323. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y. Cyclic phosphatidic acid—A unique bioactive phospholipid. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2008, 1781, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Murakami-Murofushi, K.; Uchiyama, A.; Fujiwara, Y.; Kobayashi, T.; Kobayashi, S.; Mukai, M.; Murofushi, H.; Tigyi, G. Biological functions of a novel lipid mediator, cyclic phosphatidic acid. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2002, 1582, 1–7. [Google Scholar] [CrossRef]
- Tsukahara, T. PPAR gamma Networks in Cell Signaling: Update and Impact of Cyclic Phosphatidic Acid. J. Lipids 2013, 2013, 246597. [Google Scholar] [CrossRef]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef]
- Sakai, T.; Peyruchaud, O.; Fassler, R.; Mosher, D.F. Restoration of beta1A integrins is required for lysophosphatidic acid-induced migration of beta1-null mouse fibroblastic cells. J. Biol. Chem. 1998, 273, 19378–19382. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Fu, P.; Patel, P.; Harijith, A.; Sun, T.; Zhao, Y.; Garcia, J.G.; Chun, J.; Natarajan, V. Lysophosphatidic acid receptor-2 deficiency confers protection against bleomycin-induced lung injury and fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cummings, R.; Zhao, Y.; Kazlauskas, A.; Sham, J.K.; Morris, A.; Georas, S.; Brindley, D.N.; Natarajan, V. Involvement of phospholipase D2 in lysophosphatidate-induced transactivation of platelet-derived growth factor receptor-beta in human bronchial epithelial cells. J. Biol. Chem. 2003, 278, 39931–39940. