Switching Homes: How Cancer Moves to Bone



{kind=link}
Abstract
1. Introduction
2. Cellular Players in the Bone/Bone Marrow Tumour Microenvironment
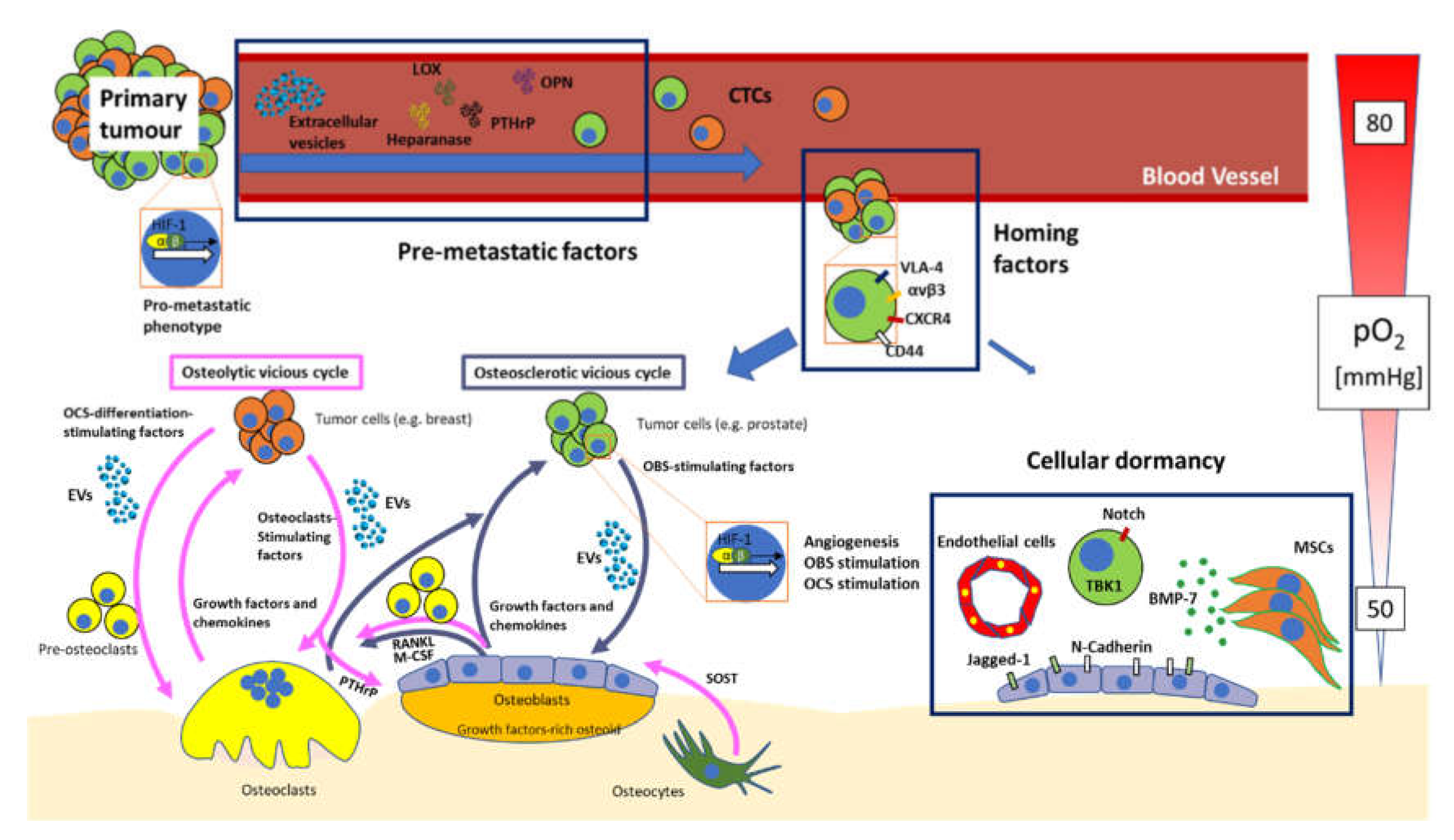
3. Bone Metastasis: Established and Novel Concepts
3.1. “Virtuous” vs. “Vicious” Cycle of the Bone
3.2. From Primary Site to Bone: A Bumpy Ride
3.3. Premetastatisation
3.4. Homing
3.5. Hypoxia, Angiogenesis and Blood Vessels in Bone Metastasis
3.6. Tumour Cellular Dormancy
3.7. Extracellular Vesicles in Bone Metastasis
3.8. Role of Immune Cells in Bone Biology and Metastases
4. Discussion and Conclusions
Funding
Conflicts of Interest
Abbreviations
| ANXA | Annexin |
| BALP | Bone-specific alkaline phosphatase |
| BM | Bone metastasis |
| BMA | Bone marrow adipocyte |
| BMM | Bone Marrow Macrophages |
| BMP | Bone morphogenetic proteins |
| BSP | Bone sialoprotein |
| CaSr | Calcium-sensing receptor |
| CCL | CC chemokine ligands |
| CCR | CC chemokine receptor |
| CDH2 | N-cadherin |
| COX | Cyclooxygenase |
| CTCs | Circulating tumour cells |
| CTGF | Connective tissue growth factor |
| CXCR | C-X-C chemokine receptor type |
| DDR | Discoidin domain receptor |
| DKK1 | Dikkopf-1 |
| DTCs | Disseminated tumour cells |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ET | Endothelin |
| EVs | Extracellular vesicles |
| FABP4 | Fatty acid-binding protein 4 |
| FAK | Focal adhesion kinase |
| FGF | Fibroblast growth factors |
| HBB | Haemoglobin beta |
| HGF | Hepatocyte growth factor |
| HIF | Hypoxia-inducible factors |
| HSC | Haematopoietic stem cells |
| IGF | Insulin-like growth factor |
| IL | Interleukin |
| LOX | Lysyl oxidase |
| MCP | Monocyte Chemoattractant Protein |
| M-CSF | Macrophage-colony stimulating factor |
| MDSC | Myeloid-derived suppressor cells |
| MITF | Microphthalmia Transcription Factor |
| MMP | Matrix metalloprotease |
| MSCs | Mesenchymal stromal cells |
| NFATc1 | Nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent |
| NfĸB | Nuclear factor kappa B |
| NICD | Notch intracellular domain |
| NO | Nitric Oxide |
| NOS | Nitric oxide synthase |
| NRDG | N-myc downstream-regulated gene |
| NSCLC | Nonsmall-cell lung cancer |
| OCN | Osteocalcin |
| OPN | Osteopontin |
| OPN | Osteopontin |
| PDGF | Platelet-derived growth factor |
| PD-L1 | Programmed death-ligand 1 |
| PMN | Premetastatic niche |
| PTHrP | Parathyroid hormone-related peptide |
| RANKL | Receptor activator of nuclear factor kappa B ligand |
| RAS | Rat sarcoma |
| Runx2 | Runt-related transcription factor 2 |
| SCF | Stem cell factor |
| SDF | Stromal-derived factor |
| SNOs | Spindle-shaped N-Cadherin-positive osteoblasts |
| SOST | Sclerostin |
| SREs | Skeletal-related events |
| TAM | Tumour-associated macrophages |
| TAN | Tumour-associated neutrophils |
| TBK1 | Tank-binding kinase 1 |
| TGF-β | Transforming growth factor-beta |
| TM4SF1 | Tetraspanin transmembrane 4 L six family member 1 |
| TNF-α | Tumour necrosis factor-alpha |
| TOR | Target of rapamycin |
| VCAM | Vascular cell adhesion molecule |
| VEGF | Vascular endothelial growth factor |
| VLA | Very late antigen |
| Wnt | Wingless-type MMTV integration |
References
- Buijs, T.J.; Kuijpers, C.H.J.C.; van der Pluijm, G. Targeted therapy options for treatment of bone metastases; beyond bisphosphonates. Curr. Pharm. Des. 2010, 16, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Aielli, F.; Ponzetti, M.; Rucci, N. Bone metastasis pain, from the bench to the bedside. Int. J. Mol. Sci. 2019, 20, 280. [Google Scholar] [CrossRef] [PubMed]
- D’Oronzo, S.; Coleman, R.; Brown, J.; Silvestris, F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J. Bone Oncol. 2019, 15, 10025. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11, 43–49. [Google Scholar] [CrossRef]
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Rucci, N.; Teti, A. Osteomimicry: How the seed grows in the soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef]
- Woodward, J.E.; Coleman, E.R. Prevention and treatment of bone metastases. Curr. Pharm. Des. 2010, 16, 2998–3006. [Google Scholar] [CrossRef]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef]
- Lee, B.; Thirunavukkarasu, K.; Zhou, L.; Pastore, L.; Baldini, A.; Hecht, J.; Geoffroy, V.; Ducy, P.; Karsenty, G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat. Genet. 1997, 16, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Galán-Díez, M.; Isa, A.; Ponzetti, M.; Nielsen, M.F.; Kassem, M.; Kousteni, S. Normal hematopoiesis and lack of β-catenin activation in osteoblasts of patients and mice harboring Lrp5 gain-of-function mutations. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef]
- Cappariello, A.; Ponzetti, M.; Rucci, N. The “soft” side of the bone: Unveiling its endocrine functions. Horm. Mol. Biol. Clin. Investig. 2016, 28, 5–20. [Google Scholar] [CrossRef]
- Clarke, B. Normal bone anatomy and physiology. Clin. J. Am. Soc. Nephrol. 2008, 3 (Suppl. 3), S131–S139. [Google Scholar] [CrossRef]
- Bonewald, L.F. Osteocytes. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism; Rosen, C.J., Ed.; Wiley: Washington, DC, USA, 2008; pp. 22–27. [Google Scholar]
- Temiyasathit, S.; Jacobs, C.R. The osteocyte primary cilium and its role in bone mechanotransduction. Ann. N. Y. Acad. Sci. 2010, 1192, 422–428. [Google Scholar] [CrossRef]
- Vaughan, T.J.; Mullen, C.A.; Verbruggen, S.W.; McNamara, L.M. Bone cell mechanosensation of fluid flow stimulation: A fluid-structure interaction model characterising the role integrin attachments and primary cilia. Biomech. Model. Mechanobiol. 2015, 14, 703–718. [Google Scholar] [CrossRef]
