Epigenetic Landscape in Pancreatic Ductal Adenocarcinoma: On the Way to Overcoming Drug Resistance?

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
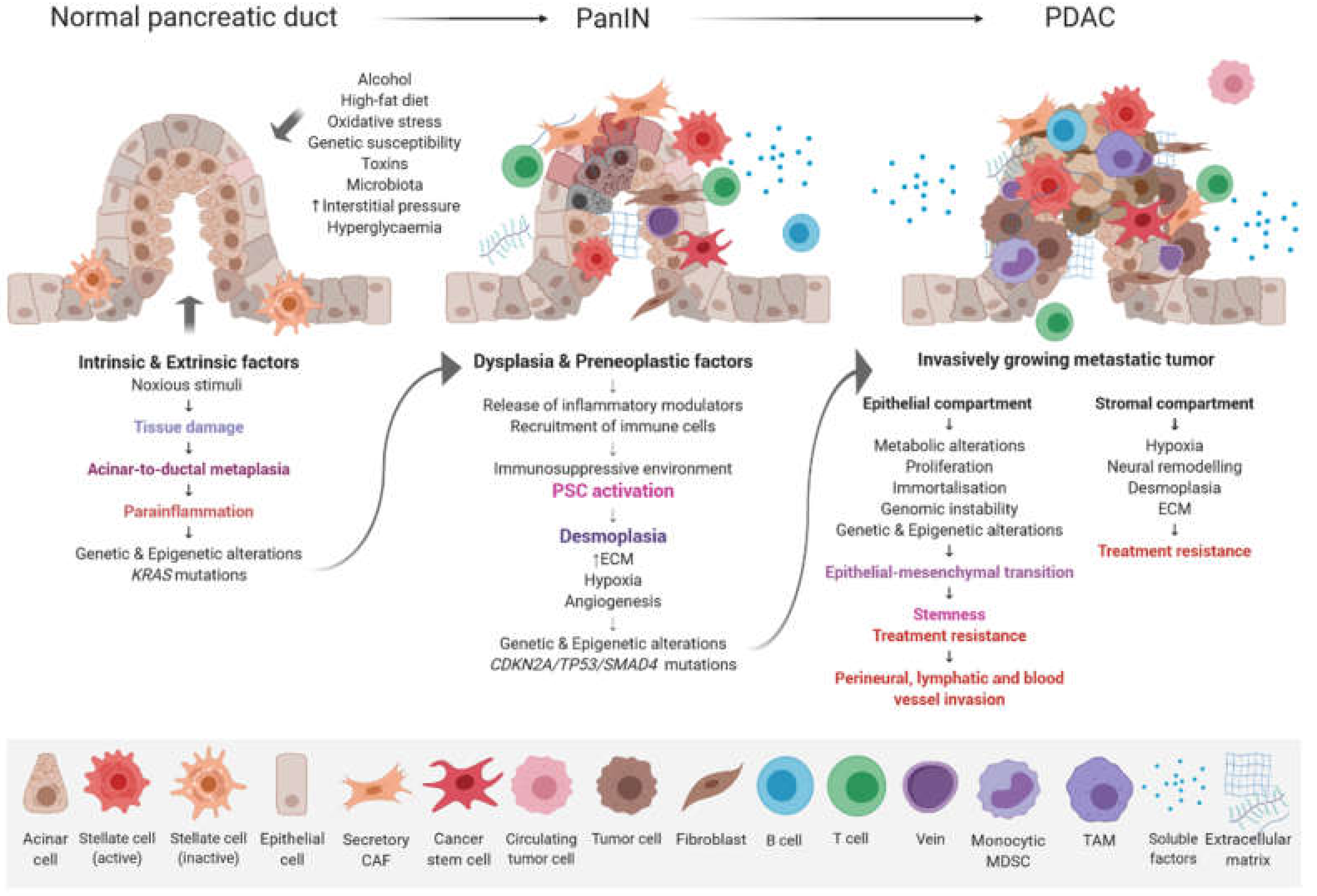
2. PDAC Genomics and its Clinical Relevance
3. Epigenetic Changes Associated with PDAC
3.1. DNA Methylation
3.2. Histone Modifications
3.3. microRNAs
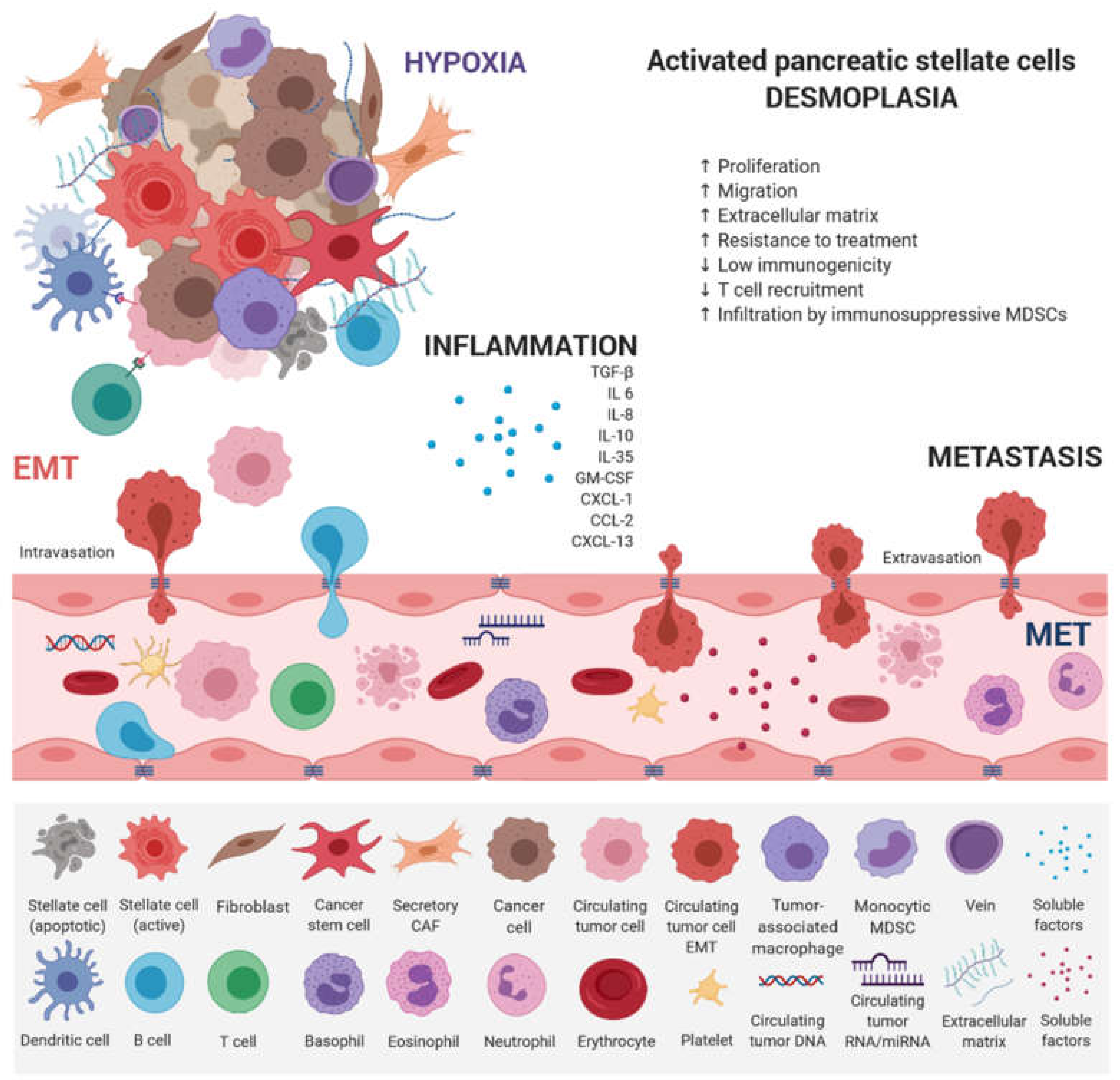
4. Epigenomic Landscapes and Molecular Tumor Heterogeneity
5. Epigenetics and Epithelial–Mesenchymal Transition in PDAC
6. Early Detection—Promises and Challenges of Liquid Biopsy
7. Search for New Epigenetic Treatments in PDAC
8. Future Directions and Clinical Implications
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ac | Acetylation |
| ADEX | Abberantly differentiated endocrine exocrine |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia mutated and Rad3 related |
| BET | Bromodomain and extra-terminal motif |
| BMP2 | Bone morphogenetic protein 2 |
| BRD4 | Bromodomain-containing 4 |
| CAF | Cancer-associated fibroblast |
| CBP | CREB-binding protein |
| CCL-2 | Chemokine (C-C motif) ligand 2 |
| CDH1 | Cadherin 1 |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CTC | Circulating tumor cell |
| CXCL-1 | Chemokine (C-X-C motif) ligand 1 |
| CXCL-13 | Chemokine (C-X-C motif) ligand 13 |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| DAB2 | DAB adaptor protein 2 |
| DNA | Deoxyribonucleic acid |
| DNMT | DNA methyltransferase |
| DOT1Li | Histone H3K79 methyltransferase inhibitor |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| EHMT2 | Histone-lysine N-methyltransferase known as G9A |
| EMT | Epithelial–mesenchymal transition |
| EZH2 | Enhancer of zeste homolog |
| FBP1 | Fructose-bisphosphatase 1 |
| FOXA1/2 | Forkhead box A1/2 |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| HDM | Histone demethylase |
| HKDC1 | Hexokinase domain-containing 1 |
| HMT | Histone methyltransferase |
| H3.3 | Histone 3.3 |
| IGF | Insulin-like growth factor |
| IL | Interleukin |
| KDM1Ai | Lysine-specific histone demethylase 1A inhibitor |
