Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals




Abstract

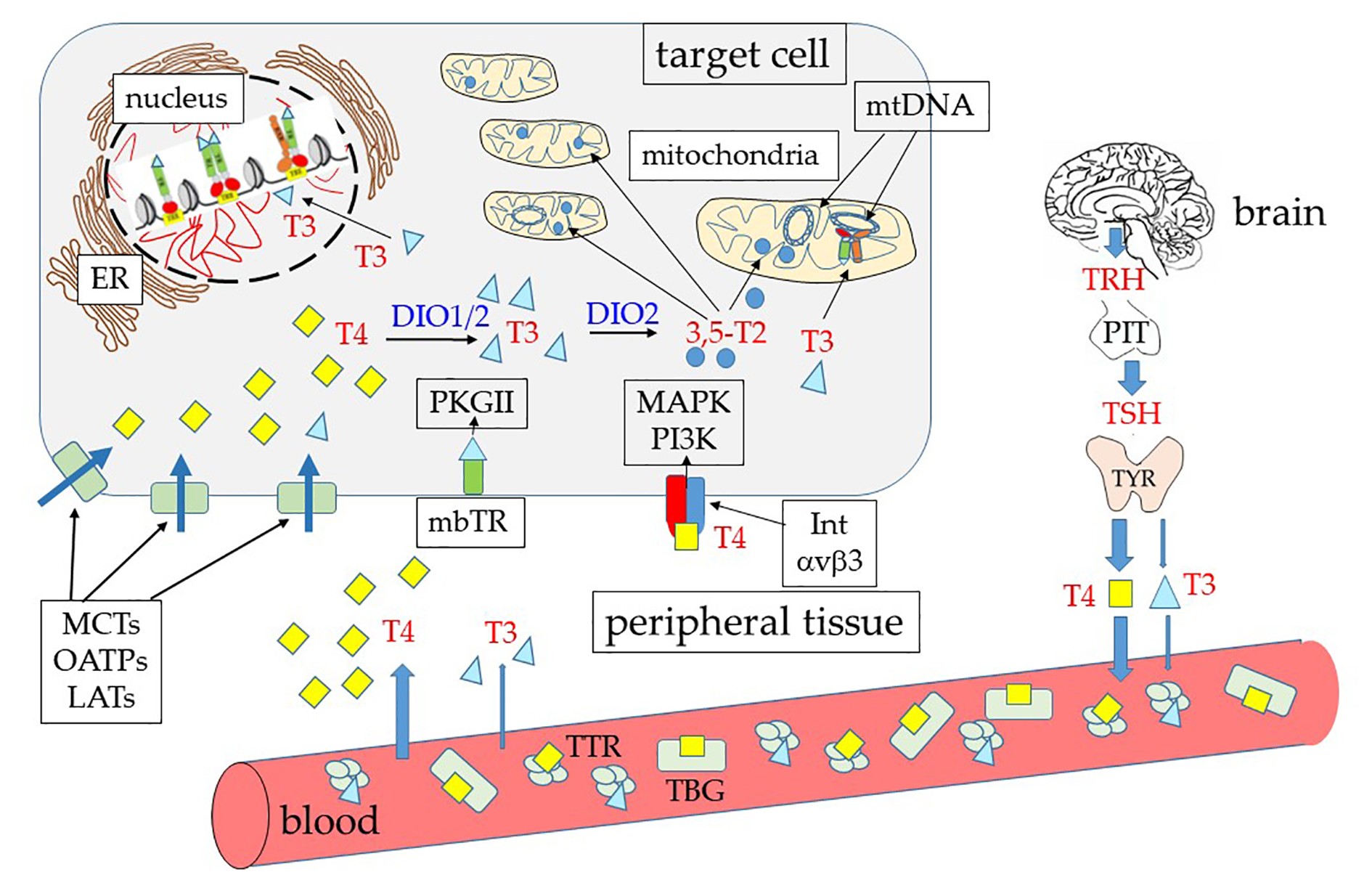{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
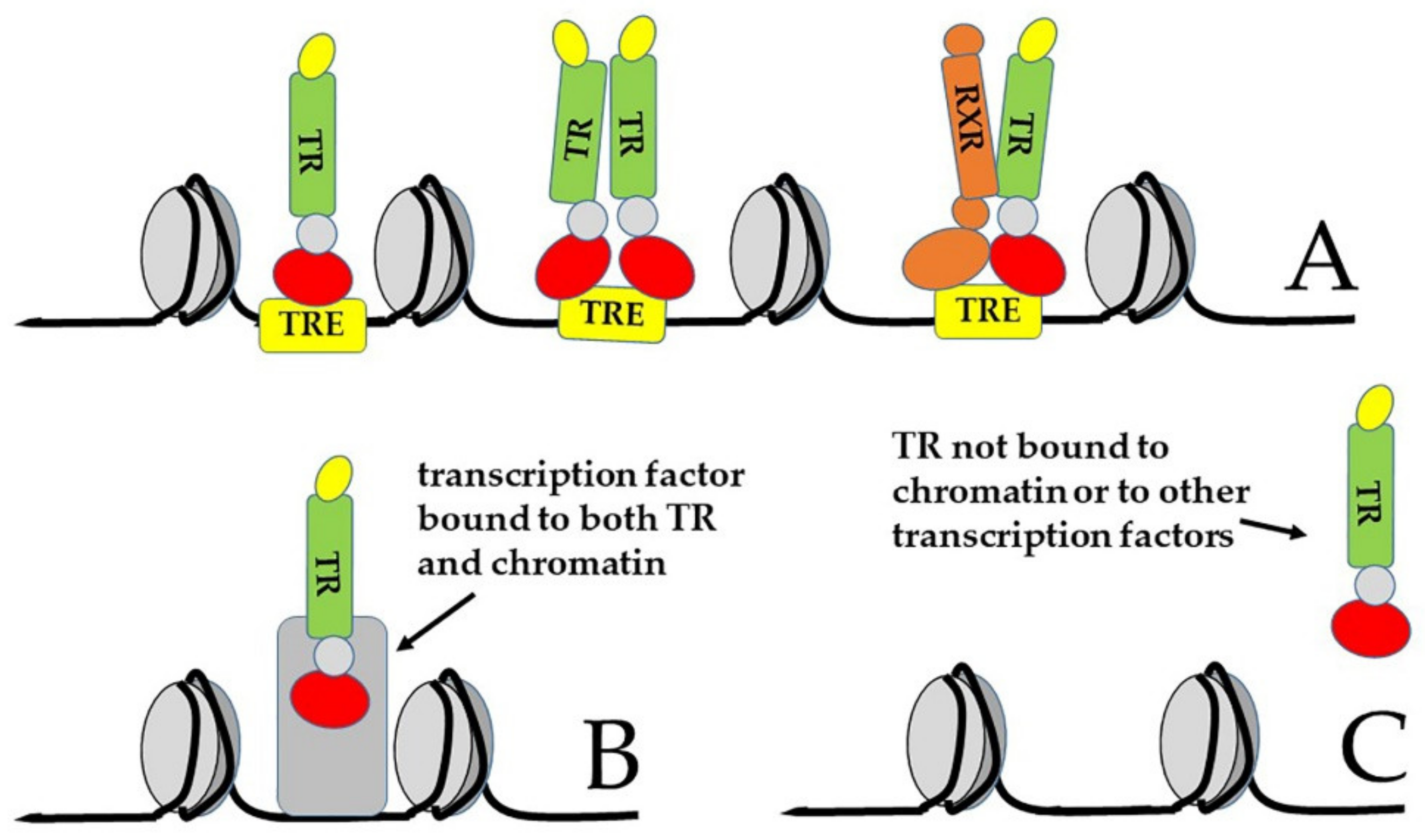{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Thyroid Hormone (TH) Metabolism
2.1. TH Synthesis, Release, and Transport to the Target Cells
2.2. Activation and Inactivation of THs by Deiodinases
3. Cellular Mechanisms of Action of THs
3.1. Thyroid Hormone Receptors (TRs)
3.2. TH Resistance
3.3. Non-Genomic TH Action
4. The 3,5-Diodothyronine (3,5-T2) as a Hormone
4.1. The 3,5-T2 Production in the Cell and Its Effects on Rest Metabolic Rate (RMR)
4.2. Cellular Targets of 3,5-T2
4.3. The 3,5-T2 and Lipid Metabolism
4.4. Effects of T2 on Lipid Peroxidation
4.5. Effects of 3,5-T2 on Adipose Tissue
4.6. The Ins and Outs of 3,5-T2 Research
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Liegro, I.; Savettieri, G.; Cestelli, A. Cellular mechanism of action of thyroid hormones. Differentiation 1987, 35, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Flamant, F.; Baxter, J.D.; Forrest, D.; Refetoff, S.; Samuels, H.; Scanlan, T.S.; Vennström, B.; Samarut, J. International Union of Pharmacology. LIX. The pharmacology and classification of the nuclear receptor superfamily: Thyroid hormone receptors. Pharmacol. Rev. 2006, 58, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Arrojo e Drigo, R.; Fonseca, T.L.; Werneck-de-Castro, J.P.; Bianco, A.C. Role of the type 2 iodothyronine deiodinase (D2) in the control of thyroid hormone signaling. Biochim. Biophys. Acta 2013, 1830, 3956–3964. [Google Scholar] [CrossRef] [PubMed]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Raja, K.; Schweizer, U.; Mugesh, G. Chemistry and Biology in the Biosynthesis and Action of Thyroid Hormones. Angew. Chem. Int. Ed. Engl. 2016, 55, 7606–7630. [Google Scholar] [CrossRef] [PubMed]
- Vella, K.R.; Hollenberg, A.N. The actions of thyroid hormone signaling in the nucleus. Mol. Cell. Endocrinol. 2017, 458, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Maia, A.L.; Kim, B.W.; Huang, S.A.; Harney, J.W.; Larsen, P.R. Type 2 iodothyronine deiodinase is the major source of plasma T3 in euthyroid humans. J. Clin. Investig. 2005, 115, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Luongo, C.; Dentice, M.; Salvatore, D. Deiodinases and their intricate role in thyroid hormone homeostasis. Nat. Rev. Endocrinol. 2019, 15, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Galton, V.A. The isolation of thyroxine (T4), the discovery of 3,5,3’-triiodothyronine (T3), and the identification of the deiodinases that generate T3 from T4: An historical review. Endocrine 2019, 66, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lazar, M.A. The mechanism of action of thyroid hormones. Annu. Rev. Physiol. 2000, 62, 439–466. [Google Scholar] [CrossRef] [PubMed]
- Di Liegro, I. Thyroid hormones and the central nervous system of mammals (Review). Mol. Med. Rep. 2008, 1, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Anyetei-Anum, C.S.; Roggero, V.R.; Allison, L.A. Thyroid hormone receptor localization in target tissues. J. Endocrinol. 2018, 237, R19–R34. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Flamant, F.; Cheng, S.Y.; Hollenberg, A.N.; Moeller, L.C.; Samarut, J.; Wondisford, F.E.; Yen, P.M.; Refetoff, S. Thyroid Hormone Signaling Pathways: Time for a More Precise Nomenclature. Endocrinology 2017, 158, 2052–2057. [Google Scholar] [CrossRef] [PubMed]
- Cody, V.; Davis, P.J.; Davis, F.B. Molecular modeling of the thyroid hormone interactions with alpha v beta 3 integrin. Steroids 2007, 72, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Mitochondrial T3 receptor and targets. Mol. Cell. Endocrinol. 2017, 458, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Steffensen, K.R.; Bruscalupi, G.; Parini, P. Emerging role of thyroid hormone metabolites. Acta Physiol. (Oxf.) 2016, 217, 184–216. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J. The Colorful Diversity of Thyroid Hormone Metabolites. Eur. Thyroid J. 2019, 8, 115–129. [Google Scholar] [CrossRef] [PubMed]
- La Guardia, M.; Giammanco, M. Breast cancer and obesity. Panminerva Med. 2001, 42, 123–133. [Google Scholar]
- Mandviwala, T.; Khalid, U.; Deswal, A. Obesity and Cardiovascular Disease: A Risk Factor or a Risk Marker? Curr. Atheroscler. Rep. 2016, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Doehner, W.; Kalantar-Zadeh, K.; Anker, S.D. The Obesity Paradigm and Lifetime Risk of Cardiovascular Disease. JAMA Cardiol. 2018, 3, 895–896. [Google Scholar] [CrossRef] [PubMed]
- Giammanco, M.; Lantieri, L.; Leto, G.; Plescia, F.; Di Majo, D. Nutrition, obesity and hormones. J. Biol. Res. 2018, 91, 108–118. [Google Scholar] [CrossRef]
- Perini, W.; van Valkengoed, I.G.M.; Snijder, M.B.; Peters, R.J.G.; Kunst, A.E. The contribution of obesity to the population burden of high metabolic cardiovascular risk among different ethnic groups. The HELIUS study. Eur. J. Public Health 2020. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- De Pauw, A.; Tejerina, S.; Raes, M.; Keijer, J.; Arnould, T. Mitochondrial dysfunction in adipocyte dedifferentiation and systemic metabolic alterations. Am. J. Pathol. 2009, 175, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Paglialunga, S.; Ludzki, A.; Root-McCaig, J.; Holloway, G.P. In adipose tissue, increased mitochondrial emission of reactive oxygen species is important for short-term high-fat diet-induced insulin resistance in mice. Diabetologia 2015, 58, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Khemka, V.K.; Chatterjee, G.; Ganguly, A.; Mukhopadhyay, S.; Chakrabarti, S. Enhanced ROS production and oxidative damage in subcutaneous white adipose tissue mitochondria in obese and type 2 diabetes subjects. Mol. Cell. Biochem. 2015, 399, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Nagata, N.; Ota, T. Impact of Glucoraphanin-Mediated Activation of Nrf2 on Non-Alcoholic Fatty Liver Disease with a Focus on Mitochondrial Dysfunction. Int. J. Mol. Sci. 2019, 20, E5920. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Lanaspa, M.A.; Kuwabara, M.; Orlicky, D.J.; Cicerchi, C.; Bales, E.; Garcia, G.E.; Roncal-Jimenez, C.A.; Sato, Y.; Johnson, R.J. Obesity causes renal mitochondrial dysfunction and energy imbalance and accelerates chronic kidney disease in mice. Am. J. Physiol. Renal Physiol. 2019, 317, F941–F948. [Google Scholar] [CrossRef]
- Obregon, M.J. Adipose tissues and thyroid hormones. Front. Physiol. 2014, 5, 479. [Google Scholar] [CrossRef]
- Vale, C.; Neves, J.S.; von Hafe, M.; Borges-Canha, M.; Leite-Moreira, A. The Role of Thyroid Hormones in Heart Failure. Cardiovasc. Drugs Ther. 2019, 33, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Barreto-Chaves, M.L.; Senger, N.; Fevereiro, M.; Parletta, A.C.; Takano, A. Impact of hyperthyroidism on cardiac hypertrophy. Endocr. Connect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fekete, C.; Lechan, R.M. Central regulation of hypothalamic-pituitary-thyroid axis under physiological and pathophysiological conditions. Endocr. Rev. 2014, 35, 159–194. [Google Scholar] [CrossRef] [PubMed]
- Ortiga-Carvalho, T.M.; Chiamolera, M.I.; Pazos-Moura, C.C.; Wondisford, F.E. Hypothalamus-Pituitary-Thyroid Axis. Compr. Physiol. 2016, 6, 1387–1428. [Google Scholar] [CrossRef]
- Mendoza, A.; Hollenberg, A.N. New insights into thyroid hormone action. Pharmacol. Ther. 2017, 173, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Fagman, H. Development of the thyroid gland. Development 2017, 144, 2123–2140. [Google Scholar] [CrossRef]
- Chiamolera, M.I.; Wondisford, F.E. Minireview: Thyrotropin-releasing hormone and the thyroid hormone feedback mechanism. Endocrinology 2009, 150, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, R.R. Iodine metabolism and thyroid physiology: Current concepts. Thyroid 1997, 7, 177–181. [Google Scholar] [CrossRef]
- Darrouzet, E.; Lindenthal, S.; Marcellin, D.; Pellequer, J.L.; Pourcher, T. The sodium/iodide symporter: State of the art of its molecular characterization. Biochim. Biophys. Acta 2014, 1838 (1 Pt B), 244–253. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.P.; Dupuy, C. Thyroid hormone biosynthesis and release. Mol. Cell. Endocrinol. 2017, 458, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.T. Fluoride Exposure Induces Inhibition of Sodium/Iodide Symporter (NIS) Contributing to Impaired Iodine Absorption and Iodine Deficiency: Molecular Mechanisms of Inhibition and Implications for Public Health. Int. J. Environ. Res. Public Health 2019, 16, E1086. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.T.; Janssen, O.E. Directional thyroid hormone distribution via the blood stream to target sites. Mol. Cell. Endocrinol. 2017, 458, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Rabah, S.A.; Gowan, I.L.; Pagnin, M.; Osman, N.; Richardson, S.J. Thyroid Hormone Distributor Proteins During Development in Vertebrates. Front. Endocrinol. (Lausanne) 2019, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- McLean, T.R.; Rank, M.M.; Smooker, P.M.; Richardson, S.J. Evolution of thyroid hormone distributor proteins. Mol. Cell. Endocrinol. 2017, 459, 43–52. [Google Scholar] [CrossRef] [PubMed]