- Nguyen, A.M.; Jacobs, C.R. Emerging role of primary cilia as mechanosensors in osteocytes. Bone 2013, 54, 196–204. [Google Scholar] [CrossRef]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Noble, B.S. The osteocyte lineage. Arch. Biochem. Biophys. 2008, 473, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Jähn, K.; Kelkar, S.; Zhao, H.; Xie, Y.; Tiede-Lewis, L.A.M.; Dusevich, V.; Dallas, S.L.; Bonewald, L.F. Osteocytes acidify their microenvironment in response to PTHrP in vitro and in lactating mice in vivo. J. Bone Miner. Res. 2017, 32, 1761–1772. [Google Scholar] [CrossRef]
- Mori, G.; D’Amelio, P.; Faccio, R.; Brunetti, G. The interplay between the bone and the immune system. Clin. Dev. Immunol. 2013, 2013, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cappariello, A.; Maurizi, A.; Veeriah, V.; Teti, A. The Great Beauty of the osteoclast. Arch. Biochem. Biophys. 2014, 558, 70–78. [Google Scholar] [CrossRef]
- Kawaguchi, N.; Noda, M. Mitf is expressed in osteoclast progenitors in vitro. Exp. Cell Res. 2000, 260, 284–291. [Google Scholar] [CrossRef]
- Tondravi, M.M.; McKercher, S.R.; Anderson, K.; Erdmann, J.M.; Quiroz, M.; Maki, R.; Teitelbaum, S.L. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature 1997, 386, 81–84. [Google Scholar] [CrossRef]
- Xu, F.; Teitelbaum, S.L. Osteoclasts: New insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef]
- Burgess, T.L.; Qian, Y.X.; Kaufman, S.; Ring, B.D.; Van, G.; Capparelli, C.; Kelley, M.; Hsu, H.; Boyle, W.J.; Dunstan, C.R.; et al. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J. Cell Biol. 1999, 145, 527–538. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 1997, 11, 3482–3496. [Google Scholar] [CrossRef] [PubMed]
- Ponzetti, M.; Rucci, N. Updates on osteoimmunology: What’s new on the cross-talk between bone and immune system. Front. Endocrinol. 2019, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef]
- Mohamad, S.F.; Xu, L.; Ghosh, J.; Childress, P.J.; Abeysekera, I.; Himes, E.R.; Wu, H.; Alvarez, M.B.; Davis, K.M.; Aguilar-Perez, A.; et al. Osteomacs interact with megakaryocytes and osteoblasts to regulate murine hematopoietic stem cell function. Blood Adv. 2017, 1, 2520–2528. [Google Scholar] [CrossRef]
- Vi, L.; Baht, G.S.; Whetstone, H.; Ng, A.; Wei, Q.; Poon, R.; Mylvaganam, S.; Grynpas, M.; Alman, B.A. Macrophages promote osteoblastic differentiation in-vivo: Implications in fracture repair and bone homeostasis. J. Bone Miner. Res. 2015, 30, 1090–1102. [Google Scholar] [CrossRef]
- Sousa, S.; Määttä, J. The role of tumour-associated macrophages in bone metastasis. J. Bone Oncol. 2016, 5, 135–138. [Google Scholar] [CrossRef]
- Soki, F.N.; Cho, S.W.; Kim, Y.W.; Jones, J.D.; Park, S.I.; Koh, A.J.; Entezami, P.; Daignault-Newton, S.; Pienta, K.J.; Roca, H.; et al. Bone marrow macrophages support prostate cancer growth in bone. Oncotarget 2015, 6, 35782–35796. [Google Scholar] [CrossRef]
- Kusumbe, A.P. Vascular niches for disseminated tumour cells in bone. J. Bone Oncol. 2016, 5, 112–116. [Google Scholar] [CrossRef][Green Version]
- Ramasamy, S.K.; Kusumbe, A.P.; Schiller, M.; Zeuschner, D.; Bixel, M.G.; Milia, C.; Gamrekelashvili, J.; Limbourg, A.; Medvinsky, A.; Santoro, M.M.; et al. Blood flow controls bone vascular function and osteogenesis. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Grosso, A.; Burger, M.G.; Lunger, A.; Schaefer, D.J.; Banfi, A.; Di Maggio, N. It takes two to tango: Coupling of angiogenesis and osteogenesis for bone regeneration. Front. Bioeng. Biotechnol. 2017, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Bei, X.; Yang, B.; Wang, X.; Jiang, C.; Shi, F.; Wang, X.; Zhu, Y.; Jing, Y.; Han, B.; et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 221. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lee, Y.C.; Yu, G.; Cheng, C.J.; Zhou, X.; Chu, K.; Murshed, M.; Le, N.T.; Baseler, L.; Abe, J.; et al. Endothelial-to-osteoblast conversion generates osteoblastic metastasis of prostate cancer. Dev. Cell 2017, 41, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Robey, P.G. Marrow stromal stem cells. J. Clin. Investig. 2000, 105, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Hardaway, A.L.; Herroon, M.K.; Rajagurubandara, E.; Podgorski, I. Bone marrow fat: Linking adipocyte-induced inflammation with skeletal metastases. Cancer Metastasis Rev. 2014, 33, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; de Paula, F.J.A.; Rosen, C.J. New insights into osteoporosis: The bone-fat connection. J. Intern. Med. 2012, 272, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.R.; Sul, O.J.; Kim, Y.Y.; Kim, H.J.; Yu, R.; Suh, J.H.; Choi, H.S. Saturated fatty acids enhance osteoclast survival. J. Lipid Res. 2010, 51, 892–899. [Google Scholar] [CrossRef]
- Corre, J.; Planat-Benard, V.; Corberand, J.X.; Pénicaud, L.; Casteilla, L.; Laharrague, P. Human bone marrow adipocytes support complete myeloid and lymphoid differentiation from human CD34+ cells. Br. J. Haematol. 2004, 127, 344–347. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; He, Y.; Yu, X. Bone marrow adipocyte: An intimate partner with tumor cells in bone metastasis. Front. Endocrinol. 2018, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Hardaway, A.L.; Powell, K.; Turchick, A.; Feldmann, D.; Podgorski, I. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget 2013, 4, 2108–2123. [Google Scholar] [CrossRef] [PubMed]
- Chi, M.; Chen, J.; Ye, Y.; Tseng, H.-Y.; Lai, F.; Tay, K.H.; Jin, L.; Guo, S.T.; Jiang, C.C.; Zhang, X.D. Adipocytes contribute to resistance of human melanoma cells to chemotherapy and targeted therapy. Curr. Med. Chem. 2014, 21, 1255–1267. [Google Scholar] [CrossRef]
- Yu, W.; Cao, D.D.; Li, Q.B.; Mei, H.L.; Hu, Y.; Guo, T. Adipocytes secreted leptin is a pro-tumor factor for survival of multiple myeloma under chemotherapy. Oncotarget 2016, 7, 86075–86086. [Google Scholar] [CrossRef]