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Natarajan, V.; Stern, R.; Gorshkova, I.A.; Solway, J.; Spannhake, E.W.; Zhao, Y. Lysophosphatidic acid-induced transactivation of epidermal growth factor receptor regulates cyclo-oxygenase-2 expression and prostaglandin E(2) release via C/EBPbeta in human bronchial epithelial cells. Biochem. J. 2008, 412, 153–162. [Google Scholar] [CrossRef]
- Ganguly, K.; Stoeger, T.; Wesselkamper, S.C.; Reinhard, C.; Sartor, M.A.; Medvedovic, M.; Tomlinson, C.R.; Bolle, I.; Mason, J.M.; Leikauf, G.D.; et al. Candidate genes controlling pulmonary function in mice: Transcript profiling and predicted protein structure. Physiol. Genom. 2007, 31, 410–421. [Google Scholar] [CrossRef]
- Van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradere, J.P.; Pettit, T.R.; Wakelam, M.J.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol. Cell. Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef]
- Tanaka, M.; Okudaira, S.; Kishi, Y.; Ohkawa, R.; Iseki, S.; Ota, M.; Noji, S.; Yatomi, Y.; Aoki, J.; Arai, H. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 2006, 281, 25822–25830. [Google Scholar] [CrossRef]
- Ferry, G.; Giganti, A.; Coge, F.; Bertaux, F.; Thiam, K.; Boutin, J.A. Functional invalidation of the autotaxin gene by a single amino acid mutation in mouse is lethal. FEBS Lett. 2007, 581, 3572–3578. [Google Scholar] [CrossRef]
- Mouratis, M.A.; Magkrioti, C.; Oikonomou, N.; Katsifa, A.; Prestwich, G.D.; Kaffe, E.; Aidinis, V. Autotaxin and Endotoxin-Induced Acute Lung Injury. PLoS ONE 2015, 10, e0133619. [Google Scholar] [CrossRef]
- Black, K.E.; Berdyshev, E.; Bain, G.; Castelino, F.V.; Shea, B.S.; Probst, C.K.; Fontaine, B.A.; Bronova, I.; Goulet, L.; Lagares, D.; et al. Autotaxin activity increases locally following lung injury, but is not required for pulmonary lysophosphatidic acid production or fibrosis. FASEB J. 2016, 30, 2435–2450. [Google Scholar] [CrossRef]
- Lee, G.; Kang, S.U.; Ryou, J.-H.; Lim, J.-J.; Lee, Y.-H. Late Breaking Abstract—BBT-877, a Potent Autotaxin Inhibitor in Clinical Development to Treat Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2019, 54, PA1293. [Google Scholar] [CrossRef]
- Maher, T.M.; van der Aar, E.M.; Van de Steen, O.; Allamassey, L.; Desrivot, J.; Dupont, S.; Fagard, L.; Ford, P.; Fieuw, A.; Wuyts, W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): A phase 2a randomised placebo-controlled trial. Lancet Respir. Med. 2018, 6, 627–635. [Google Scholar] [CrossRef]