- de Souza, E.C.; Dias, G.R.; Cardoso, R.C.; Lima, L.P.; Fortunato, R.S.; Visser, T.J.; Vaisman, M.; Ferreira, A.C.; Carvalho, D.P. MCT8 is Downregulated by Short Time Iodine Overload in the Thyroid Gland of Rats. Horm. Metab. Res. 2015, 47, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.C.; Dumitrescu, A.; Gereben, B.; Ribeiro, M.O.; Fonseca, T.L.; Fernandes, G.W.; Bocco, B.M.L.C. Paradigms of Dynamic Control of Thyroid Hormone Signaling. Endocr. Rev. 2019, 40, 1000–1047. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, S.; van Geest, F.S.; Peeters, R.P.; Heuer, H.; Visser, W.E. Thyroid Hormone Transporters. Endocr. Rev. 2020, 41, bnz008. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, A.M.; Liao, X.H.; Best, T.B.; Brockmann, K.; Refetoff, S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am. J. Hum. Genet. 2004, 74, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Strømme, P.; Groeneweg, S.; Lima de Souza, E.C.; Zevenbergen, C.; Torgersbråten, A.; Holmgren, A.; Gurcan, E.; Meima, M.E.; Peeters, R.P.; Visser, W.E.; et al. Mutated Thyroid Hormone Transporter OATP1C1 Associates with Severe Brain Hypometabolism and Juvenile Neurodegeneration. Thyroid 2018, 28, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Steegborn, C.; Schweizer, U. Structure and Mechanism of Iodothyronine Deiodinases - What We Know, What We Don’t Know, and What Would Be Nice to Know. Exp. Clin. Endocrinol. Diabetes 2019. [Google Scholar] [CrossRef] [PubMed]
- Heuer, H. The importance of thyroid hormone transporters for brain development and function. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Stohn, J.P. The Type 3 Deiodinase: Epigenetic Control of Brain Thyroid Hormone Action and Neurological Function. Int. J. Mol. Sci. 2018, 19, E1804. [Google Scholar] [CrossRef] [PubMed]
- Fekete, C.; Lechan, R.M. Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: Role of neuronal afferents and type 2 deiodinase. Front. Neuroendocrinol. 2007, 28, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Fonseca, T.L.; Bocco, B.M.L.C.; Fernandes, G.W.; McAninch, E.A.; Bolin, A.P.; Da Conceição, R.R.; Werneck-de-Castro, J.P.; Ignacio, D.L.; Egri, P.; et al. Type 2 deiodinase polymorphism causes ER stress and hypothyroidism in the brain. J. Clin. Investig. 2019, 129, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, J.; Han, W.; Jiang, Y.; Peng, S.; Shan, Z.; Teng, W. The Type 2 Deiodinase TR92Ala Polymorphism Is Associated with Worse Glycemic Control in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. J. Diabetes Res. 2016, 2016, 5928726. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.M.; Pessin, J.E. Identification of a skeletal muscle-specific regulatory domain in the rat GLUT4/muscle-fat gene. J. Biol. Chem. 1993, 268, 21021–21027. [Google Scholar] [PubMed]
- Louzada, R.A.; Carvalho, D.P. Similarities and Differences in the Peripheral Actions of Thyroid Hormones and Their Metabolites. Front. Endocrinol. (Lausanne) 2018, 9, 394. [Google Scholar] [CrossRef] [PubMed]
- Galton, V.A.; Schneider, M.J.; Clark, A.S.; St Germain, D.L. Life without thyroxine to 3,5,3’-triiodothyronine conversion: Studies in mice devoid of the 5’-deiodinases. Endocrinology 2009, 150, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, J.H.; Koerner, D.; Schwartz, H.L.; Surks, M.I. Specific nuclear triiodothyronine binding sites in rat liver and kidney. J. Clin. Endocrinol. Metab. 1972, 35, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Schadlow, A.R.; Surks, M.I.; Schwartz, H.L.; Oppenheimer, J.H. Specific triiodothyronine binding sites in the anterior pituitary of the rat. Science 1972, 176, 1252–1254. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nature 1986, 320, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Sap, J.; Muñoz, A.; Damm, K.; Goldberg, Y.; Ghysdael, J.; Leutz, A.; Beug, H.; Vennström, B. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature 1986, 324, 635–640. [Google Scholar] [CrossRef]
- Weinberger, C.; Thompson, C.C.; Ong, E.S.; Lebo, R.; Gruol, D.J.; Evans, R.M. The c-erb-A gene encodes a thyroid hormone receptor. Nature 1986, 324, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Pascual, A.; Aranda, A. Thyroid hormone receptors, cell growth and differentiation. Biochim. Biophys. Acta 2013, 1830, 3908–3916. [Google Scholar] [CrossRef] [PubMed]
- Izumo, S.; Mahdavi, V. Thyroid hormone receptor alpha isoforms generated by alternative splicing differentially activate myosin HC gene transcription. Nature 1988, 334, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.A.; Hodin, R.A.; Darling, D.S.; Chin, W.W. Identification of a rat c-erbA alpha-related protein which binds deoxyribonucleic acid but does not bind thyroid hormone. Mol. Endocrinol. 1988, 2, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, T.; Tennyson, G.E.; Nikodem, V.M. Alternative splicing generates messages encoding rat c-erbA proteins that do not bind thyroid hormone. Proc. Natl. Acad. Sci. USA 1988, 85, 5804–5808. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.A. Thyroid hormone receptors: Multiple forms, multiple possibilities. Endocr. Rev. 1993, 14, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Yen, P.M. A clinician’s guide to understanding resistance to thyroid hormone due to receptor mutations in the TRα and TRβ isoforms. Clin. Diabetes Endocrinol. 2017, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.B.; Koenig, R.J. Expression of erbA alpha and beta mRNAs in regions of adult rat brain. Mol. Cell. Endocrinol. 1990, 70, 13–20. [Google Scholar] [CrossRef]
- Castiglia, D.; Cestelli, A.; Di Liegro, C.; Bonfanti, L.; Di Liegro, I. Accumulation of different c-erbA transcripts during rat brain development and in cortical neurons cultured in a synthetic medium. Cell. Mol. Neurobiol. 1992, 12, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar] [CrossRef] [PubMed]
- McKenna, N.J.; O’Malley, B.W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 108, 465–474. [Google Scholar] [CrossRef]
- Ortiga-Carvalho, T.M.; Sidhaye, A.R.; Wondisford, F.E. Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat. Rev. Endocrinol. 2014, 10, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Yeh, C.T.; Lin, K.H. Molecular Functions of Thyroid Hormone Signaling in Regulation of Cancer Progression and Anti-Apoptosis. Int. J. Mol. Sci. 2019, 20, E4986. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Brent, G.A. Posttranslational Modification of Thyroid Hormone Nuclear Receptor by Phosphorylation. Methods Mol. Biol. 2018, 1801, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Anyetei-Anum, C.S.; Evans, R.M.; Back, A.M.; Roggero, V.R.; Allison, L.A. Acetylation modulates thyroid hormone receptor intracellular localization and intranuclear mobility. Mol. Cell. Endocrinol. 2019, 495, 110509. [Google Scholar] [CrossRef] [PubMed]
- Umesono, K.; Murakami, K.K.; Thompson, C.C.; Evans, R.M. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 1991, 65, 1255–1266. [Google Scholar] [CrossRef]
- Nascimento, A.S.; Gomes Dias, S.M.; Nunes, F.M.; Aparício, R.; Ambrosio, A.L.B.; Bleicher, L.; Figueira, A.C.M.; Santos, M.A.M.; De Oliveira Neto, M.; Fischer, H.; et al. Structural rearrangements in the thyroid hormone receptor hinge domain and their putative role in the receptor function. J. Mol. Biol. 2006, 360, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, B.J.; Lee, S.; Privalsky, M.L. Coactivator recruitment is enhanced by thyroid hormone receptor trimers. Mol. Cell. Endocrinol. 2008, 280, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Zechel, C.; Shen, X.Q.; Chen, J.Y.; Chen, Z.P.; Chambon, P.; Gronemeyer, H. The dimerization interfaces formed between the DNA binding domains of RXR, RAR and TR determine the binding specificity and polarity of the full-length receptors to direct repeats. EMBO J. 1994, 13, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-Y. Multiple mechanisms for regulation of the transcriptional activity of thyroid hormone receptors. Rev. Endocr. Metab. Disord. 2000, 1, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.A.; Berrodin, T.J.; Harding, H.P. Differential DNA binding by monomeric, homodimeric, and potentially heteromeric forms of the thyroid hormone receptor. Mol. Cell. Biol. 1991, 11, 5005–5015. [Google Scholar] [CrossRef] [PubMed]
- Velasco, L.F.; Togashi, M.; Walfish, P.G.; Pessanha, R.P.; Moura, F.N.; Barra, G.B.; Nguyen, P.; Rebong, R.; Yuan, C.; Simeoni, L.A.; et al. Thyroid hormone response element organization dictates the composition of active receptor. J. Biol. Chem. 2007, 282, 12458–12466. [Google Scholar] [CrossRef] [PubMed]
- Ramadoss, P.; Abraham, B.J.; Tsai, L.; Zhou, Y.; Costa-e-Sousa, R.H.; Ye, F.; Bilban, M.; Zhao, K.; Hollenberg, A.N. Novel mechanism of positive versus negative regulation by thyroid hormone receptor β1 (TRβ1) identified by genome-wide profiling of binding sites in mouse liver. J. Biol. Chem. 2014, 289, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.R. Cloning and characterization of two novel thyroid hormone receptor β isoforms. Mol. Cell. Biol. 2000, 20, 8329–8342. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Koenig, R.J. Gene regulation by thyroid hormones. Trends Endocrinol. Metab. 2000, 11, 207–211. [Google Scholar] [CrossRef]