- Haider, M.T.; Smit, D.J.; Taipaleenmäki, H. The endosteal niche in breast cancer bone metastasis. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Fattore, A.D.; Teti, A.; Rucci, N. Bone cells and the mechanisms of bone remodelling. Front. Biosci. 2012, 4, 2302–2321. [Google Scholar] [CrossRef]
- Maurizi, A.; Rucci, N. The osteoclast in bone metastasis: Player and target. Cancers 2018, 10, 218. [Google Scholar] [CrossRef]
- Rucci, N.; Teti, A. The “love-hate” relationship between osteoclasts and bone matrix. Matrix Biol. 2016, 52–54, 176–190. [Google Scholar] [CrossRef]
- Rucci, N. Molecular biology of bone remodelling. Clin. Cases Miner. Bone Metab. 2008, 5, 49–56. [Google Scholar] [PubMed]
- Raisz, L.G. Physiology and pathophysiology of bone remodeling. Clin. Chem. 1999, 45, 1353–1358. [Google Scholar] [PubMed]
- Harada, S.I.; Rodan, G.A. Control of osteoblast function and regulation of bone mass. Nature 2003, 423, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.; Flamini, E.; Mercatali, L.; Sacanna, E.; Serra, P.; Amadori, D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 2010, 116, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Mohammad, K.S.; Käkönen, S.M.; Harris, S.; Wu-Wong, J.R.; Wessale, J.L.; Padley, R.J.; Garrett, I.R.; Chirgwin, J.M.; Guise, T.A. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 10954–10959. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.L.; Kang, S.; MacDougald, O.A.; Keller, E.T. Role of Wnts in prostate cancer bone metastases. J. Cell. Biochem. 2006, 97, 661–672. [Google Scholar] [CrossRef]
- Clines, G.A.; Mohammad, K.S.; Bao, Y.; Stephens, O.W.; Suva, L.J.; Shaughnessy, J.D.; Fox, J.W.; Chirgwin, J.M.; Guise, T.A. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol. Endocrinol. 2007, 21, 486–498. [Google Scholar] [CrossRef]
- Ottewell, P.D. The role of osteoblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124–127. [Google Scholar] [CrossRef]
- Kim, W.; Takyar, F.M.; Swan, K.; Jeong, J.; Vanhouten, J.; Sullivan, C.; Dann, P.; Yu, H.; Fiaschi-Taesch, N.; Chang, W.; et al. Calcium-sensing receptor promotes breast cancer by stimulating intracrine actions of parathyroid hormone-related protein. Cancer Res. 2016, 76, 5348–5360. [Google Scholar] [CrossRef]
- Kim, W.; Wysolmerski, J.J. Calcium-sensing receptor in breast physiology and cancer. Front. Physiol. 2016, 7, 440. [Google Scholar] [CrossRef]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Waning, D.L.; Guise, T.A. Molecular mechanisms of bone metastasis and associated muscle weakness. Clin. Cancer Res. 2014, 20, 3071–3077. [Google Scholar] [CrossRef]
- Van Kempen, L.C.L.; Coussens, L.M. MMP9 potentiates pulmonary metastasis formation. Cancer Cell 2002, 2, 251–252. [Google Scholar] [CrossRef]
- Mendes, O.; Kim, H.T.; Stoica, G. Expression of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat model. Clin. Exp. Metastasis 2005, 22, 237–246. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Nakamura, K.; Iwai, S.; Murakami, M.; Itoh, T.; Kijima, H.; Shipley, J.M.; Senior, R.M.; Shibuya, M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2002, 2, 289–300. [Google Scholar] [CrossRef]
- Owyong, M.; Chou, J.; van den Bijgaart, R.J.E.; Kong, N.; Efe, G.; Maynard, C.; Talmi-Frank, D.; Solomonov, I.; Koopman, C.; Hadler-Olsen, E.; et al. MMP9 modulates the metastatic cascade and immune landscape for breast cancer anti-metastatic therapy. Life Sci. Alliance 2019, 2, e201800226. [Google Scholar] [CrossRef]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Li, C.J.; Yiang, G.T.; Cheng, Y.L.; Tsai, A.P.Y.; Hou, Y.T.; Ho, Y.C.; Hou, M.F.; Chu, P.Y. Molecular Regulation of bone metastasis pathogenesis. Cell. Physiol. Biochem. 2018, 46, 1423–1438. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef]
- Reynaud, C.; Ferreras, L.; Di Mauro, P.; Kan, C.; Croset, M.; Bonnelye, E.; Pez, F.; Thomas, C.; Aimond, G.; Karnoub, A.E.; et al. Lysyl oxidase is a strong determinant of tumor cell colonization in bone. Cancer Res. 2017, 77, 268–278. [Google Scholar] [CrossRef]
- Cox, T.R.; Rumney, R.M.H.; Schoof, E.M.; Perryman, L.; Høye, A.M.; Agrawal, A.; Bird, D.; Latif, N.A.; Forrest, H.; Evans, H.R.; et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 2015, 522, 106–110. [Google Scholar] [CrossRef]
- Kelly, T.; Suva, L.J.; Huang, Y.; MacLeod, V.; Miao, H.Q.; Walker, R.C.; Sanderson, R.D. Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Res. 2005, 65, 5778–5784. [Google Scholar] [CrossRef]
- Martin, T.J.; Johnson, R.W. Multiple actions of parathyroid hormone-related protein in breast cancer bone metastasis. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massagué, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Psaila, B.; Lyden, D. Bone marrow cells in the “pre-metastatic niche”: Within bone and beyond. Cancer Metastasis Rev. 2006, 25, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y. The lung metastatic niche. J. Mol. Med. 2015, 93, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Doglioni, G.; Parik, S.; Fendt, S.M. Interactions in the (pre)metastatic niche support metastasis formation. Front. Oncol. 2019, 9, 219–291. [Google Scholar] [CrossRef]
- Pang, X.; Gong, K.; Zhang, X.; Wu, S.; Cui, Y.; Qian, B.Z. Osteopontin as a multifaceted driver of bone metastasis and drug resistance. Pharmacol. Res. 2019, 144, 235–244. [Google Scholar] [CrossRef]
- Shevde, L.; Das, S.; Clark, D.; Samant, R. Osteopontin: An effector and an effect of tumor metastasis. Curr. Mol. Med. 2010, 10, 71–81. [Google Scholar] [CrossRef]