- Zemski Berry, K.A.; Murphy, R.C.; Kosmider, B.; Mason, R.J. Lipidomic characterization and localization of phospholipids in the human lung. J. Lipid Res. 2017, 58, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Funke, M.; Zhao, Z.; Xu, Y.; Chun, J.; Tager, A.M. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.M.; Snyder, L.; Todd, J.L.; Soule, B.; Christian, R.; Anstrom, K.; Luo, Y.; Gagnon, R.; Rosen, G. Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial of BMS-986020, a Lysophosphatidic Acid Receptor Antagonist for the Treatment of Idiopathic Pulmonary Fibrosis. Chest 2018, 154, 1061–1069. [Google Scholar] [CrossRef]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef]
- Gianazza, E.; Brioschi, M.; Fernandez, A.M.; Banfi, C. Lipoxidation in cardiovascular diseases. Redox Biol. 2019, 23, 101119. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Gang, X.; Kim, J.H.; Sussan, T.E.; Witztum, J.L.; Biswal, S. Oxidized phospholipids impair pulmonary antibacterial defenses: Evidence in mice exposed to cigarette smoke. Biochem. Biophys. Res. Commun. 2012, 426, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Paliogiannis, P.; Fois, A.G.; Collu, C.; Bandinu, A.; Zinellu, E.; Carru, C.; Pirina, P.; Mangoni, A.A.; Zinellu, A. Oxidative stress-linked biomarkers in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Biomark. Med. 2018, 12, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Tashiro, Y.; Chibana, S.; Yamashita, A.; Karakawa, T.; Kohrogi, H. Role of lipid-derived free radical in bleomycin-induced lung injury in mice: Availability for ESR spin trap method with organic phase extraction. Biol. Pharm. Bull. 2008, 31, 1855–1859. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suntres, Z.E.; Shek, P.N. Protective effect of liposomal alpha-tocopherol against bleomycin-induced lung injury. Biomed. Environ. Sci. 1997, 10, 47–59. [Google Scholar] [PubMed]
- Sener, G.; Topaloglu, N.; Sehirli, A.O.; Ercan, F.; Gedik, N. Resveratrol alleviates bleomycin-induced lung injury in rats. Pulm. Pharmacol. Ther. 2007, 20, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, K.C.; Soares, F.S.; Rocha, L.G.; Silveira, P.C.; Silva, L.A.; Valenca, S.S.; Dal Pizzol, F.; Streck, E.L.; Pinho, R.A. Attenuation of bleomycin-induced lung injury and oxidative stress by N-acetylcysteine plus deferoxamine. Pulm. Pharmacol. Ther. 2008, 21, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Araya, J.; Yoshida, M.; Sakamoto, T.; Koumura, T.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; et al. Involvement of GPx4-Regulated Lipid Peroxidation in Idiopathic Pulmonary Fibrosis Pathogenesis. J. Immunol. 2019, 203, 2076–2087. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Yasuda, K.; Sato, A.; Nishimura, K.; Chida, K.; Hayakawa, H. Phospholipid analysis of alveolar macrophages and bronchoalveolar lavage fluid following bleomycin administration to rabbits. Lung 1994, 172, 91–102. [Google Scholar] [CrossRef]