- Cheng, S.-Y. Isoform-dependent action of thyroid hormone nuclear receptors: Lessons from knockin mutant mice. Steroids 2005, 70, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Roeder, R.G. Mediator-dependent nuclear receptor function. Semin. Cell. Dev. Biol. 2011, 22, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Astapova, I.; Hollenberg, A.N. The in vivo role of nuclear receptor corepressors in thyroid hormone action. Biochim. Biophys. Acta 2013, 1830, 3876–3881. [Google Scholar] [CrossRef] [PubMed]
- Vella, K.R.; Ramadoss, P.; Costa-E-Sousa, R.H.; Astapova, I.; Ye, F.D.; Holtz, K.A.; Harris, J.C.; Hollenberg, A.N. Thyroid hormone signaling in vivo requires a balance between coactivators and corepressors. Mol. Cell. Biol. 2014, 34, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, K.; Hermanson, O.; Onami, T.M.; Gleiberman, A.S.; Lunyak, V.; McEvilly, R.J.; Kurokawa, R.; Kumar, V.; Liu, F.; Seto, E.; et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell 2000, 102, 753–763. [Google Scholar] [CrossRef]
- Nevado, J.; Tenbaum, S.P.; Aranda, A. hSrb7, an essential human Mediator component, acts as a coactivator for the thyroid hormone receptor. Mol. Cell. Endocrinol. 2004, 222, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Heinrich, R. Biological control TRough regulated transcriptional coactivators. Cell 2004, 119, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; O’Malley, B.W. Coregulator function: A key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 2004, 25, 45–71. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.G.; Lunyak, V.V.; Glass, C.K. Sensors and signals: A coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006, 20, 1405–1428. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Auwerx, J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007, 17, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Spencer, T.E.; Jenster, G.; Burcin, M.M.; Allis, C.D.; Zhou, J.; Mizzen, C.A.; McKenna, N.J.; Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 1997, 389, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Oetting, A.; Yen, P.M. New insights into thyroid hormone action. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Selvi, R.B.; Kundu, T.K. Reversible acetylation of chromatin: Implication in regulation of gene expression, disease and therapeutics. Biotechnol. J. 2009, 4, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Imhof, A.; Yang, X.J.; Ogryzko, V.V.; Nakatani, Y.; Wolffe, A.P.; Ge, H. Acetylation of general transcription factors by histone acetyltransferases. Curr. Biol. 1997, 7, 689–692. [Google Scholar] [CrossRef]
- Grøntved, L.; Waterfall, J.J.; Kim, D.W.; Baek, S.; Sung, M.H.; Zhao, L.; Park, J.W.; Nielsen, R.; Walker, R.L.; Zhu, Y.J.; et al. Transcriptional activation by the thyroid hormone receptor TRough ligand-dependent receptor recruitment and chromatin remodelling. Nat. Commun. 2015, 6, 7048. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, H.; Sasaki, S.; Matsushita, A.; Ohba, K.; Matsunaga, H.; Misawa, H.; Oki, Y.; Ishizuka, K.; Nakamura, H.; Suda, T. Essential role of TEA domain transcription factors in the negative regulation of the MYH 7 gene by thyroid hormone and its receptors. PLoS ONE 2014, 9, e88610. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Matsushita, A.; Sasaki, S.; Misawa, H.; Nishiyama, K.; Kashiwabara, Y.; Nakamura, H. Thyroid-hormone-dependent negative regulation of thyrotropin beta gene by thyroid hormone receptors: Study with a new experimental system using CV1 cells. Biochem. J. 2004, 378, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, J.M. To bind or not to bind—How to down-regulate target genes by liganded thyroid hormone receptor? Thyroid Res. 2008, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Kashiwabara, Y.; Sasaki, S.; Matsushita, A.; Nagayama, K.; Ohba, K.; Iwaki, H.; Matsunaga, H.; Suzuki, S.; Misawa, H.; Ishizuka, K.; et al. Functions of PIT1 in GATA2-dependent transactivation of the thyrotropin beta promoter. J. Mol. Endocrinol. 2009, 42, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, T.L.; Correa-Medina, M.; Campos, M.P.; Wittmann, G.; Werneck-de-Castro, J.P.; Arrojo e Drigo, R.; Mora-Garzon, M.; Ueta, C.B.; Caicedo, A.; Fekete, C.; et al. Coordination of hypothalamic and pituitary T3 production regulates TSH expression. J. Clin. Investig. 2013, 123, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, H.; Sasaki, S.; Suzuki, S.; Matsushita, A.; Nakamura, H.; Nakamura, H.M.; Hirahara, N.; Kuroda, G.; Iwaki, H.; Ohba, K.; et al. Essential Role of GATA2 in the Negative Regulation of Type 2 Deiodinase Gene by Liganded Thyroid Hormone Receptor β2 in Thyrotroph. PLoS ONE 2015, 10, e0142400. [Google Scholar] [CrossRef] [PubMed]
- Lazcano, I.; Hernández-Puga, G.; Robles, J.P.; Orozco, A. Alternative ligands for thyroid hormone receptors. Mol. Cell. Endocrinol. 2019, 493, 110448. [Google Scholar] [CrossRef] [PubMed]
- Ayers, S.; Switnicki, M.P.; Angajala, A.; Lammel, J.; Arumanayagam, A.S.; Webb, P. Genome-wide binding patterns of thyroid hormone receptor beta. PLoS ONE 2014, 9, e81186. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, H.; Schwappacher, R.; Joshua, J.; Zhuang, S.; Scott, B.T.; Klos, M.; Casteel, D.E.; Frangos, J.A.; Dillmann, W.; Boss, G.R.; et al. Nongenomic thyroid hormone signaling occurs through a plasma membrane-localized receptor. Sci. Signal. 2014, 7, ra48. [Google Scholar] [CrossRef] [PubMed]
- Roggero, V.R.; Zhang, J.; Parente, L.E.; Doshi, Y.; Dziedzic, R.C.; McGregor, E.L.; Varjabedian, A.D.; Schad, S.E.; Bondzi, C.; Allison, L.A. Nuclear import of the thyroid hormone receptor α1 is mediated by importin 7, importin β1, and adaptor importin α1. Mol. Cell. Endocrinol. 2016, 419, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.S.; Dziedzic, R.C.; Nelson, H.N.; Stern, M.E.; Roggero, V.R.; Bondzi, C.; Allison, L.A. Multiple exportins influence thyroid hormone receptor localization. Mol. Cell. Endocrinol. 2015, 411, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Roggero, V.R.; Allison, L.A. Nuclear Import and Export of the Thyroid Hormone Receptor. Vitam. Horm. 2018, 106, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Brent, G.A. Posttranslational Modification of Thyroid Hormone Nuclear Receptor by Sumoylation. Methods Mol. Biol. 2018, 1801, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, M.; Fayad, T.; Langlois, M.F. Degradation of thyroid hormone receptor beta 1: Existence of stable and unstable forms. Thyroid 2011, 21, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.; Chatterjee, K. Resistance to thyroid hormone due to defective thyroid receptor alpha. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Boers, M.E.; Pazos-Moura, C.; Moura, E.; Kaulbach, H.; Zakaria, M.; Lowell, B.; Radovick, S.; Liberman, M.C.; Wondisford, F. Divergent roles for thyroid hormone receptor beta isoforms in the endocrine axis and auditory system. J. Clin. Investig. 1999, 104, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Refetoff, S.; DeWind, L.T.; DeGroot, L.J. Familial syndrome combining deaf-mutism, stuppled epiphyses, goiter and abnormally high PBI: Possible target organ refractoriness to thyroid hormone. J. Clin. Endocrinol. Metab. 1967, 27, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Forrest, D.; Hanebuth, E.; Smeyne, R.J.; Everds, N.; Stewart, C.L.; Wehner, J.M.; Curran, T. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor beta: Evidence for tissue-specific modulation of receptor function. EMBO J. 1996, 15, 3006–3015. [Google Scholar] [CrossRef] [PubMed]
- Refetoff, S.; Bassett, J.H.; Beck-Peccoz, P.; Bernal, J.; Brent, G.; Chatterjee, K.; De Groot, L.J.; Dumitrescu, A.M.; Jameson, J.L.; Kopp, P.A.; et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. Thyroid 2014, 24, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, T.; Jameson, J.L. Thyroid hormone receptor dimerization is required for dominant negative inhibition by mutations that cause thyroid hormone resistance. J. Biol. Chem. 1993, 268, 15766–15771. [Google Scholar] [PubMed]
- Collingwood, T.N.; Wagner, R.; Matthews, C.H.; Clifton-Bligh, R.J.; Gurnell, M.; Rajanayagam, O.; Agostini, M.; Fletterick, R.J.; Beck-Peccoz, P.; Reinhardt, W.; et al. A role for helix 3 of the TRbeta ligand-binding domain in coactivator recruitment identified by characterization of a third cluster of mutations in resistance to thyroid hormone. EMBO J. 1998, 17, 4760–4770. [Google Scholar] [CrossRef]
- Yen, P.M. Molecular basis of resistance to thyroid hormone. Trends Endocrinol. Metab. 2003, 14, 327–333. [Google Scholar] [CrossRef]
- Nagaya, T.; Madison, L.D.; Jameson, J.L. Thyroid hormone receptor mutants that cause resistance to thyroid hormone. Evidence for receptor competition for DNA sequences in target genes. J. Biol. Chem. 1992, 267, 13014–13019. [Google Scholar]
- Fozzatti, L.; Kim, D.W.; Park, J.W.; Willingham, M.C.; Hollenberg, A.N.; Cheng, S.Y. Nuclear receptor corepressor (NCOR1) regulates in vivo actions of a mutated thyroid hormone receptor α. Proc. Natl. Acad. Sci. USA 2013, 110, 7850–7855. [Google Scholar] [CrossRef]
- Kim, D.W.; Park, J.W.; Willingham, M.C.; Cheng, S.Y. A histone deacetylase inhibitor improves hypothyroidism caused by a TRα1 mutant. Hum. Mol. Genet. 2014, 23, 2651–2664. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, B.; Shetty, S.; Butterfield, N.C.; Logan, J.G.; Han, C.R.; Zhu, X.; Astapova, I.; Hollenberg, A.N.; Cheng, S.Y.; Bassett, J.H.D.; et al. Genetic and Pharmacological Targeting of Transcriptional Repression in Resistance to Thyroid Hormone Alpha. Thyroid 2019, 29, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hayashi, Y.; Tsucamoto, Y.; Okamoto, Y. The Mutant Thyroid Hormone Receptor Beta R320P Causes Syndrome of Resistance to Thyroid Hormone. Case Rep. Endocrinol. 2018, 2018, 4081769. [Google Scholar] [CrossRef] [PubMed]