- Wang, L.; Song, L.; Li, J.; Wang, Y.; Yang, C.; Kou, X.; Xiao, B.; Zhang, W.; Li, L.; Liu, S.; et al. Bone sialoprotein-αvβ3 integrin axis promotes breast cancer metastasis to the bone. Cancer Sci. 2019, 110, 3157–3172. [Google Scholar] [CrossRef] [PubMed]
- Kwakwa, K.A.; Sterling, J.A. Integrin αvβ3 signaling in tumor-induced bone disease. Cancers 2017, 9, 84. [Google Scholar] [CrossRef]
- Sloan, E.K.; Pouliot, N.; Stanley, K.L.; Chia, J.; Moseley, J.M.; Hards, D.K.; Anderson, R.L. Tumor-specific expression of αvβ3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006, 8. [Google Scholar] [CrossRef]
- Karadag, A.; Ogbureke, K.U.E.; Fedarko, N.S.; Fisher, L.W. Bone sialoprotein, matrix metalloproteinase 2, and αvβ3 integrin in osteotropic cancer cell invasion. J. Natl. Cancer Inst. 2004, 96, 956–965. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchaud, O.; Baron, R.; Clément-Lacroix, P.; Clézardin, P. Tumor αvβ3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007, 67, 5821–5830. [Google Scholar] [CrossRef]
- Schneider, J.G.; Amend, S.R.; Weilbaecher, K.N. Integrins and bone metastasis: Integrating tumor cell and stromal cell interactions. Bone 2011, 48, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Ren, W.; Sun, X.; Wang, K.; Feng, H.; Liu, Y.; Fei, C.; Wan, S.; Wang, W.; Luo, J.; Shi, Q.; et al. BMP9 inhibits the bone metastasis of breast cancer cells by downregulating CCN2 (connective tissue growth factor, CTGF) expression. Mol. Biol. Rep. 2014, 41, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Kubota, S.; Yoshioka, N.; Ibaragi, S.; Isowa, S.; Eguchi, T.; Sasaki, A.; Takigawa, M. Pathogenic role of connective tissue growth factor (CTGF/CCN2) in osteolytic metastasis of breast cancer. J. Bone Miner. Res. 2006, 21, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, H.; Jung, S.; Moon, A.; Noh, D.Y.; Lee, Z.H.; Kim, H.J.; Kim, H.H. A CTGF-RUNX2-RANKL axis in breast and prostate cancer cells promotes tumor progression in bone. J. Bone Miner. Res. 2020, 35, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Allocca, G.; Hughes, R.; Wang, N.; Brown, H.K.; Ottewell, P.D.; Brown, N.J.; Holen, I. The bone metastasis niche in breast cancer-potential overlap with the haematopoietic stem cell niche in vivo. J. Bone Oncol. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Sharma, M.; Afrin, F.; Satija, N.; Tripathi, R.P.; Gangenahalli, G.U. Stromal-derived factor-1/CXCR4 signaling: Indispensable role in homing and engraftment of hematopoietic stem cells in bone marrow. Stem Cells Dev. 2011, 20, 933–946. [Google Scholar] [CrossRef]
- Kucia, M.; Jankowski, K.; Reca, R.; Wysoczynski, M.; Bandura, L.; Allendorf, D.J.; Zhang, J.; Ratajczak, J.; Ratajczak, M.Z. CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J. Mol. Histol. 2004, 35, 233–245. [Google Scholar] [CrossRef]
- Zhou, Y.; Cao, H.B.; Li, W.J.; Zhao, L. The CXCL12 (SDF-1)/CXCR4 chemokine axis: Oncogenic properties, molecular targeting, and synthetic and natural product CXCR4 inhibitors for cancer therapy. Chin. J. Nat. Med. 2018, 16, 801–810. [Google Scholar] [CrossRef]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2014; Volume 124, pp. 31–82. [Google Scholar]
- Cho, Y.; Lee, H.W.; Kang, H.G.; Kim, H.Y.; Kim, S.J.; Chun, K.H. Cleaved CD44 intracellular domain supports activation of stemness factors and promotes tumorigenesis of breast cancer. Oncotarget 2015, 6, 8709–8721. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as cancer stem cell markers: An enduring ambiguity. Clin. Dev. Immunol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Stivarou, T.; Patsavoudi, E. Extracellular molecules involved in cancer cell invasion. Cancers 2015, 7, 238–265. [Google Scholar] [CrossRef] [PubMed]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 2017, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999, 13, 35–48. [Google Scholar] [CrossRef]
- Zöller, M. CD44, hyaluronan, the hematopoietic stem cell, and leukemia-initiating cells. Front. Immunol. 2015, 6, 235. [Google Scholar]
- Jung, Y.; Shiozawa, Y.; Wang, J.; Patel, L.R.; Havens, A.M.; Song, J.; Krebsbach, P.H.; Roodman, G.D.; Taichman, R.S. Annexin-2 is a regulator of stromal cell-derived factor-1/CXCL12 function in the hematopoietic stem cell endosteal niche. Exp. Hematol. 2011, 39. [Google Scholar] [CrossRef]
- Jung, Y.; Wang, J.; Song, J.; Shiozawa, Y.; Wang, J.; Havens, A.; Wang, Z.; Sun, Y.X.; Emerson, S.G.; Krebsbach, P.H.; et al. Annexin II expressed by osteoblasts and endothelial cells regulates stem cell adhesion, homing, and engraftment following transplantation. Blood 2007, 110, 82–90. [Google Scholar] [CrossRef][Green Version]
- Shiozawa, Y.; Havens, A.M.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, J.; Lu, G.; Roodman, G.D.; Loberg, R.D.; et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell. Biochem. 2008, 105, 370–380. [Google Scholar] [CrossRef]
- Jung, Y.; Wang, J.; Lee, E.; McGee, S.; Berry, J.E.; Yumoto, K.; Dai, J.; Keller, E.T.; Shiozawa, Y.; Taichman, R.S. Annexin 2-CXCL12 interactions regulate metastatic cell targeting and growth in the bone marrow. Mol. Cancer Res. 2015, 13, 197–207. [Google Scholar] [CrossRef]
- Zheng, H.; Bae, Y.; Kasimir-Bauer, S.; Tang, R.; Chen, J.; Ren, G.; Yuan, M.; Esposito, M.; Li, W.; Wei, Y.; et al. Therapeutic antibody targeting tumor- and osteoblastic niche-derived jagged1 sensitizes bone metastasis to chemotherapy. Cancer Cell 2017, 32, 731–747. [Google Scholar] [CrossRef]