- Romero, F.; Shah, D.; Duong, M.; Penn, R.B.; Fessler, M.B.; Madenspacher, J.; Stafstrom, W.; Kavuru, M.; Lu, B.; Kallen, C.B.; et al. A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 74–86. [Google Scholar] [CrossRef]
- Kim, K.K.; Dotson, M.R.; Agarwal, M.; Yang, J.; Bradley, P.B.; Subbotina, N.; Osterholzer, J.J.; Sisson, T.H. Efferocytosis of apoptotic alveolar epithelial cells is sufficient to initiate lung fibrosis. Cell Death Dis. 2018, 9, 1056. [Google Scholar] [CrossRef]
- Parks, B.W.; Black, L.L.; Zimmerman, K.A.; Metz, A.E.; Steele, C.; Murphy-Ullrich, J.E.; Kabarowski, J.H. CD36, but not G2A, modulates efferocytosis, inflammation, and fibrosis following bleomycin-induced lung injury. J. Lipid Res. 2013, 54, 1114–1123. [Google Scholar] [CrossRef]
- Bradley, P.; Subbotina, N.; Dotson, M.; Teitz-Tennenbaum, S.; Roussey, J.; Osterholzer, J.; Sisson, T. CD36 Scavenger Receptor Promotes Pulmonary Fibrosis in Response to Oropharyngeal Administration of Oxidized Phospholipid. In C72. Pulmonary Fibrosis: Mechanisms and Models. Presented at American Thoracic Society 2018 Conference, San Diego, CA, USA, 18–23 May 2018; p. A5759. [Google Scholar]