| LCN2 | Lipocalin-2 |
| MDC1 | Mediator of DNA damage checkpoint 1 |
| MDSC | Myeloid-derived suppressor cell |
| Me | Methylation |
| MET | Mesenchymal–epithelial transition |
| miRNA | microRNA |
| mTOR | Mammalian (or Mechanistic) target of rapamycin |
| MUC2 | Mucin 2 |
| NPTX2 | Neuronal pentraxin 2 |
| PanIN | Pancreatic intraepithelial neoplasia |
| PARP | Poly (ADP-ribose) polymerase |
| PCAF | P300/CBP-associated factor |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDX | Patient-derived xenograft |
| PDX1 | Pancreatic and duodenal homeobox 1 |
| PENK | Proenkephalin |
| PI3K | Phosphatidylinositol 3-kinase |
| PRMT | Protein arginine methyltransferase |
| PSC | Pancreatic stellate cell |
| RASSF1 | Ras association domain family member 1 |
| RB | Retinoblastoma |
| RNA | Ribonucleic acid |
| S100P | S100 calcium-binding protein P |
| SERPINB5 | Serpin peptidase inhibitor, clade B, member 5 |
| SFN | Stratifin |
| SHH | Sonic hedgehog |
| SPARC | Secreted protein acidic and cysteine rich |
| SUV39H1 | Histone-lysine N-methyltransferase |
| SWI/SNF | ATP-dependent chromatin remodeling complex |
| TAM | Tumor-associated macrophage |
| TET | Ten-eleven translocation protein |
| TFF2 | Trefoil factor 2 |
| TGF-β | Transforming growth factor-beta |
| VEGFA | Vascular endothelial growth factor A |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar]
- Siegel, R.L.; Miller, K.D. Jemal ACancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar]
- Zhang, Q.; Zeng, L.; Chen, Y.; Lian, G.; Qian, C.; Chen, S.; Li, J.; Huang, K. Pancreatic cancer epidemiology, detection, and management. Gastroenterol. Res. Pract. 2016, 2016. [Google Scholar] [CrossRef]
- Hong, S.-M.; Park, J.Y.; Hruban, R.H.; Goggins, M. Molecular signatures of pancreatic cancer. Arch. Pathol. Lab. Med. 2011, 135, 716–727. [Google Scholar]
- Hruban, R.H.; Adsay, N.V.; Albores–Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Klöppel, G.; Longnecker, D.S. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 2001, 25, 579–586. [Google Scholar]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.-M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.-L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar]