- Sethi, N.; Kang, Y. Notch signalling in cancer progression and bone metastasis. Br. J. Cancer 2011, 105, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Sethi, N.; Dai, X.; Winter, C.G.; Kang, Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 2011, 19, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhang, H.; Li, X.; Li, X.; Cong, M.; Peng, F.; Yu, J.; Zhang, X.; Yang, Q.; Hu, G. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat. Cell Biol. 2017, 19, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Ricarte, F.R.; Le Henaff, C.; Kolupaeva, V.G.; Gardella, T.J.; Partridge, N.C. Parathyroid hormone(1–34) and its analogs differentially modulate osteoblastic Rankl expression via PKA/SIK2/SIK3 and PP1/PP2A-CRTC3 signaling. J. Biol. Chem. 2018, 293, 20200–20213. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Loberg, R.; Liao, J.; Ying, C.; Snyder, L.A.; Pienta, K.J.; McCauley, L.K. A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res. 2009, 69, 1685–1692. [Google Scholar] [CrossRef]
- Mulholland, B.S.; Forwood, M.R.; Morrison, N.A. Monocyte Chemoattractant Protein-1 (MCP-1/CCL2) drives activation of bone remodelling and skeletal metastasis. Curr. Osteoporos. Rep. 2019, 17, 538–547. [Google Scholar] [CrossRef]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, V.; Sahebkar, A.; Atkin, S.L.; Pirro, M. The regulation and importance of monocyte chemoattractant protein-1. Curr. Opin. Hematol. 2018, 25, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; Ratcliffe, P.J.; Semenza, G.L. Pathways for oxygen regulation and homeostasis: The 2016 albert lasker basic medical research award. JAMA J. Am. Med. Assoc. 2016, 316, 1252–1253. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Dutta, B.; Laha, A.; Bandopadhyay, R. 2019 Nobel Prize in Physiology and Medicine: A long road from bench to bedside of cell in oxygen sensing and adaptive responses. Natl. Acad. Sci. Lett. 2020. [Google Scholar] [CrossRef]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51. [Google Scholar] [CrossRef]
- Pezzuto, A.; Carico, E. Role of HIF-1 in cancer progression: Novel insights. A review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, E.S.; Sund, S.; Homsi, E.N.; Challenger, L.F.; Rameshwar, P. A theoretical simulation of hematopoietic stem cells during oxygen fluctuations: Prediction of bone marrow responses during hemorrhagic shock. Shock 2004, 22, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Hulley, P.A.; Bishop, T.; Vernet, A.; Schneider, J.E.; Edwards, J.R.; Athanasou, N.A.; Knowles, H.J. Hypoxia-inducible factor 1-alpha does not regulate osteoclastogenesis but enhances bone resorption activity via prolyl-4-hydroxylase 2. J. Pathol. 2017, 242, 322–333. [Google Scholar] [CrossRef]
- Bentovim, L.; Amarilio, R.; Zelzer, E. HIF1α is a central regulator of collagen hydroxylation and secretion under hypoxia during bone development. Development 2012, 139, 4473–4483. [Google Scholar] [CrossRef]
- Wang, Y.; Wan, C.; Deng, L.; Liu, X.; Cao, X.; Gilbert, S.R.; Bouxsein, M.L.; Faugere, M.C.; Guldberg, R.E.; Gerstenfeld, L.C.; et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Investig. 2007, 117, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Devignes, C.S.; Aslan, Y.; Brenot, A.; Devillers, A.; Schepers, K.; Fabre, S.; Chou, J.; Casbon, A.J.; Werb, Z.; Provot, S. HIF signaling in osteoblast-lineage cells promotes systemic breast cancer growth and metastasis in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E992–E1001. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef]
- Pez, F.; Dayan, F.; Durivault, J.; Kaniewski, B.; Aimond, G.; Le Provost, G.S.; Deux, B.; Clézardin, P.; Sommer, P.; Pouysségur, J.; et al. The HIF-1-inducible lysyl oxidase activates HIF-1 via the akt pathway in a positive regulation loop and synergizes with HIF-1 in promoting tumor cell growth. Cancer Res. 2011, 71, 1647–1657. [Google Scholar] [CrossRef]
- Pezzuto, A.; Perrone, G.; Orlando, N.; Citarella, F.; Ciccozzi, M.; Scarlata, S.; Tonini, G. A close relationship between HIF-1α expression and bone metastases in advanced NSCLC, a retrospective analysis. Oncotarget 2019, 10, 7071–7079. [Google Scholar] [CrossRef]
- Woelfle, U.; Cloos, J.; Sauter, G.; Riethdorf, L.; Jänicke, F.; Van Diest, P.; Brakenhoff, R.; Pantel, K. Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res. 2003, 63, 5679–5684. [Google Scholar]
- Hiraga, T.; Kizaka-Kondoh, S.; Hirota, K.; Hiraoka, M.; Yoneda, T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007, 67, 4157–4163. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Hiraoka, M.; Kizaka-Kondoh, S. Antitumor effect of TAT-oxygen-dependent degradation-caspase-3 fusion protein specifically stabilized and activated in hypoxic tumor cells. Cancer Res. 2002, 62, 2013–2018. [Google Scholar] [PubMed]
- Capulli, M.; Angelucci, A.; Driouch, K.; Garcia, T.; Clement-Lacroix, P.; Martella, F.; Ventura, L.; Bologna, M.; Flamini, S.; Moreschini, O.; et al. Increased expression of a set of genes enriched in oxygen binding function discloses a predisposition of breast cancer bone metastases to generate metastasis spread in multiple organs. J. Bone Miner. Res. 2012, 27, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Ponzetti, M.; Capulli, M.; Angelucci, A.; Ventura, L.; Monache, S.D.; Mercurio, C.; Calgani, A.; Sanità, P.; Teti, A.; Rucci, N. Non-conventional role of haemoglobin beta in breast malignancy. Br. J. Cancer 2017, 117, 994–1006. [Google Scholar] [CrossRef]
- Zheng, Y.; Miyamoto, D.T.; Wittner, B.S.; Sullivan, J.P.; Aceto, N.; Jordan, N.V.; Yu, M.; Karabacak, N.M.; Comaills, V.; Morris, R.; et al. Expression of β-globin by cancer cells promotes cell survival during blood-borne dissemination. Nat. Commun. 2017, 8, 14344. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Hiasa, M.; Nagata, Y.; Okui, T.; White, F.A. Acidic microenvironment and bone pain in cancer-colonized bone. Bonekey Rep. 2015, 4. [Google Scholar] [CrossRef]