- Boorjian, S. Commentary on “Conditional survival of patients with metastatic renal cell carcinoma treated with VEGF-targeted therapy: A population-based study.” Harshman LC, Xie W, Bjarnason GA, Knox JJ, MacKenzie M, Wood L, Srinivas S, Vaishampayan UN, Tan MH, Rha SY, Donskov F, Agarwal N, Kollmannsberger C, North S, Rini BI, Heng DY, Choueiri TK, Stanford Cancer Institute, Stanford University School of Medicine, Stanford, CA: Lancet Oncol 2012;13(9):927-35 (Epub 2012 Aug 8). Urol. Oncol. 2013, 31, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Osterholzer, J.J.; Subbotina, N.; Dotson, M.; Teitz-Tennenbaum, S.; Sisson, T. The Plasminogen Activator Inhibitor-1 Promotes Pulmonary Fibrosis In Response To Intratracheal Administration Of Oxidized Phospholipid. In C78. Fibrosis: Mediators and Modulators. Presented at American Thoracic Society 2017 International Conference, Washington, DC, USA, 19–24 May 2017; p. A6438. [Google Scholar]







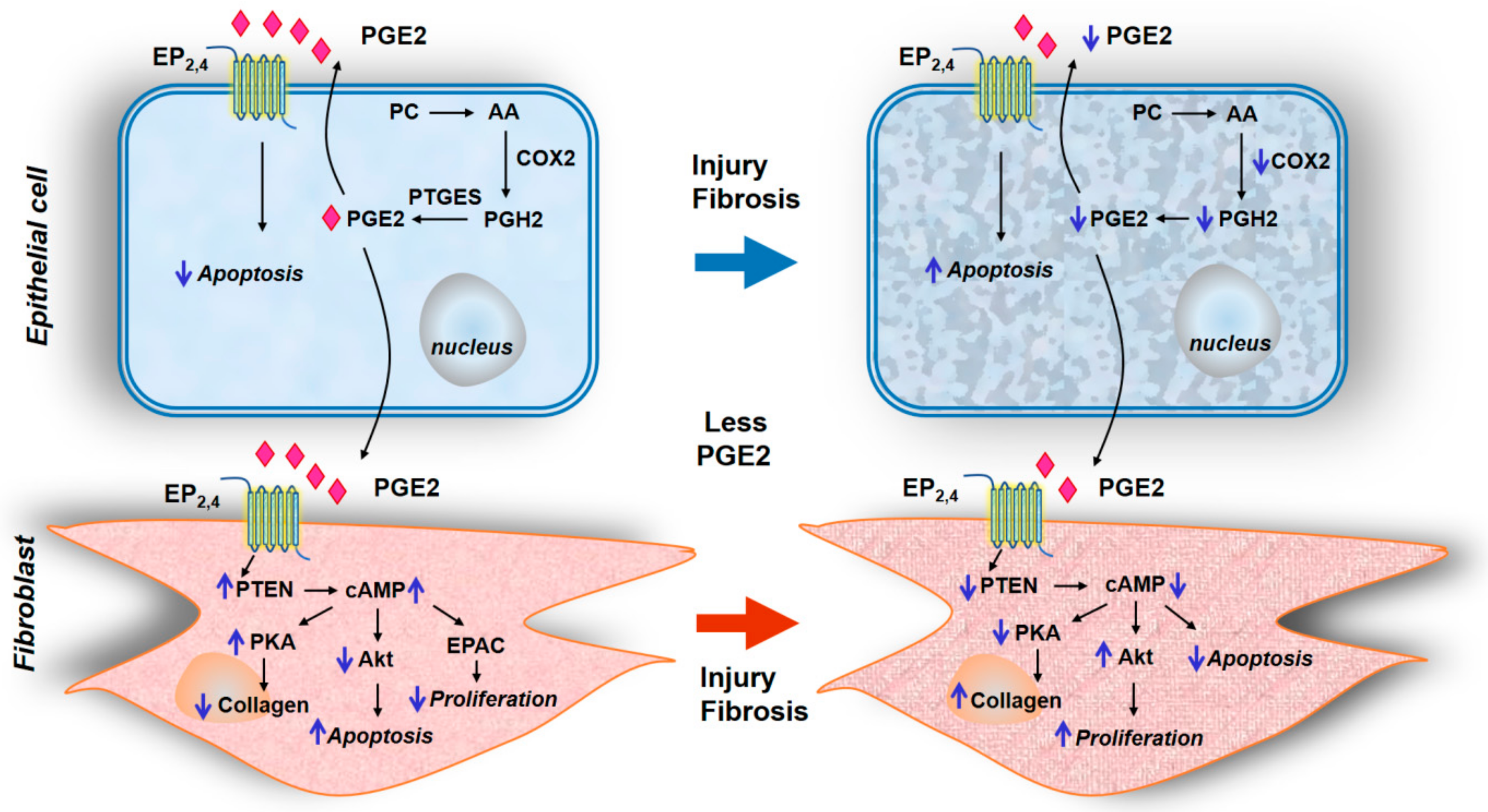{kind=link}
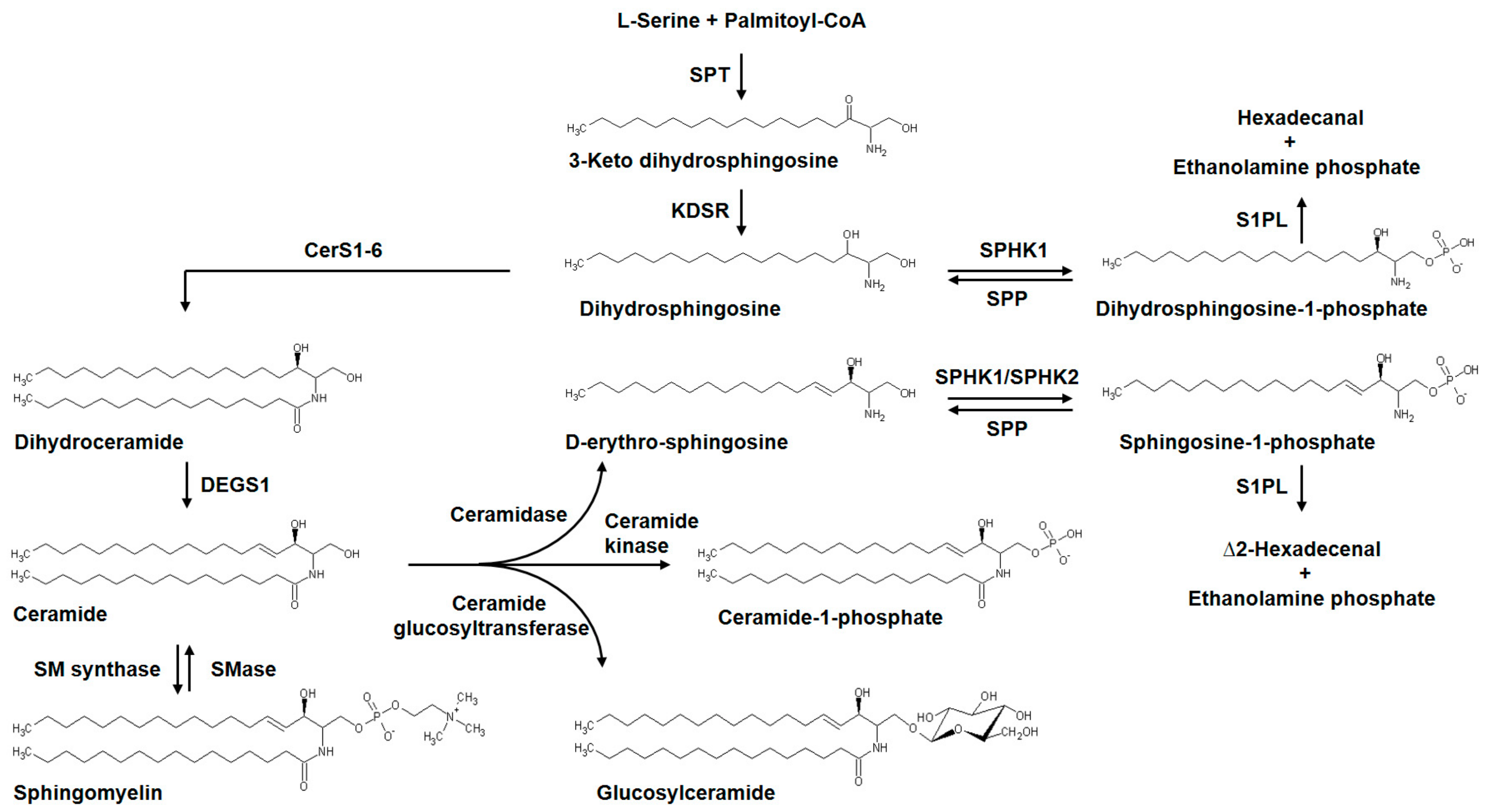{kind=link}
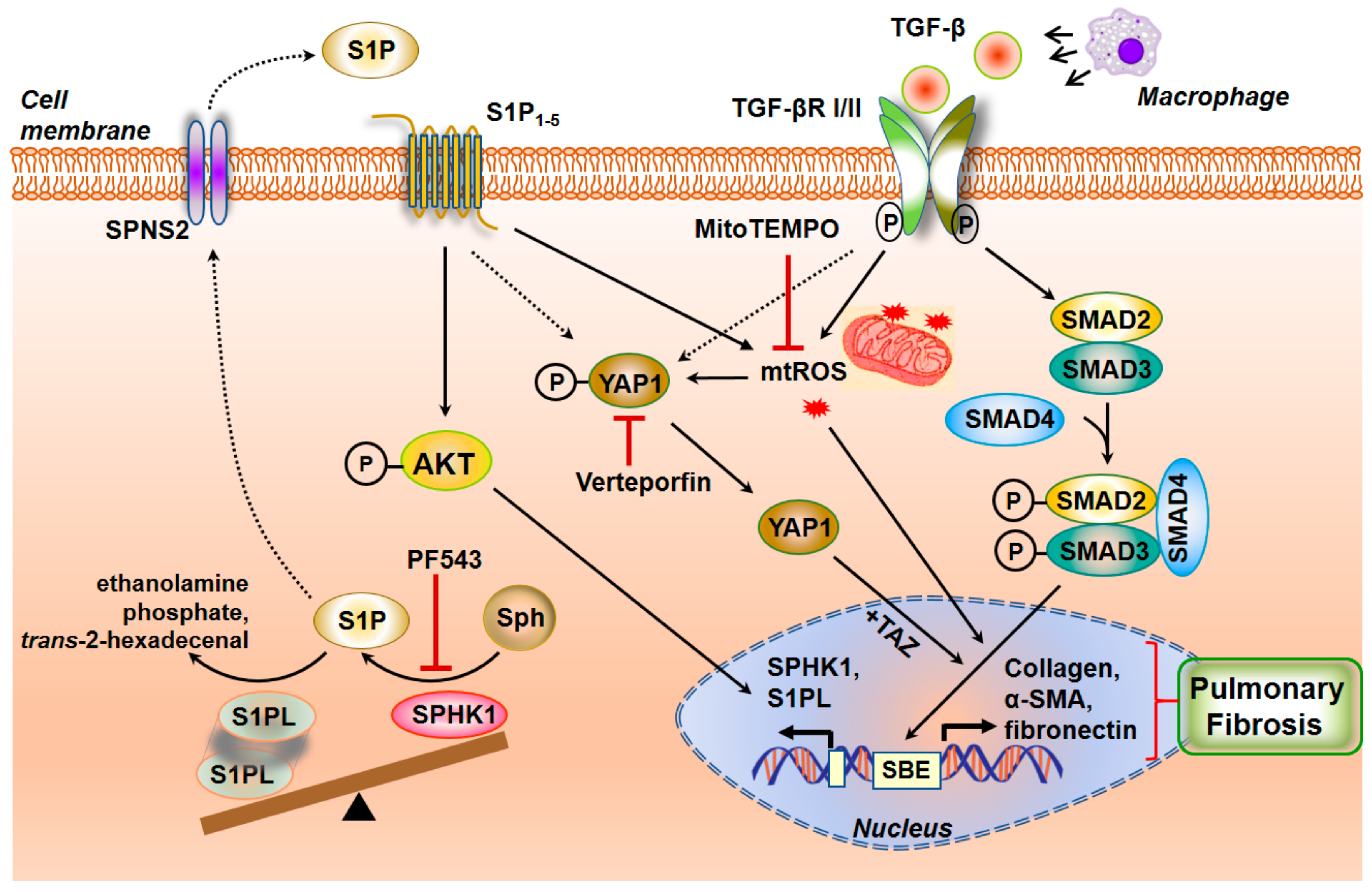{kind=link}
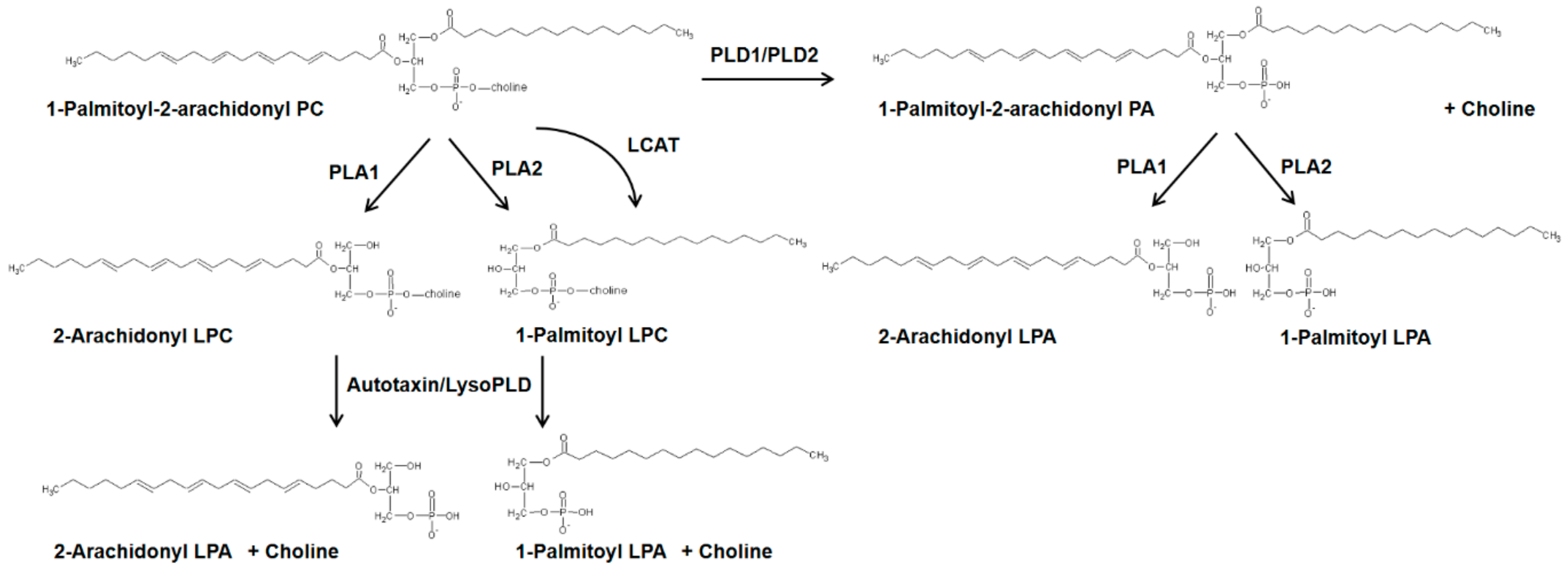{kind=link}
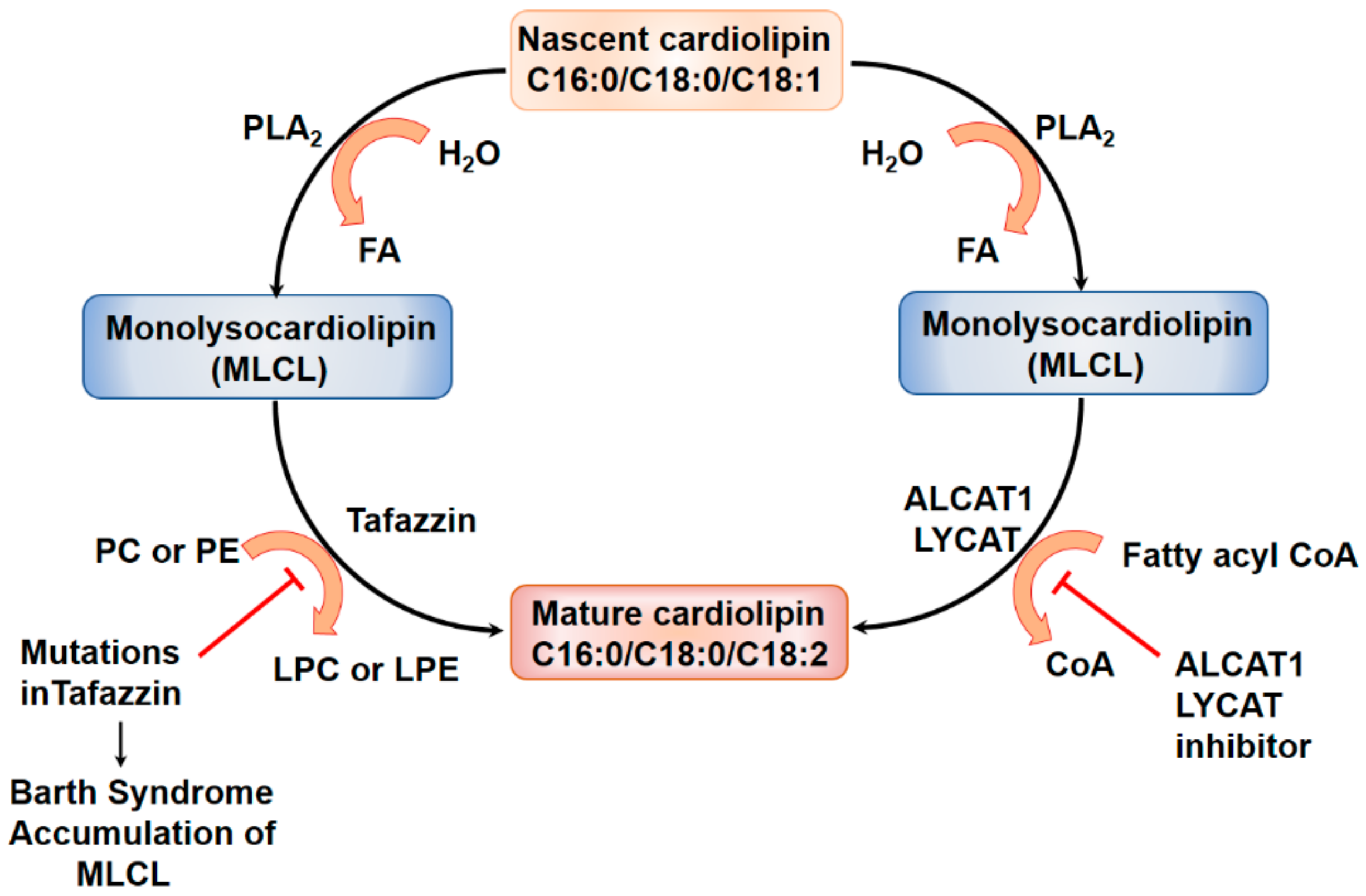{kind=link}
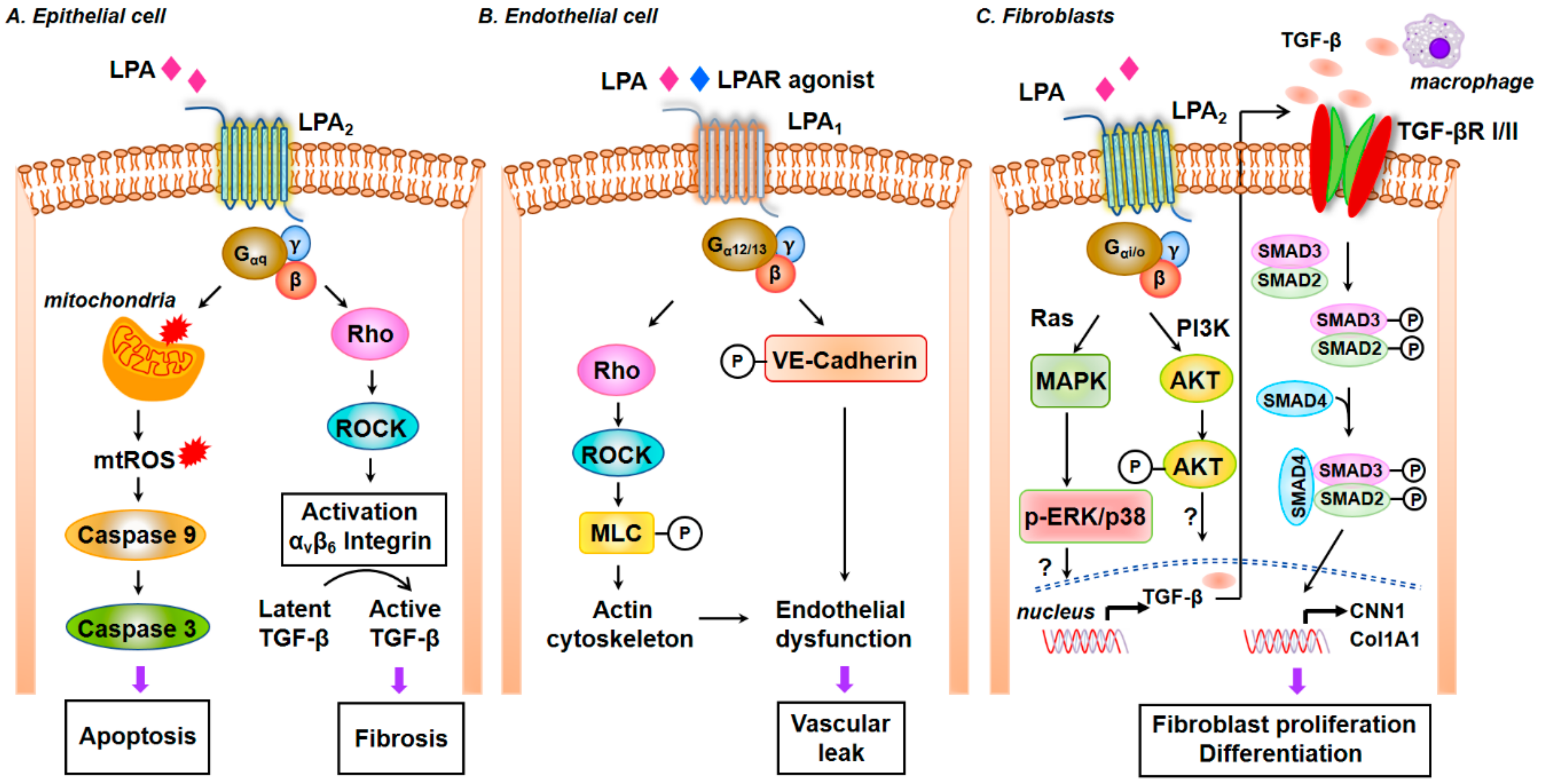{kind=link}
| Drug | Target Specificity | Identifier a | Stage |
|---|---|---|---|
| BMS-986278 | LPA1 receptor antagonist | NCT04308681 | Phase II |
| BMS-986020 | LPA1 receptor antagonist | NCT01766817 | Phase II |
| 18F-BMS-986327 | LPA1 ligand for PET | NCT04069143 | Phase I |