- Ramakrishnan, S. Sno (RNA) wing and Pancreatic Cancer Metastasis. Gastroenterology 2017, 153, 12–14. [Google Scholar]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar]
- Sohn, T.A.; Yeo, C.J.; Cameron, J.L.; Koniaris, L.; Kaushal, S.; Abrams, R.A.; Sauter, P.K.; Coleman, J.; Hruban, R.H.; Lillemoe, K.D. Resected adenocarcinoma of the pancreas—616 patients: Results, outcomes, and prognostic indicators. J. Gastrointest. Surg. 2000, 4, 567–579. [Google Scholar]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic Cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar]
- Waddell, N.; Pajic, M.; Patch, A.; Chang, D.; Kassahn, K.; Bailey, P.; Johns, A.; Miller, D.; Nones, K.; Quek, K. Australian Pancreatic Cancer Genome Initiative Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar]
- Lowery, M.A.; Jordan, E.J.; Basturk, O.; Ptashkin, R.N.; Zehir, A.; Berger, M.F.; Leach, T.; Herbst, B.; Askan, G.; Maynard, H. Real-time genomic profiling of pancreatic ductal adenocarcinoma: Potential actionability and correlation with clinical phenotype. Clin. Cancer Res. 2017, 23, 6094–6100. [Google Scholar]
- Golan, T.; Kanji, Z.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Bowen, K.; Fielding, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germ-line BRCA1/2 mutation: An open-label phase II study. J. Clin. Oncol. 2013, 31, 11024. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Lowery, M.A.; Capanu, M.; Stadler, Z.K.; Moore, M.J.; Dhani, N.; Kindler, H.L.; Estrella, H.; Maynard, H.; et al. Phase 1 trial evaluating cisplatin, gemcitabine, and veliparib in 2 patient cohorts: Germline BRCA mutation carriers and wild-type BRCA pancreatic ductal adenocarcinoma. Cancer 2018, 124, 1374–1382. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L. ATM mutations in hereditary pancreatic cancer patients. Cancer Discov. 2012, 2, 41. [Google Scholar]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar]