- Falk, S.; Dickenson, A.H. Pain and nociception: Mechanisms of cancer-induced bone pain. J. Clin. Oncol. 2014, 32, 1647–1654. [Google Scholar] [CrossRef]
- Di Pompo, G.; Lemma, S.; Canti, L.; Rucci, N.; Ponzetti, M.; Errani, C.; Donati, D.M.; Russell, S.; Gillies, R.; Chano, T.; et al. Intratumoral acidosis fosters cancer-induced bone pain through the activation of the mesenchymal tumor-associated stroma in bone metastasis from breast carcinoma. Oncotarget 2017, 8, 54478–54496. [Google Scholar] [CrossRef]
- Yoneda, T.; Hiasa, M.; Nagata, Y.; Okui, T.; White, F. Contribution of acidic extracellular microenvironment of cancer-colonized bone to bone pain. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2677–2684. [Google Scholar] [CrossRef]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002, 16, 1151–1162. [Google Scholar] [CrossRef]
- Maroni, P.; Matteucci, E.; Luzzati, A.; Perrucchini, G.; Bendinelli, P.; Desiderio, M.A. Nuclear co-localization and functional interaction of COX-2 and HIF-1α characterize bone metastasis of human breast carcinoma. Breast Cancer Res. Treat. 2011, 129, 433–450. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Luzzati, A.; Perrucchini, G.; Desiderio, M.A. Microenvironmental stimuli affect Endothelin-1 signaling responsible for invasiveness and osteomimicry of bone metastasis from breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 815–826. [Google Scholar] [CrossRef]
- Raymaekers, K.; Stegen, S.; van Gastel, N.; Carmeliet, G. The vasculature: A vessel for bone metastasis. Bonekey Rep. 2015. [Google Scholar] [CrossRef]
- Cook, L.M.; Shay, G.; Aruajo, A.; Lynch, C.C. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metastasis Rev. 2014, 33, 511–525. [Google Scholar] [CrossRef]
- Hu, K.; Olsen, B.R. Osteoblast-derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J. Clin. Investig. 2016, 126, 509–526. [Google Scholar] [CrossRef]
- Dai, J.; Kitagawa, Y.; Zhang, J.; Yao, Z.; Mizokami, A.; Cheng, S.; Nör, J.; McCauley, L.K.; Taichman, R.S.; Keller, E.T. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res. 2004, 64, 994–999. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Dai, J.; Zhang, J.; Keller, J.M.; Nor, J.; Yao, Z.; Keller, E.T. Vascular endothelial growth factor contributes to prostate cancer-mediated osteoblastic activity. Cancer Res. 2005, 65, 10921–10929. [Google Scholar] [CrossRef]
- Yang, Q.; McHugh, K.P.; Patntirapong, S.; Gu, X.; Wunderlich, L.; Hauschka, P.V. VEGF enhancement of osteoclast survival and bone resorption involves VEGF receptor-2 signaling and β3-integrin. Matrix Biol. 2008, 27, 589–599. [Google Scholar] [CrossRef]
- Hurley, M.M.; Lee, S.K.; Raisz, L.G.; Bernecker, P.; Lorenzo, J. Basic fibroblast growth factor induces osteoclast formation in murine bone marrow cultures. Bone 1998, 22, 309–316. [Google Scholar] [CrossRef]
- Collin-Osdoby, P.; Rothe, L.; Bekker, S.; Anderson, F.; Huang, Y.; Osdoby, P. Basic fibroblast growth factor stimulates osteoclast recruitment, development, and bone pit resorption in association with angiogenesis in vivo on the chick chorioallantoic membrane and activates isolated avian osteoclast resorption in vitro. J. Bone Miner. Res. 2002, 17, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Vignani, F.; Bertaglia, V.; Buttigliero, C.; Tucci, M.; Scagliotti, G.V.; Di Maio, M. Skeletal metastases and impact of anticancer and bone-targeted agents in patients with castration-resistant prostate cancer. Cancer Treat. Rev. 2016, 44, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Felber, K.; Elks, P.M.; Lecca, M.; Roehl, H.H. Expression of osterix is regulated by FGF and Wnt/β-catenin signalling during osteoblast differentiation. PLoS ONE 2015, 10, e0144982. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Inoue, M. Dormancy in cancer. Cancer Sci. 2019, 110, 474–480. [Google Scholar] [CrossRef]
- Jahanban-Esfahlan, R.; Seidi, K.; Manjili, M.H.; Jahanban-Esfahlan, A.; Javaheri, T.; Zare, P. Tumor cell dormancy: Threat or opportunity in the fight against cancer. Cancers 2019, 11, 1207. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Kyriakou, T.C.; Papageorgis, P. Mechanisms of metastatic tumor dormancy and implications for cancer therapy. Int. J. Mol. Sci. 2019, 20, 6158. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef]
- Byrne, N.M.; Summers, M.A.; McDonald, M.M. Tumor cell dormancy and reactivation in bone: Skeletal biology and therapeutic opportunities. JBMR Plus 2019, 3, e10125. [Google Scholar] [CrossRef]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Xing, F.; Liu, Y.; Wu, K.; Said, N.; Pochampally, R.; Shiozawa, Y.; Lin, H.K.; Balaji, K.C.; Watabe, K. Secreted protein acidic and rich in cysteine (sparc) mediates metastatic dormancy of prostate cancer in bone. J. Biol. Chem. 2016, 291, 19351–19363. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jung, Y.; Wang, J.; Joseph, J.; Mishra, A.; Hill, E.E.; Krebsbach, P.H.; Pienta, K.J.; Shiozawa, Y.; Taichman, R.S. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia 2013, 15, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Capulli, M.; Hristova, D.; Valbret, Z.; Carys, K.; Arjan, R.; Maurizi, A.; Masedu, F.; Cappariello, A.; Rucci, N.; Teti, A. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br. J. Cancer 2019, 121, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Asada, N.; Katayama, Y. Regulation of hematopoiesis in endosteal microenvironments. Int. J. Hematol. 2014, 99, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Zhao, M.; Tao, F.; Venkatraman, A.; Li, Z.; Smith, S.E.; Unruh, J.; Chen, S.; Ward, C.; Qian, P.; Perry, J.M.; et al. N-cadherin-expressing bone and marrow stromal progenitor cells maintain reserve hematopoietic stem cells. Cell Rep. 2019, 26, 652–669. [Google Scholar] [CrossRef]