- Refetoff, S. Resistance to thyroid hormone: One of several defects causing reduced sensitivity to thyroid hormone. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lv, H. Identification of a novel mutation in the thyroid hormone receptor β gene that causes thyroid hormone resistance syndrome: A case report. Mol. Med. Rep. 2019, 20, 4683–4687. [Google Scholar] [CrossRef] [PubMed]
- Bochukova, E.; Schoenmakers, N.; Agostini, M.; Schoenmakers, E.; Rajanayagam, O.; Keogh, J.M.; Henning, E.; Reinemund, J.; Gevers, E.; Sarri, M.; et al. A mutation in the thyroid hormone receptor alpha gene. N. Engl. J. Med. 2012, 366, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Demir, K.; van Gucht, A.L.; Buyukinan, M.; Catli, G.; Ayhan, Y.; Bas, V.N.; Dundar, B.; Ozkan, B.; Meima, M.E.; Visser, W.E.; et al. Diverse genotypes and phenotypes of three novel thyroid hormone receptor-alpha mutations. J. Clin. Endocrinol. Metab. 2016, 101, 2945–2954. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yu, M.; Lian, X. Resistance to thyroid hormone alpha, revelation of basic study to clinical consequences. J. Pediatr. Endocrinol. Metab. 2016, 29, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wu, H.; Xie, R.; Wang, F.; Chen, T.; Chen, X.; Wang, X.; Flamant, F.; Chen, L. New Case of Thyroid Hormone Resistance α Caused by a Mutation of THRA /TRα1. J. Endocr. Soc. 2019, 3, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Le Maire, A.; Bouhours-Nouet, N.; Soamalala, J.; Delphine, M.P.; Paloni, M.; Guee, L.; Heron, D.; Mignot, C.; Illouz, F.; Joubert, F.; et al. Two novel cases of resistance to thyroid hormone due to THRA mutation (RTHα). Thyroid 2020. [Google Scholar] [CrossRef] [PubMed]
- Krieger, T.G.; Moran, C.M.; Frangini, A.; Visser, W.E.; Schoenmakers, E.; Muntoni, F.; Clark, C.A.; Gadian, D.; Chong, W.K.; Kuczynski, A.; et al. Mutations in thyroid hormone receptor α1 cause premature neurogenesis and progenitor cell depletion in human cortical development. Proc. Natl. Acad. Sci. USA 2019, 116, 22754–22763. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.; Schoenmakers, N.; Agostini, M.; Schoenmakers, E.; Offiah, A.; Kydd, A.; Kahaly, G.; Mohr-Kahaly, S.; Rajanayagam, O.; Lyons, G.; et al. An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor α. J. Clin. Endocrinol. Metab. 2013, 98, 4254–4261. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.; Merico, D.; Cao, H.; Pellecchia, G.; Alipanahi, B.; Thiruvahindrapuram, B.; Tong, X.; Sun, Y.; Cao, D.; Zhang, T.; et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom. Med. 2016, 1, 160271–1602710. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Shieh, H.Y.; Chen, S.L.; Hsu, H.C. Expression of mutant thyroid hormone nuclear receptors in human hepatocellular carcinoma cells. Mol. Carcinog. 1999, 26, 53–61. [Google Scholar] [CrossRef]
- Kamiya, Y.; Puzianowska-Kuznicka, M.; McPhie, P.; Nauman, J.; Cheng, S.Y.; Nauman, A. Expression of mutant thyroid hormone nuclear receptors is associated with human renal clear cell carcinoma. Carcinogenesis 2002, 23, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Iglesias, O.; Garcia-Silva, S.; Tenbaum, S.P.; Regadera, J.; Larcher, F.; Paramio, J.M.; Vennström, B.; Aranda, A. Thyroid hormone receptor beta1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer Res. 2009, 69, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.G.; Cheng, S.Y. Thyroid hormone receptors and cancer. Biochim. Biophys. Acta 2013, 1830, 3928–3936. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Ando, S.; Schneider, A.; Skarulis, M.C.; Sarlis, N.J.; Yen, P.M. Resistance to thyroid hormone in a patient without thyroid hormone receptor mutations. Thyroid 2002, 12, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Reutrakul, S.; Sadow, P.M.; Pannain, S.; Pohlenz, J.; Carvalho, G.A.; Macchia, P.E.; Weiss, R.E.; Refetoff, S. Search for abnormalities of nuclear corepressors, coactivators, and a coregulator in families with resistance to thyroid hormone without mutations in thyroid hormone receptor beta or alpha genes. J. Clin. Endocrinol. Metab. 2000, 85, 3609–3617. [Google Scholar] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Wojcicka, A.; Piekielko-Witkowska, A.; Kedzierska, H.; Rybicka, B.; Poplawski, P.; Boguslawska, J.; Master, A.; Nauman, A. Epigenetic regulation of thyroid hormone receptor beta in renal cancer. PLoS ONE 2014, 9, e97624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, Y.; Liu, S.; Wang, Y.C.; Guo, F.; Zhai, Q.; Jiang, J.; Ying, H. microRNA and thyroid hormone signaling in cardiac and skeletal muscle. Cell Biosci. 2017, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Nicolini, G.; Pitto, L.; Iervasi, G. Novel Insight Into the Epigenetic and Post-transcriptional Control of Cardiac Gene Expression by Thyroid Hormone. Front. Endocrinol. (Lausanne) 2019, 10, 601. [Google Scholar] [CrossRef] [PubMed]
- Segal, J. In vivo effect of 3,5,3′-triiodothyronine on calcium uptake in several tissues in the rat: Evidence for a physiological role of calcium as a first messenger for the prompt action of thyroid hormone at the level of the plasma membrane. Endocrinology 1990, 127, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Kavok, N.S.; Krasilnikova, O.A.; Babenko, N.A. Thyroxine signal transduction in liver cells involves phospholipase C and phospholipase D activation. Genomic independent action of thyroid hormone. BMC Cell. Biol. 2001, 2, 5–12. [Google Scholar] [CrossRef] [PubMed]
- D’Arezzo, S.; Incerpi, S.; Davis, F.B.; Filippo, A.; Marino, M.; Farias, R.N.; Davis, P.J. Rapid nongenomic effects of 3,5,3′-triiodo-Lthyronine on the intracellular pH of L-6 myoblasts are mediated by intracellular calcium mobilization and kinase pathways. Endocrinology 2004, 145, 5694–5703. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Davis, F.B.; Cody, V. Membrane receptors mediating thyroid hormone action. Trends Endocrinol. Metab. 2005, 16, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Farwell, A.P.; Dubord-Tomasetti, S.A.; Pietrzykowski, A.Z.; Leonard, J.L. Dynamic nongenomic actions of thyroid hormone in the developing rat brain. Endocrinology 2006, 147, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Adamson, L.F.; Ingbar, S.H. Some properties of the stimulatory effect of thyroid hormones on amino acid transport by embryonic chick bone. Endocrinology 1967, 81, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, I.D.; Simons, C.G.; Smith, G.J.; Ingbar, S.H. Cycloleucine transport in isolated rat thymocytes: In vitro effects of triiodothyronine and thyroxine. Endocrinology 1975, 96, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Segal, J.; Gordon, A. The effects of actinomycin D, puromycin, cycloheximide and hydroxyurea on 3′,5,3-triiodo-L-thyronine stimulated 2-deoxy-D-glucose uptake in chick embryo heart cells in vitro. Endocrinology 1977, 101, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Davis, F.B.; Lawrence, W.D. Thyroid hormone regulation of membrane Ca2+-ATPase activity. Endocr. Res. 1989, 15, 651–682. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.; Zhang, S.; Cao, H.J.; Tang, H.Y.; Davis, F.B.; Davis, P.J.; Lin, H.Y. Disparate effects of thyroid hormone on actions of epidermal growth factor and transforming growth factor-alpha are mediated by 3’,5’-cyclic adenosine 5’-monophosphate-dependent protein kinase II. Endocrinology 2004, 145, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Bergh, J.J.; Lin, H.Y.; Lansing, L.; Mohamed, N.S.; Davis, F.B.; Moura, S.; Davis, J.P. Integrin αVß3 contains a cell surface receptor site for thyroid hormone that is linked to activation of MAPK and induction of angiogenesis. Endocrinology 2005, 146, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- Hercbergs, A. Clinical Implications and Impact of Discovery of the Thyroid Hormone Receptor on Integrin αvβ3-A Review. Front. Endocrinol. (Lausanne) 2019, 10, 565. [Google Scholar] [CrossRef]
- Cayrol, F.; Sterle, H.A.; Diaz Flaque, M.C.; Barreiro Arcos, M.L.; Cremaschi, G.A. Non-genomic actions of thyroid hormones regulate the growth and angiogenesis of T cell lymphomas. Front. Endocrinol. 2019, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Ashur-Fabian, O.; Incerpi, S.; Mousa, S.A. Editorial: Non Genomic Actions of Thyroid Hormones in Cancer. Front. Endocrinol. (Lausanne). 2019, 10, 847. [Google Scholar] [CrossRef]
- Davis, P.J.; Tang, H.Y.; Hercbergs, A.; Lin, H.Y.; Keating, K.A.; Mousa, S.A. Bioactivity of Thyroid Hormone Analogs at Cancer Cells. Front. Endocrinol. (Lausanne) 2018, 9, 739. [Google Scholar] [CrossRef] [PubMed]