- Petersen, G.M. Familial pancreatic cancer. Semin. Oncol. 2016, 43, 548–553. [Google Scholar] [CrossRef]
- Earl, J.; Galindo-Pumariño, C.; Encinas, J.; Barreto, E.; Castillo, M.E.; Pachón, V.; Ferreiro, R.; Rodríguez-Garrote, M.; González-Martínez, S.; Ramon, Y.C.T.; et al. A comprehensive analysis of candidate genes in familial pancreatic cancer families reveals a high frequency of potentially pathogenic germline variants. EBioMedicine 2020, 53, 102675. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, H.; Yan, H. Gene expression profiling of 1200 pancreatic ductal adenocarcinoma reveals novel subtypes. BMC Cancer 2018, 18, 603. [Google Scholar]
- Thompson, M.J.; Rubbi, L.; Dawson, D.W.; Donahue, T.R.; Pellegrini, M. Pancreatic Cancer Patient Survival Correlates with DNA Methylation of Pancreas Development Genes. PLoS ONE 2015, 10, e0128814. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, Y.; Li, X. Cancer epigenetics and the potential of epigenetic drugs for treating solid tumors. Expert Rev. Anticancer Ther. 2019, 19, 139–149. [Google Scholar] [CrossRef]
- Henriksen, S.D.; Madsen, P.H.; Krarup, H.; Thorlacius-Ussing, O. DNA Hypermethylation as a Blood-Based Marker for Pancreatic Cancer: A Literature Review. Pancreas 2015, 44, 1036–1045. [Google Scholar] [CrossRef]
- Syren, P.; Andersson, R.; Bauden, M.; Ansari, D. Epigenetic alterations as biomarkers in pancreatic ductal adenocarcinoma. Scand. J. Gastroenterol. 2017, 52, 668–673. [Google Scholar] [CrossRef]
- Tchio Mantho, C.I.; Harbuzariu, A.; Gonzalez-Perez, R.R. Histone deacetylases, microRNA and leptin crosstalk in pancreatic cancer. World J. Clin. Oncol. 2017, 8, 178–189. [Google Scholar] [CrossRef]
- McCleary-Wheeler, A.L.; Lomberk, G.A.; Weiss, F.U.; Schneider, G.; Fabbri, M.; Poshusta, T.L.; Dusetti, N.J.; Baumgart, S.; Iovanna, J.L.; Ellenrieder, V.; et al. Insights into the epigenetic mechanisms controlling pancreatic carcinogenesis. Cancer Lett. 2013, 328, 212–221. [Google Scholar] [CrossRef]