- Greenbaum, A.M.; Revollo, L.D.; Woloszynek, J.R.; Civitelli, R.; Link, D.C. N-cadherin in osteolineage cells is not required for maintenance of hematopoietic stem cells. Blood 2012, 120, 295–302. [Google Scholar] [CrossRef]
- Abravanel, D.L.; Belka, G.K.; Pan, T.C.; Pant, D.K.; Collins, M.A.; Sterner, C.J.; Chodosh, L.A. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J. Clin. Investig. 2015, 125, 2484–2496. [Google Scholar] [CrossRef]
- Nobutani, K.; Shimono, Y.; Mizutani, K.; Ueda, Y.; Suzuki, T.; Kitayama, M.; Minami, A.; Momose, K.; Miyawaki, K.; Akashi, K.; et al. Downregulation of CXCR4 in metastasized breast cancer cells and implication in their dormancy. PLoS ONE 2015, 10, e0130032. [Google Scholar] [CrossRef]
- Gao, X.L.; Zheng, M.; Wang, H.F.; Dai, L.L.; Yu, X.H.; Yang, X.; Pang, X.; Li, L.; Zhang, M.; Wang, S.S.; et al. NR2F1 contributes to cancer cell dormancy, invasion and metastasis of salivary adenoid cystic carcinoma by activating CXCL12/CXCR4 pathway. BMC Cancer 2019, 19, 743. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, D.; Maier, P.; Laufs, S.; Wenz, F.; Zeller, W.J.; Fruehauf, S.; Allgayer, H. CXCR4 expression and treatment with SDF-1α or plerixafor modulate proliferation and chemosensitivity of colon cancer cells. Transl. Oncol. 2013, 6, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, S.; Lan, Q.; Lorusso, G.; Rüegg, C. Chemotherapy-induced immunological breast cancer dormancy: A new function for old drugs? J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38α/β signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef]
- White, D.E.; Kurpios, N.A.; Zuo, D.; Hassell, J.A.; Blaess, S.; Mueller, U.; Muller, W.J. Targeted disruption of β1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 2004, 6, 159–170. [Google Scholar] [CrossRef]
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the malignant phenotype of human breast cells in three- dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 1997, 137, 231–245. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar] [CrossRef]
- Park, S.Y.; Nam, J.S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020. [CrossRef]
- Barkan, D.; Chambers, A.F. β1-integrin: A potential therapeutic target in the battle against cancer recurrence. Clin. Cancer Res. 2011, 17, 7219–7223. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chakraborty, G.; Zhang, Z.; Akalay, I.; Gadiya, M.; Gao, Y.; Sinha, S.; Hu, J.; Jiang, C.; Akram, M.; et al. Multi-organ Site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell 2016, 166, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Kaldis, P.; Pagano, M. Wnt signaling in mitosis. Dev. Cell 2009, 17, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Malladi, S.; MacAlinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; De Stanchina, E.; Massagué, J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.L.; Vallée, A. Multiple targets of the canonical WNT/β-catenin signaling in cancers. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.W. Chronic inflammation and breast cancer recurrence. J. Clin. Oncol. 2009, 27, 3418–3419. [Google Scholar] [CrossRef]
- Cappariello, A.; Rucci, N. Tumour-derived extracellular vesicles (EVs): A dangerous “message in a bottle” for bone. Int. J. Mol. Sci. 2019, 20, 4805. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Dudás, J.; Riechelmann, H.; Skvortsova, I.I. The role of exosomes in cancer metastasis. Semin. Cancer Biol. 2017, 44, 170–181. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Cappariello, A.; Loftus, A.; Muraca, M.; Maurizi, A.; Rucci, N.; Teti, A. Osteoblast-derived extracellular vesicles are biological tools for the delivery of active molecules to bone. J. Bone Miner. Res. 2018, 33, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Loftus, A.; Cappariello, A.; George, C.; Ucci, A.; Shefferd, K.; Green, A.; Paone, R.; Ponzetti, M.; Delle Monache, S.; Muraca, M.; et al. Extracellular vesicles from osteotropic breast cancer cells affect bone resident cells. J. Bone Miner. Res. 2020, 35, 396–412. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Taverna, S.; Pucci, M.; Giallombardo, M.; Di Bella, M.A.; Santarpia, M.; Reclusa, P.; Gil-Bazo, I.; Rolfo, C.; Alessandro, R. Amphiregulin contained in NSCLC-exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, X.; Wang, H.; Li, J.; Dai, L.; Li, J.; Dong, C. Lung adenocarcinoma cell-derived exosomal miR-21 facilitates osteoclastogenesis. Gene 2018, 666, 116–122. [Google Scholar] [CrossRef]
- Valencia, K.; Luis-Ravelo, D.; Bovy, N.; Antón, I.; Martínez-Canarias, S.; Zandueta, C.; Ormazábal, C.; Struman, I.; Tabruyn, S.; Rebmann, V.; et al. MiRNA cargo within exosome-like vesicle transfer influences metastatic bone colonization. Mol. Oncol. 2014, 8, 689–703. [Google Scholar] [CrossRef]
- Karlsson, T.; Lundholm, M.; Widmark, A.; Persson, E. Tumor cell-derived exosomes from the prostate cancer cell line TRAMP-C1 impair osteoclast formation and differentiation. PLoS ONE 2016, 11, e0166284. [Google Scholar] [CrossRef]
- Inder, K.L.; Ruelcke, J.E.; Petelin, L.; Moon, H.; Choi, E.; Rae, J.; Blumenthal, A.; Hutmacher, D.; Saunders, N.A.; Stow, J.L.; et al. Cavin-1/PTRF alters prostate cancer cell-derived extracellular vesicle content and internalization to attenuate extracellular vesicle-mediated osteoclastogenesis and osteoblast proliferation. J. Extracell. Vesicles 2014, 3, 23784. [Google Scholar] [CrossRef]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of bone resorption in periodontitis. J. Immunol. Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Moutsopoulos, N.M.; Hajishengallis, E.; Chavakis, T. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin. Immunol. 2016, 28, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Roato, I.; Grano, M.; Brunetti, G.; Colucci, S.; Mussa, A.; Bertetto, O.; Ferracini, R. Mechanisms of spontaneous osteoclastogenesis in cancer with bone involvement. FASEB J. 2005, 19. [Google Scholar] [CrossRef]