| Tipelukast/MN-001 | Orphan drug (LT receptor, PDE inhibitor, 5-LO pathway) | NCT02503657 | Phase II |
| GLPG1690 | ATX inhibitor | NCT03733444 | Phase III |
| BBT-877 | ATX inhibitor | NCT03830125 | Phase I |
| X-165 | ATX inhibitor | Preclinical, IND | - |
| GSK2126458 | PI3K/mTOR inhibitor | NCT01725139 | Phase I |
| Sirolimus | mTOR inhibitor | NCT01462006 | Phase I |
| HEC68498 | PI3K/mTOR inhibitor | NCT03502902 | Phase I |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suryadevara, V.; Ramchandran, R.; Kamp, D.W.; Natarajan, V. Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 4257. https://doi.org/10.3390/ijms21124257
Suryadevara V, Ramchandran R, Kamp DW, Natarajan V. Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways. International Journal of Molecular Sciences. 2020; 21(12):4257. https://doi.org/10.3390/ijms21124257
Chicago/Turabian StyleSuryadevara, Vidyani, Ramaswamy Ramchandran, David W. Kamp, and Viswanathan Natarajan. 2020. "Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways" International Journal of Molecular Sciences 21, no. 12: 4257. https://doi.org/10.3390/ijms21124257
APA StyleSuryadevara, V., Ramchandran, R., Kamp, D. W., & Natarajan, V. (2020). Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways. International Journal of Molecular Sciences, 21(12), 4257. https://doi.org/10.3390/ijms21124257