- Uzair, I.D.; Conte Grand, J.; Flamini, M.I.; Sanchez, A.M. Molecular Actions of Thyroid Hormone on Breast Cancer Cell Migration and Invasion via Cortactin/N-WASP. Front. Endocrinol. (Lausanne) 2019, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Sun, M.; Tang, H.Y.; Lin, C.; Luidens, M.K.; Mousa, S.A.; Incerpi, S.; Drusano, G.L.; Davis, F.B.; Davis, P.J. L-Thyroxine vs. 3,5,3’-triiodo-L-thyronine and cell proliferation: Activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am. J. Physiol. Cell. Physiol. 2009, 296, C980–C991. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Kambe, F.; Moeller, L.C.; Refetoff, S.; Seo, H. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol. Endocrinol. 2005, 19, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Hiroi, Y.; Kim, H.H.; Ying, H.; Furuya, F.; Huang, Z.; Simoncini, T.; Noma, K.; Ueki, K.; Nguyen, N.H.; Scanlan, T.S.; et al. Rapid nongenomic actions of thyroid hormone. Proc. Natl. Acad. Sci. USA 2006, 103, 14104–14109. [Google Scholar] [CrossRef] [PubMed]
- Bigler, J.; Eisenman, R.N. c-erbA encodes multiple proteins in chicken erythroid cells. Mol. Cell. Biol. 1988, 8, 4155–4161. [Google Scholar] [CrossRef] [PubMed]
- Carazo, A.; Levin, J.; Casas, F.; Seyer, P.; Grandemange, S.; Busson, M.; Pessemesse, L.; Wrutniak-Cabello, C.; Cabello, G. Protein sequences involved in the mitochondrial import of the 3,5,3′-L-triiodothyronine receptor p43. J. Cell. Physiol. 2012, 227, 3768–3777. [Google Scholar] [CrossRef] [PubMed]
- Wrutniak, C.; Cassar-Malek, I.; Marchal, S.; Rascle, A.; Heusser, S.; Keller, J.M.; Fléchon, J.; Dauça, M.; Samarut, J.; Ghysdael, J.; et al. A 43-kDa protein related to c-Erb A alpha 1 is located in the mitochondrial matrix of rat liver. J. Biol. Chem. 1995, 270, 16347–16354. [Google Scholar] [CrossRef] [PubMed]
- Morrish, F.; Buroker, N.E.; Ge, M.; Ning, X.H.; Lopez-Guisa, J.; Hockenbery, D.; Portman, M.A. Thyroid hormone receptor isoforms localize to cardiac mitochondrial matrix with potential for binding to receptor elements on mtDNA. Mitochondrion 2006, 6, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Casas, F.; Daury, L.; Grandemange, S.; Busson, M.; Seyer, P.; Hatier, R.; Carazo, A.; Cabello, G.; Wrutniak-Cabello, C. Endocrine regulation of mitochondrial activity: Involvement of truncated RXRalpha and c-Erb Aalpha1 proteins. FASEB J. 2003, 17, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Wirth, E.K.; Meyer, F. Neuronal effects of thyroid hormone metabolites. Mol. Cell. Endocrinol. 2017, 458, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Bahn, R.S.; Burch, H.B.; Cooper, D.S.; Garber, J.R.; Greenlee, M.C.; Klein, I.; Laurberg, P.; McDougall, I.R.; Montori, V.M.; Rivkees, S.A.; et al. Hyperthyroidism and other causes of thyrotoxicosis: Management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr. Pract. 2011, 17, 456–520. [Google Scholar] [CrossRef] [PubMed]
- Lanni, A.; Moreno, M.; Lombardi, A.; Goglia, F. Calorigen effect of diiodothyronines in the rat. J. Physiol. 1996, 494, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.G.; Sokolov, J.; Chin, W.W. 3,5-Diiodo-L-thyronine (T2) has selective thyromimetic effects in vivo and in vitro. J. Mol. Endocrinol. 1997, 19, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Baur, A.; Bauer, K.; Jarry, H.; Köhrle, J. 3,5-diiodo-L-thyronine stimulates type 1 5’deiodinase activity in rat anterior pituitaries in vivo and in reaggregate cultures and GH3 cells in vitro. Endocrinology 1997, 138, 3242–3248. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Lombardi, A.; Beneduce, L.; Silvestri, E.; Pinna, G.; Goglia, F.; Lanni, A. Are the effects of T3 on resting metabolic rate in euthyroid rats entirely caused by T3 itself? Endocrinology 2002, 143, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Leonard, J.L.; Davis, F.B. Mechanisms of nongenomic actions of thyroid hormone. Front. Neuroendocrinol. 2008, 29, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Lietzow, J.; Golchert, J.; Homuth, G.; Völker, U.; Jonas, W.; Köhrle, J. 3,5-T2 alters murine genes relevant for xenobiotic, steroid, and thyroid hormone metabolism. J. Mol. Endocrinol. 2016, 56, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Giacco, A.; Di Munno, C.; Goglia, F. Direct and rapid effects of 3,5-diiodo-L-thyronine (T2). Mol. Cell. Endocrinol. 2017, 458, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Padron, A.S.; Neto, R.A.; Pantaleão, T.U.; de Souza dos Santos, M.C.; Araujo, R.L.; de Andrade, B.M.; da Silva Leandro, M.; de Castro, J.P.; Ferreira, A.C.; de Carvalho, D.P. Administration of 3,5-diiodothyronine (3,5-T2) causes central hypothyroidism and stimulates thyroid-sensitive tissues. J. Endocrinol. 2014, 221, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Jonas, W.; Lietzow, J.; Wohlgemuth, F.; Hoefig, C.S.; Wiedmer, P.; Schweizer, U.; Köhrle, J.; Schürmann, A. 3,5-Diiodo-L-thyronine (3,5-t2) exerts thyromimetic effects on hypothalamus-pituitary-thyroid axis, body composition, and energy metabolism in male diet-induced obese mice. Endocrinology 2015, 156, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Giammanco, M.; Aiello, S.; Casuccio, A.; La Guardia, M.; Cicero, L.; Puleio, R.; Vazzana, I.; Tomasello, G.; Cassata, G.; Leto, G.; et al. Effects of 3,5-diiodo-L-thyronine on the liver of high fat diet fed rats. J. Biol. Res. 2016, 89, 5667. [Google Scholar] [CrossRef]
- Cimmino, M.; Mion, F.; Goglia, F.; Minaire, Y.; Géloen, A. Demostration of in vivo metabolic effects of 3,5-diiodothyronine. J. Endocrinol. 1996, 149, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Lanni, A.; Lombardi, A.; Goglia, F. How the thyroid controls metabolism in the rat: Different roles for triiodothyronine and diiodothyronines. J. Physiol. 1997, 505, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Goglia, F.; Moreno, M.; Lanni, A. Action of thyroid hormones at the cellular level: The mitochondrial target. FEBS Lett. 1999, 452, 115–120. [Google Scholar] [CrossRef]
- Hulbert, A.J. Thyroid hormones and their effects: A new perspective. Biol. Rev. 2000, 75, 519–631. [Google Scholar] [CrossRef] [PubMed]
- Lanni, A.; Moreno, M.; Lombardi, A.; De Lange, P.; Goglia, F. Control of energy metabolism by iodothyronines. J. Endocrinol. Investig. 2001, 24, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Goglia, F. The effects of 3,5-diiodothyronine on energy balance. Front. Physiol, 2005; 5, 528. [Google Scholar] [CrossRef]
- Lanni, A.; Moreno, M.; Lombardi, A.; De Lange, P.; Silvestri, E.; Ragni, M.; Farina, P.; Baccari, G.C.; Fallahi, P.; Antonelli, A.; et al. 3,5-Diiodo-L-thyronine powerfully reduces adiposity in rats by increasing the burning of fats. FASEB J. 2005, 19, 1552–1554. [Google Scholar] [CrossRef] [PubMed]
- de Lange, P.; Cioffi, F.; Senese, R.; Moreno, M.; Lombardi, A.; Silvestri, E.; De Matteis, R.; Lionetti, L.; Mollica, M.P.; Goglia, F.; et al. Non-Thyrotoxic Prevention of Diet-Induced Insulin Resistance by 3,5-Diiodo-L-Thyronine in Rats. Diabetes 2011, 60, 2730–2739. [Google Scholar] [CrossRef]
- Vatner, D.F.; Snikeris, J.; Popov, V.; Perry, R.J.; Rahimi, Y.; Samuel, V.T. 3,5 Diiodo-L-Thyronine (T2) Does Not Prevent Hepatic Steatosis or Insulin Resistance in Fat-Fed Sprague Dawley Rats. PLoS ONE 2015, 10, e0140837. [Google Scholar] [CrossRef] [PubMed]
- da Silva Teixeira, S.; Filgueira, C.; Sieglaff, D.H.; Benod, C.; Villagomez, R.; Minze, L.J.; Zhang, A.; Webb, P.; Nunes, M.T. 3,5-diiodothyronine (3,5-T2) reduces blood glucose independently of insulin sensitization in obese mice. Acta Physiol. (Oxf.) 2017, 220, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Lehmphul, I.; Brabant, G.; Wallaschofski, H.; Ruchala, M.; Strasburger, C.J.; Köhrle, J.; Wu, Z. Detection of 3,5-diiodothyronine in sera of patients with altered thyroid status using a new monoclonal antibody-based chemiluminescence immunoassay. Thyroid 2014, 24, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, L.; Nguyen, N.M.; Sacripanti, G.; Serni, E.; Borsò, M.; Saponaro, F.; Cecchi, E.; Simoncini, T.; Ghelardoni, S.; Zucchi, R.; et al. Assay of Endogenous 3,5-diiodo-L-thyronine (3,5-T2) and 3,3′-diiodo-L-thyronine (3,3′-T2) in Human Serum: A Feasibility Study. Front. Endocrinol. (Lausanne) 2019, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J.; Lehmphul, I.; Pietzner, M.; Renko, K.; Rijntjes, E.; Richards, K.; Anselmo, J.; Danielsen, M.; Jonklaas, J. 3,5-T2-A Janus-Faced Thyroid Hormone Metabolite Exerts Both Canonical T3-Mimetic Endocrine and Intracrine Hepatic Action. Front. Endocrinol. (Lausanne) 2020, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- Pietzner, M.; Lehmphul, I.; Friedrich, N.; Schurmann, C.; Ittermann, T.; Dörr, M.; Nauck, M.; Laqua, R.; Völker, U.; Brabant, G. Translating pharmacological findings from hypothyroid rodents to euthyroid humans: Is there a functional role of endogenous 3,5-T2? Thyroid 2015, 25, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Pietzner, M.; Homuth, G.; Budde, K.; Lehmphul, I.; Völker, U.; Völzke, H.; Nauck, M.; Köhrle, J.; Friedrich, N. Urine Metabolomics by (1)H-NMR Spectroscopy Indicates Associations between Serum 3,5-T2 Concentrations and Intermediary Metabolism in Euthyroid Humans. Eur. Thyroid J. 2015, 4, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.W.; Müller, P.; Schiedat, F.; Schlömicher, M.; Strauch, J.; Chatzitomaris, A.; Klein, H.H.; Mügge, A.; Köhrle, J.; Rijntjes, E.; et al. Nonthyroidal Illness Syndrome in Cardiac Illness Involves Elevated Concentrations of 3,5-Diiodothyronine and Correlates with Atrial Remodeling. Eur. Thyroid J. 2015, 4, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Senese, R.; de Lange, P.; Petito, G.; Moreno, M.; Goglia, F.; Lanni, A. 3,5-Diiodothyronine: A Novel Thyroid Hormone Metabolite and Potent Modulator of Energy Metabolism. Front. Endocrinol. (Lausanne) 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Gentile, A.; Silvestri, E.; Goglia, F.; Lombardi, A. Effect of Iodothyronines on Thermogenesis: Focus on Brown Adipose Tissue. Front. Endocrinol. (Lausanne) 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, R.; Rutigliano, G.; Saponaro, F. Novel thyroid hormones. Endocrine 2019, 66, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Ellis, E.C.S.; Johansson, H.; Eriksson, M.; Bruscalupi, G.; Steffensen, K.R.; Parini, P. Diiodothyronines regulate metabolic homeostasis in primary human hepatocytes by modulating mTORC1 and mTORC2 activity. Mol. Cell. Endocrinol. 2020, 499, 110604. [Google Scholar] [CrossRef] [PubMed]