- Roato, I.; Brunetti, G.; Gorassini, E.; Grano, M.; Colucci, S.; Bonello, L.; Buffoni, L.; Manfredi, R.; Ruffini, E.; Ottaviani, D.; et al. IL-7 up-regulates TNF-α-dependent osteoclastogenesis in patients affected by solid tumor. PLoS ONE 2006, 1, e124. [Google Scholar] [CrossRef]
- Roato, I.; Gorassini, E.; Brunetti, G.; Grano, M.; Ciuffreda, L.; Mussa, A.; Ferracini, R. IL-7 modulates osteoclastogenesis in patients affected by solid tumors. Ann. New York Acad. Sci. 2007, 1117, 377–384. [Google Scholar] [CrossRef]
- Roato, I.; Caldo, D.; Godio, L.; D’Amico, L.; Giannoni, P.; Morello, E.; Quarto, R.; Molfetta, L.; Buracco, P.; Mussa, A.; et al. Bone invading NSCLC cells produce IL-7: Mice model and human histologic data. BMC Cancer 2010, 10, 12. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancermetastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef]
- Zhang, K.; Kim, S.; Cremasco, V.; Hirbe, A.C.; Novack, D.V.; Weilbaecher, K.; Faccio, R. CD8+ T cells regulate bone tumor burden independent of osteoclast resorption. Cancer Res. 2011, 71, 4799–4808. [Google Scholar] [CrossRef]
- Yoon, S.H.; Lee, Y.; Kim, H.J.; Lee, Z.H.; Hyung, S.W.; Lee, S.W.; Kim, H.H. Lyn inhibits osteoclast differentiation by interfering with PLCγ1-mediated Ca2+ signaling. FEBS Lett. 2009, 583, 1164–1170. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, A.; Bronte, V.; Visintin, A.; Spitzer, J.H.; Apolloni, E.; Serafini, P.; Zanovello, P.; Segal, D.M. Myeloid suppressor lines inhibit T cell responses by an no-dependent mechanism. J. Immunol. 2002, 168, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Deshane, J.; Jules, J.; Lee, C.M.; Harris, B.A.; Feng, X.; Ponnazhagan, S. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res. 2013, 73, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Ponnazhagan, S. Myeloid-derived suppressor cells as osteoclast progenitors: A novel target for controlling osteolytic bone metastasis. Cancer Res. 2013, 73, 4606–4610. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef]
- Wang, H.; Wang, S.; Huang, M.; Liang, X.; Tang, Y.-J.; Tang, Y. Targeting Immune-Mediated Dormancy: A Promising Treatment of Cancer. Front. Oncol. 2019, 9, 489–498. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef]
- Shaul, M.E.; Levy, L.; Sun, J.; Mishalian, I.; Singhal, S.; Kapoor, V.; Horng, W.; Fridlender, G.; Albelda, S.M.; Fridlender, Z.G. Tumor-associated neutrophils display a distinct N1 profile following TGFβ modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology 2016, 5, e1232221. [Google Scholar] [CrossRef]
- Capietto, A.H.; Faccio, R. Immune regulation of bone metastasis. Bonekey Rep. 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Pantel, K. Liquid biopsies, what we do not know (Yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Bardelli, A. Blood circulating tumor DNA for non-invasive genotyping of colon cancer patients. Mol. Oncol. 2016, 10, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Bardelli, A. Genotyping cell-free tumor DNA in the blood to detect residual disease and drug resistance. Genome Biol. 2014, 15, 1–6. [Google Scholar] [CrossRef]
- Stevic, I.; Buescher, G.; Ricklefs, F.L. Monitoring therapy efficiency in cancer through extracellular vesicles. Cells 2020, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Rosenow, M.; Xiao, N.; Spetzler, D. Profiling plasma extracellular vesicle by pluronic block-copolymer based enrichment method unveils features associated with breast cancer aggression, metastasis and invasion. J. Extracell. Vesicles 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K. ISEV2018 abstract book. J. Extracell. Vesicles 2018, 7, 1461450. [Google Scholar] [CrossRef]
- Srinivasan, S.; Duval, M.X.; Kaimal, V.; Cuff, C.; Clarke, S.H. Assessment of methods for serum extracellular vesicle small RNA sequencing to support biomarker development. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef]
- Oeyen, E.; Willems, H.; ’T Kindt, R.; Sandra, K.; Boonen, K.; Hoekx, L.; De Wachter, S.; Ameye, F.; Mertens, I. Determination of variability due to biological and technical variation in urinary extracellular vesicles as a crucial step in biomarker discovery studies. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef]
- Ramirez, M.I.; Amorim, M.G.; Gadelha, C.; Milic, I.; Welsh, J.A.; Freitas, V.M.; Nawaz, M.; Akbar, N.; Couch, Y.; Makin, L.; et al. Technical challenges of working with extracellular vesicles. Nanoscale 2018, 10, 881–906. [Google Scholar] [CrossRef]
- Fizazi, K.; Carducci, M.; Smith, M.; Damião, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef]
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; De Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponzetti, M.; Rucci, N. Switching Homes: How Cancer Moves to Bone. Int. J. Mol. Sci. 2020, 21, 4124. https://doi.org/10.3390/ijms21114124
Ponzetti M, Rucci N. Switching Homes: How Cancer Moves to Bone. International Journal of Molecular Sciences. 2020; 21(11):4124. https://doi.org/10.3390/ijms21114124
Chicago/Turabian StylePonzetti, Marco, and Nadia Rucci. 2020. "Switching Homes: How Cancer Moves to Bone" International Journal of Molecular Sciences 21, no. 11: 4124. https://doi.org/10.3390/ijms21114124
APA StylePonzetti, M., & Rucci, N. (2020). Switching Homes: How Cancer Moves to Bone. International Journal of Molecular Sciences, 21(11), 4124. https://doi.org/10.3390/ijms21114124