- Horst, C.; Rokos, H.; Seitz, H.J. Rapid stimulation of oxygen consumption by 3,5-diiodo-L-thyronine. Biochem. J. 1989, 261, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Lanni, A.; Moreno, M.; Cioffi, M.; Goglia, F. Effect of 3,3′-diiodothyronine and 3,5-diiodothyronine on rat liver oxidative capacity. Mol. Cell. Endocrinol. 1992, 86, 143–148. [Google Scholar] [CrossRef]
- Cavallo, A.; Taurino, F.; Damiano, F.; Siculella, L.; Sardanelli, A.M.; Gnoni, A. Acute administration of 3,5-diiodo-L-thyronine to hypothyroid rats stimulates bioenergetic parameters in liver mitochondria. J. Bioenerg. Biomembr. 2016, 48, 521–529. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, I.; Murphy, M.P. Studies on the rapid stimulation of mitochondrial respiration by thyroid hormones. Acta Endocrinol. (Copenh) 1992, 127, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Tata, J.R.; Ernster, L.; Lindberg, O. Control of basal metabolic rate by thyroid hormone and cellular function. Nature 1962, 198, 1058–1060. [Google Scholar] [CrossRef] [PubMed]
- Tata, J.R. Inhibition of the biological action of thyroid hormones by actinomycin D and puromycin. Nature 1963, 197, 1167–1168. [Google Scholar] [CrossRef] [PubMed]
- Tata, J.R.; Ernster, L.; Lindberg, O.; Arrhenius, E.; Pedersen, S.; Hedman, R. The action of thyroid hormone at the cell level. Biochem. J. 1963, 86, 408–428. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.L.; Koehrle, J. Intracellular pathways of iodothyronine metabolism. In Werner & Ingbar’s The thyroid: A fundamental and clinical text; Braverman, L.E., Utiger, R.D., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2002; pp. 136–173. [Google Scholar]
- Lanni, A.; Moreno, M.; Lombardi, A.; Goglia, F. 3,5-diiodo-L-thyronine and 3,5,3′-triiodo-L-thyronine both improve the cold tolerance of hypothyroid rats, but possibly via different mechanisms. Pflugers Arch. 1998, 436, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Goglia, F.; Lanni, A.; Horst, C.; Moreno, M.; Thoma, R. In vitro binding of 3,5-diiodo-L-thyronine to rat liver mitochondria. J. Mol. Endocrinol. 1994, 13, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lanni, A.; Moreno, M.; Cioffi, M.; Goglia, F. Effect of 3,3′-di-iodothyronine and 3,5-di-iodothyronine on rat liver mitochondria. J. Endocrinol. 1993, 136, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Goglia, F.; Lanni, A.; Barth, J.; Kadenbach, B. Interaction of diiodothyronines with isolated cytochrome c oxidase. FEBS Lett. 1994, 346, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Lanni, A.; Moreno, M.; Brand, M.D.; Goglia, F. Effect of 3,5-diiodo-L-thyronine on the mitocondrial energy-transduction apparatus. Biochem. J. 1998, 330, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; Goglia, F.; Kadenbach, B. 3,5-Diiodothyronine binds to subunit Va of cytochrome-c oxidase and abolishes the allosteric inhibition of respiration by ATP. Eur. J. Biochem. 1998, 252, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Silvestri, E.; Lombardi, A.; Visser, T.J.; Goglia, F.; Lanni, A. Identification of 3,5-diiodo-L-thyronine-binding proteins in rat liver cytosol by photoaffinity labeling. Endocrinology 2003, 144, 2297–2303. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hummerich, H.; Soboll, S. Rapid stimulation of calcium uptake into rat liver by L-tri-iodothyronine. Biochem. J. 1989, 258, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M.; McCormack, J.G. Ca2+ transport by mammalian mitochondria and its role in hormone action. Am. J. Physiol. 1985, 249, E543–E554. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; Geller, H.M.; Green, W.L.; Craeliu, W. Acute effects of thyroid hormone analogs on sodium currents in neonatal rat myocytes. J. Mol. Cell. Cardiol. 1999, 31, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Incerpi, S.; De Vito, P.; Luly, P.; Spagnolo, S.; Leoni, S. Short-term effects of thyroid hormones and 3,5-diiodothyronine on membrane transport systems in chick embryo hepatocytes. Endocrinology 2002, 143, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, A.; Siculella, L.; Paglialonga, G.; Damiano, F.; Giudetti, A.M. 3,5-diiodo-L-thyronine increases de novo lipogenesis in liver from hypothyroid rats by SREBP-1 and ChREBP-mediated transcriptional mechanisms. IUBMB Life 2019, 71, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Sacripanti, G.; Nguyen, N.M.; Lorenzini, L.; Frascarelli, S.; Saba, A.; Zucchi, R.; Ghelardoni, S. 3,5-Diiodo-l-Thyronine Increases Glucose Consumption in Cardiomyoblasts Without Affecting the Contractile Performance in Rat Heart. Front. Endocrinol. (Lausanne) 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Silvestri, E.; De Matteis, R.; de Lange, P.; Lombardi, A.; Glinni, D.; Senese, R.; Cioffi, F.; Salzano, A.M.; Scaloni, A.; et al. 3,5-Diiodo-L-thyronine prevents high-fat-diet-induced insulin resistance in rat skeletal muscle through metabolic and structural adaptations. FASEB J. 2011, 25, 3312–3324. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, M.W. Lipid metabolism and liver infiammation. Hepatic fatty acid uptake: Possible role in steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, 194–198. [Google Scholar] [CrossRef]
- Hribal, M.L.; Procopio, T.; Petta, S.; Sciacqua, A.; Grimaudo, S.; Pipitone, R.M.; Perticone, F.; Sesti, G. Insulin-Like Growth Factor-I, inflammatory proteins, and fibrosis in subjects with Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2013, 98, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: Real-world study of 18 million patients in four European cohorts. BMC Med. 2019, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Toan, S.; Zhou, H. Role of mitochondrial quality control in the pathogenesis of nonalcoholic fatty liver disease. Aging (Albany NY) 2020, 12, 6467–6485. [Google Scholar] [CrossRef] [PubMed]
- Brady, L.J.; Brady, P.S.; Romsos, D.R.; Hoppel, C.L. Elevated hepatic mitochondrial and peroxisomal oxidative capacities in fed and starved adult obese (ob/ob) mice. Biochem. J. 1985, 231, 439–444. [Google Scholar] [CrossRef]
- Jasper, R.T.; Zillikens, M.C.; Friesema, E.C.; delli Paoli, G.; Bloch, W.; Uitterlinden, A.G.; Goglia, F.; Lanni, A.; de Lange, P. Exercise, fasting, and mimetics: Toward beneficial combinations? FASEB J. 2017, 31, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Di Liegro, I. Genetic and Epigenetic Modulation of Cell Functions by Physical Exercise. Genes (Basel) 2019, 10, E1043. [Google Scholar] [CrossRef] [PubMed]
- Glass, O.; Filozof, C.; Noureddin, M.; Berner-Hansen, M.; Schabel, E.; Omokaro, S.O.; Schattenberg, J.M.; Barradas, K.; Miller, V.; Francque, S.; et al. Standardization of Diet and Exercise in Clinical Trials of NAFLD-NASH: Recommendations from the Liver Forum. J. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Li, Y.L.; Mellström, K.; Mellin, C.; Bladh, L.G.; Koehler, K.; Garg, N.; Garcia Collazo, A.M.; Litten, C.; Husman, B.; et al. Thyroid receptor ligands. 1. Agonist ligands selective for the thyroid receptor beta1. J. Med. Chem. 2003, 46, 1580–1588. [Google Scholar] [CrossRef]
- Grover, G.J.; Mellstrom, K.; Malm, J. Development of the Thyroid Hormone Receptor β-Subtype Agonist KB-141: A Strategy for Body Weight Reduction and Lipid Lowering with Minimal Cardiac Side Effects. Cardiovasc. Drug Rev. 2005, 23, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Duntas, L.H.; Brenta, G. A Renewed Focus on the Association Between Thyroid Hormones and Lipid Metabolism. Front. Endocrinol. (Lausanne) 2018, 9, 511. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.A.; Columbano, A.; Perra, A. Thyroid Hormones, Thyromimetics and Their Metabolites in the Treatment of Liver Disease. Front. Endocrinol. (Lausanne) 2018, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic Fatty Liver Disease and Hypercholesterolemia: Roles of Thyroid Hormones, Metabolites, and Agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Zambad, S.P.; Chhipa, L.; Senese, R.; Busiello, R.A.; Tuli, D.; Munshi, S.; Moreno, M.; Lombardi, A.; Gupta, R.C.; et al. TRC150094, a novel functional analog of iodothyronines reduces adiposity by increasing energy expenditure and fatty acid oxidation in rats receiving a high-fat diet. FASEB J. 2010, 24, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Lionetti, L.; Moreno, M.; Lombardi, A.; De Lange, P.; Antonelli, A.; Lanni, A.; Cavaliere, G.; Barletta, A.; Goglia, F. 3,5-diiodo-L-thyronine, by modulating mithocondrial function, reverses hepatic fat accumulation in rats fed a higt-fat diet. J. Hepatol. 2009, 51, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Fleury, C.; Sanchis, D. The mitochondrial uncoupling protein-2: Current status. Int. J. Biochem. Cell. Biol. 1999, 31, 1261–1278. [Google Scholar] [CrossRef]
- Jezek, P. Possible physiological roles of mitochondrial uncoupling proteins—UCPn. Int. J. Biochem. Cell Biol. 2002, 34, 1190–1206. [Google Scholar] [CrossRef]
- Kadenbach, B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim. Biophys. Acta 2003, 1604, 77–94. [Google Scholar] [CrossRef]
- Rousset, S.; Alves-Guerra, M.C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.M.; Bouillaud, F.; Ricquier, D. The biology of mitochondrial uncoupling proteins. Diabetes 2004, 53 (Suppl. 1), S130–S135. [Google Scholar] [CrossRef] [PubMed]
- Krauss, S.; Zhang, C.Y.; Lowell, B.B. The mitochondrial uncoupling-protein homologues. Nat. Rev. Mol. Cell. Biol. 2005, 6, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, A.; Gnoni, A.; Conte, E.; Siculella, L.; Zanotti, F.; Papa, S.; Gnoni, G.V. 3,5-diiodo-L-thyronine increases FoF1-ATP synthase activity and cardiolipin level in liver mitochondria of hypothyroid rats. J. Bioenerg. Biomembr. 2011, 43, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Mangiullo, R.; Gnoni, A.; Damiano, F.; Siculella, L.; Zanotti, F.; Papa, S.; Gnoni, G.V. 3,5-diiodo-L-thyronine upregulates rat-liver mitochondrial FoF1-ATP synthase by GA-binding protein/nuclear respiratory factor-2. Biochim. Biophys. Acta 2010, 1797, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, E.; Lombardi, A.; Coppola, M.; Gentile, A.; Cioffi, F.; Senese, R.; Goglia, F.; Lanni, A.; Moreno, M.; de Lange, P. Differential Effects of 3,5-Diiodo-L-Thyronine and 3,5,3′-Triiodo-L-Thyronine On Mitochondrial Respiratory Pathways in Liver from Hypothyroid Rats. Cell. Physiol. Biochem. 2018, 47, 2471–2483. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Geelen, M.J.; Guzmán, M. Control of hepatic fatty acid oxidation by 5′-AMP-activated protein kinase involves a malonyl-CoA-dependent and a malonyl-CoA-independent mechanism. Arch. Biochem. Biophys. 1997, 337, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Geelen, M.J.; Gómez del Pulgar, T.; Guzmán, M. Malonyl-CoA-independent acute control of hepatic carnitine palmitoyltransferase I activity. Role of Ca2+/calmodulin-dependent protein kinase II and cytoskeletal components. J. Biol. Chem. 1998, 273, 21497–21504. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.K.; Brand, M.D. Body mass dependence of H+ leak in mitochondria and its relevance to metabolic rate. Nature 1993, 362, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Munoz, A.; Hoppner, W.; Sap, J.; Brady, G.; Nordstrom, K.; Seitz, H.J.; Vennstrom, B. The chicken c-erbA alpha-product induces expression of thyroid hormone-responsive genes in 3,5,3′-triiodothyronine receptor-deficient rat hepatoma cells. Mol. Endocrinol. 1990, 4, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Canesi, L.; Goglia, F.; Ravera, S.; Panfoli, I.; Gallo, G.; Vergani, L. Non-receptor-mediated actions are responsible for the lipid-lowering effects of iodothyronines in Fat rat hepatoma cells. J. Endocrinol. 2011, 210, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Canesi, L.; Salis, A.; Damonte, G.; Compalati, A.D.; Goglia, F.; Gallo, G.; Vergani, L. 3,5-diiodo-L-thyronine modifies the lipid droplet composition in a model of hepatosteatosis. Cell Physiol. Biochem. 2014, 33, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Vock, C.; Gleissner, M.; Klapper, M.; Döring, F. Oleate regulates genes controlled by signaling pathways of mitogen activated protein kinase, insulin, and hypoxia. Nutr. Res. 2008, 28, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Londos, C.; Sztalryd, C.; Tansey, J.T.; Kimmel, A.R. Role of PAT proteins in lipid metabolism. Biochimie 2005, 87, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Bickel, P.E.; Tansey, J.T.; Welte, M.A. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta 2009, 1791, 419–440. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Pesce, C.; Canesi, L.; Fugassa, E.; Gallo, G.; Vergani, L. PAT protein mRNA expression in primary rat hepatocytes: Effects of exposure to fatty acids. Int. J. Mol. Med. 2010, 25, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Motomura, W.; Inoue, M.; Ohtake, T.; Takahashi, N.; Nagamine, M.; Tanno, S.; Kohgo, Y.; Okumura, T. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem. Biophys. Res. Commun. 2006, 340, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Targett-Adams, P.; McElwee, M.J.; Ehrenborg, E.; Gustafsson, M.C.; Palmer, C.N.; McLauchlan, J. A PPAR response element regulates transcription of the gene for human adipose differentiation-related protein. Biochim. Biophys. Acta 2005, 1728, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Rajak, S.; Singh, B.K.; Yen, P.M. Hepatic Lipid Catabolism via PPARα-Lysosomal Crosstalk. Int. J. Mol. Sci. 2020, 21, E2391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, E2061. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M. The role of PPARalpha in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARalpha actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed]
- Schadinger, S.E.; Bucher, N.L.; Schreiber, B.M.; Farmer, S.R. PPAR gamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-López, T.; Escolà-Gil, J.C.; Cedó, L.; Zali, M.R.; Molaei, M.; et al. Hepatic regulation of VLDL receptor by PPARβ/δ and FGF21 modulates non-alcoholic fatty liver disease. Mol. Metab. 2018, 8, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Nagasawa, T.; Inada, Y.; Nakano, S.; Tamura, T.; Takahashi, T.; Maruyama, K.; Yamazaki, Y.; Kuroda, J.; Shibata, N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine and choline-deficient diet. Eur. J. Pharmacol. 2006, 536, 182–191. [Google Scholar] [CrossRef] [PubMed]
- de Lange, P.; Lombardi, A.; Silvestri, E.; Goglia, F.; Lanni, A.; Moreno, M. Peroxisome Proliferator-Activated Receptor Delta: A Conserved Director of Lipid Homeostasis TRough Regulation of the Oxidative Capacity of Muscle. PPAR Res. 2008, 2008, 172676. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.D.; Hassall, D.G.; Winegar, D.A.; Haselden, J.N.; Nicholls, A.W.; Griffin, J.L. Increased hepatic oxidative metabolism distinguishes the action of Peroxisome proliferator-activated receptor delta from Peroxisome proliferator-activated receptor gamma in the ob/ob mouse. Genome Med. 2009, 1, 115. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Canesi, L.; Portincasa, P.; Voci, A.; Vergani, L.; Demori, I. Models of non-Alcoholic Fatty Liver Disease and Potential Translational Value: The Effects of 3,5-L-diiodothyronine. Ann. Hepatol. 2017, 16, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Canesi, L.; De Matteis, R.; Goglia, F.; Cioffi, F.; Fugassa, E.; Gallo, G.; Vergani, L. Direct effects of iodothyronines on excess fat storage in rat hepatocytes. J. Hepatol. 2011, 54, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Demori, I.; Canesi, L.; De Matteis, R.; Goglia, F.; Lanni, A.; Gallo, G.; Vergani, L. 3,5-Diiodo-L-thyronine modulates the expression of genes of lipid metabolism in a rat model of fatty liver. J. Endocrinol. 2012, 212, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Demori, I.; Vecchione, G.; Compalati, A.D.; Gallo, G.; Goglia, F.; De Matteis, R.; Silvestri, E.; Vergani, L. Triglyceride Mobilization from Lipid Droplets Sustains the Anti-Steatotic Action of Iodothyronines in Cultured Rat Hepatocytes. Front. Physiol. 2016, 6, 418. [Google Scholar] [CrossRef] [PubMed]
- Iannucci, L.F.; Cioffi, F.; Senese, R.; Goglia, F.; Lanni, A.; Yen, P.M.; Sinha, R.A. Metabolomic analysis shows differential hepatic effects of T2 and T3 in rats after short-term feeding with high fat diet. Sci. Rep. 2017, 7, 2023. [Google Scholar] [CrossRef] [PubMed]
- Damiano, F.; Rochira, A.; Gnoni, A.; Siculella, L. Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms. Int. J. Mol. Sci. 2017, 18, E744. [Google Scholar] [CrossRef] [PubMed]
- Senese, R.; Cioffi, F.; de Lange, P.; Leanza, C.; Iannucci, L.F.; Silvestri, E.; Moreno, M.; Lombardi, A.; Goglia, F.; Lanni, A. Both 3,5-Diiodo-L-Thyronine and 3,5,3’-Triiodo-L-Thyronine Prevent Short-term Hepatic Lipid Accumulation via Distinct Mechanisms in Rats Being Fed a High-Fat Diet. Front. Physiol. 2017, 8, 706. [Google Scholar] [CrossRef] [PubMed]
- Rochira, A.; Damiano, F.; Marsigliante, S.; Gnoni, G.V.; Siculella, L. 3,5-Diiodo-l-thyronine induces SREBP-1 proteolytic cleavage block and apoptosis in human hepatoma (Hepg2) cells. Biochim. Biophys. Acta 2013, 1831, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K. Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal beta-oxidation, PPARalpha and steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Rao, M.S. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G852–G858. [Google Scholar] [CrossRef] [PubMed]
- Akbiyik, F.; Cinar, K.; Demirpence, E.; Ozsullu, T.; Tunca, R.; Haziroglu, R.; Yurdaydin, C.; Uzunalimoglu, O.; Bozkaya, H. Ligand-induced expression of peroxisome proliferator-activated receptor alpha and activation of fatty acid oxidation enzymes in fatty liver. Eur. J. Clin. Investig. 2004, 34, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Reddy, J.K.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 2006, 147, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.C.; Huang, C.J. Changes in liver PPAR alpha mRNA expression in response to two levels of high-safflower-oil diets correlate with changes in adiposity and serum leptin in rats and mice. J. Nutr. Biochem. 2007, 18, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Nakamuta, M.; Kohjima, M.; Morizono, S.; Kotoh, K.; Yoshimoto, T.; Miyagi, I.; Enjoji, M. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2005, 16, 631–635. [Google Scholar] [PubMed]
- Yang, S.; Zhu, H.; Li, Y.; Lin, H.; Gabrielson, K.; Trush, M.A.; Diehl, A.M. Mitochondrial adaptations to obesity-related oxidant stress. Arch. Biochem. Biophys. 2000, 378, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Passayre, D.; Fromenty, B.; Mansouri, A. Mitochondrial injury in steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2004, 16, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Canesi, L.; Voci, A.; De Matteis, R.; Demori, I.; Fugassa, E.; Vergani, L. Effects of 3,5-Diiodo-L-Thyronine Administration on the Liver of High Fat Diet-Fed Rats. Exp. Biol. Med. 2008, 233, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Pastore, A.; Gaeta, L.M.; Tozzi, G.; Comparcola, D.; Sartorelli, M.R.; Marcellini, M.; Bertini, E.; Piemonte, F. Glutathione metabolism and antioxidant enzymes in patients affected by nonalcoholic steatohepatitis. Clin. Chim. Acta 2005, 355, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Winge, D.R.; Nielson, K.B.; Zeikus, R.D.; Gray, W.R. Structural characterization of the isoforms of neonatal and adult rat liver metallothionein. J. Biol. Chem. 1984, 259, 11419–11425. [Google Scholar] [PubMed]
- Kumari, M.V.; Hiramatsu, M.; Ebadi, M. Free radical scavenging actions of metallothionein isoforms I and II. Free Radic. Res. 1998, 29, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D. The elusive function of metallothioneins. Proc. Natl. Acad. Sci. USA 1998, 95, 8428–8430. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Maret, W. Different redox states of metallothionein/thionein in biological tissue. Biochem. J. 2007, 402, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Babula, P.; Masarik, M.; Adam, V.; Eckschlager, T.; Stiborova, M.; Trnkova, L.; Skutkova, H.; Provaznik, I.; Hubalek, J.; Kizek, R. Mammalian metallothioneins: Properties and functions. Metallomics 2012, 4, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Vergani, L.; Lanza, C.; Borghi, C.; Scarabelli, L.; Panfoli, I.; Burlando, B.; Dondero, F.; Viarengo, A.; Gallo, G. Effects of growth hormone and cadmium on the transcription regulation of two metallothionein isoforms. Mol. Cell. Endocrinol. 2007, 263, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sun, X.; James Kang, Y. Metallothionein protection against alcoholic liver injury TRough inhibition of oxidative stress. Exp. Biol. Med. 2002, 227, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Beattie, J.H.; Wood, A.M.; Newman, A.M.; Bremner, I.; Choo, K.H.; Michalska, A.E.; Duncan, J.S.; Trayhurn, P. Obesity and hyperleptinemia in metallothionein (-I and -II) null mice. Proc. Natl. Acad. Sci. USA 1998, 95, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Cherian, M.G.; Kang, Y.J. Metallothionin and liver cell regeneration. Exp. Biol. Med. 2006, 231, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.R.; Jiang, S.; Cherian, M.G. Augmented hepatic injury followed by impaired regeneration in metallothionein-I/II knockout mice after treatment with thioacetamide. Toxicol. Appl. Pharmacol. 2006, 210, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Do, M.S.; Nam, S.Y.; Hong, S.E.; Kim, K.W.; Duncan, J.S.; Beattie, J.H.; Trayhurn, P. Metallothionein gene expression in human adipose tissue from lean and obese subjects. Horm. Metab. Res. 2002, 34, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Andrews, G.K. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem. Pharmacol. 2000, 59, 95–104. [Google Scholar] [CrossRef]
- Haq, F.; Mahoney, M.; Koropatnick, J. Signaling events for metallothionein induction. Mutat. Res. 2003, 533, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Jeong, J. Synergistic cellular responses to heavy metal exposure: A minireview. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Maret, W.; Vallee, B.L. Zinc metallothionein imported into liver mitochondria modulates respiration. Proc. Natl. Acad. Sci. USA 2001, 98, 2317–2322. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Senese, R.; Petito, G.; Lasala, P.; de Lange, P.; Silvestri, E.; Lombardi, A.; Moreno, M.; Goglia, F.; Lanni, A. Both 3,3’,5-triiodothyronine and 3,5-diodo-L-thyronine Are Able to Repair Mitochondrial DNA Damage but by Different Mechanisms. Front. Endocrinol. (Lausanne) 2019, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Parrettini, S.; Cavallo, M.; Gaggia, F.; Calafiore, R.; Luca, G. Adipokines: A Rainbow of Proteins with Metabolic and Endocrine Functions. Protein Pept. Lett 2020. May 5. [Google Scholar] [CrossRef]
- Angilletta, M.J., Jr.; Cooper, B.S.; Schuler, M.S.; Boyles, J.G. The evolution of thermal physiology in endotherms. Front. Biosci. (Elite Ed.) 2010, 2, 861–881. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, J.; Cannon, B. Brown adipose tissue as a heat-producing thermoeffector. Handb. Clin. Neurol. 2018, 156, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019, 29, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.E.; Bianco, S.D. Thyroid-adrenergic interactions: Physiological and clinical implications. Thyroid 2008, 18, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, cellular and molecular biology and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Senese, R.; De Matteis, R.; Busiello, R.A.; Cioffi, F.; Goglia, F.; Lanni, A. 3,5-Diiodo-L-thyronine activates brown adipose tissue thermogenesis in hypothyroid rats. PLoS ONE 2015, 10, e0116498. [Google Scholar] [CrossRef] [PubMed]
- Weiner, J.; Hankir, M.; Heiker, J.T.; Fenske, W.; Krause, K. Thyroid hormones and browning of adipose tissue. Mol. Cell. Endocrinol. 2017, 458, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Senese, R.; Cioffi, F.; De Matteis, R.; Petito, G.; de Lange, P.; Silvestri, E.; Lombardi, A.; Moreno, M.; Goglia, F.; Lanni, A. 3,5 Diiodo-l-Thyronine (T2) Promotes the Browning of White Adipose Tissue in High-Fat Diet-Induced Overweight Male Rats Housed at Thermoneutrality. Cells 2019, 8, E256. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, E.; Senese, R.; Cioffi, F.; De Matteis, R.; Lattanzi, D.; Lombardi, A.; Giacco, A.; Salzano, A.M.; Scaloni, A.; Ceccarelli, M.; et al. 3,5-Diiodo-L-Thyronine Exerts Metabolically Favorable Effects on Visceral Adipose Tissue of Rats Receiving a High-Fat Diet. Nutrients 2019, 11, E278. [Google Scholar] [CrossRef] [PubMed]
- Pietzner, M.; Köhrle, J.; Lehmphul, I.; Budde, K.; Kastenmüller, G.; Brabant, G.; Völzke, H.; Artati, A.; Adamski, J.; Völker, U.; et al. A Thyroid Hormone-Independent Molecular Fingerprint of 3,5-Diiodothyronine Suggests a Strong Relationship with Coffee Metabolism in Humans. Thyroid 2019, 29, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, K.A.; Müller, A.M.; Wechselberger, A.; Rühland, S.; Salb, N.; Schwenk, N.; Heuer, H.; Carlsen, J.; Göke, B.; Nelson, P.J.; et al. Thyroid hormones and tetrac: New regulators of tumour stroma formation via integrin αvβ3. Endocr. Relat. Cancer 2015, 22, 941–952. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zanatta, A.P.; Gonçalves, R.; Zanatta, L.; de Oliveria, G.T.; Ludwig Moraes, A.L.; Zamoner, A.; Fernández-Dueñas, V.; Lanznaster, D.; Ciruela, F.; Tasca, C.I.; et al. New ionic targets of 3,3’,5’-triiodothyronine at the plasma membrane of rat Sertoli cells. Biochim. Biophys. Acta Biomembr. 2019, 1861, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Hoefig, C.S.; Wuensch, T.; Rijntjes, E.; Lehmphul, I.; Daniel, H.; Schweizer, U.; Mittag, J.; Köhrle, J. Biosynthesis of 3-iodothyronamine from T4 in murine intestinal tissue. Endocrinology 2015, 156, 4356–4364. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, G.; Erba, P.; Carnicelli, V.; Manfredi, C.; Frascarelli, S.; Ghelardoni, S.; Mariani, G.; Zucchi, R. Distribution of exogenous [125I]-3-iodothyronamine in mouse in vivo: Relationship with trace amine-associated receptors. J. Endocrinol. 2012, 213, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Rutigliano, G.; Zucchi, R. Cardiac actions of thyroid hormone metabolites. Mol. Cell. Endocrinol. 2017, 458, 76–81. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giammanco, M.; Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals. Int. J. Mol. Sci. 2020, 21, 4140. https://doi.org/10.3390/ijms21114140
Giammanco M, Di Liegro CM, Schiera G, Di Liegro I. Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals. International Journal of Molecular Sciences. 2020; 21(11):4140. https://doi.org/10.3390/ijms21114140
Chicago/Turabian StyleGiammanco, Marco, Carlo Maria Di Liegro, Gabriella Schiera, and Italia Di Liegro. 2020. "Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals" International Journal of Molecular Sciences 21, no. 11: 4140. https://doi.org/10.3390/ijms21114140
APA StyleGiammanco, M., Di Liegro, C. M., Schiera, G., & Di Liegro, I. (2020). Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals. International Journal of Molecular Sciences, 21(11), 4140. https://doi.org/10.3390/ijms21114140