- Antequera, F.; Bird, A. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr. Biol. 1999, 9, R661–R667. [Google Scholar] [CrossRef]
- Cheng, X.; Blumenthal, R.M. Mammalian DNA methyltransferases: A structural perspective. Structure 2008, 16, 341–350. [Google Scholar] [CrossRef]
- Li, A.; Omura, N.; Hong, S.M.; Goggins, M. Pancreatic cancer DNMT1 expression and sensitivity to DNMT1 inhibitors. Cancer Biol. Ther. 2010, 9, 321–329. [Google Scholar] [CrossRef]
- Tan, A.C.; Jimeno, A.; Lin, S.H.; Wheelhouse, J.; Chan, F.; Solomon, A.; Rajeshkumar, N.V.; Rubio-Viqueira, B.; Hidalgo, M. Characterizing DNA methylation patterns in pancreatic cancer genome. Mol. Oncol. 2009, 3, 425–438. [Google Scholar] [CrossRef]
- Hong, L.; Sun, G.; Peng, L.; Tu, Y.; Wan, Z.; Xiong, H.; Li, Y.; Xiao, W. The interaction between miR-148a and DNMT1 suppresses cell migration and invasion by reactivating tumor suppressor genes in pancreatic cancer. Oncol. Rep. 2018, 40, 2916–2925. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Pradhan, K.; Campbell, N.; Mazdo, J.; Vasantkumar, A.; Maqbool, S.; Bhagat, T.D.; Gupta, S.; Suzuki, M.; Yu, Y.; et al. Altered hydroxymethylation is seen at regulatory regions in pancreatic cancer and regulates oncogenic pathways. Genome Res. 2017, 27, 1830–1842. [Google Scholar] [CrossRef]
- Singh, R.R.; Goldberg, J.; Varghese, A.M.; Yu, K.H.; Park, W.; O’Reilly, E.M. Genomic profiling in pancreatic ductal adenocarcinoma and a pathway towards therapy individualization: A scoping review. Cancer Treat. Rev. 2019, 75, 27–38. [Google Scholar] [CrossRef]
- Zagorac, S.; Alcala, S.; Fernandez Bayon, G.; Bou Kheir, T.; Schoenhals, M.; González-Neira, A.; Fernandez Fraga, M.; Aicher, A.; Heeschen, C.; Sainz, B., Jr. DNMT1 Inhibition Reprograms Pancreatic Cancer Stem Cells via Upregulation of the miR-17-92 Cluster. Cancer Res. 2016, 76, 4546–4558. [Google Scholar] [CrossRef]
- Lomberk, G.A.; Iovanna, J.; Urrutia, R. The promise of epigenomic therapeutics in pancreatic cancer. Epigenomics 2016, 8, 831–842. [Google Scholar] [CrossRef]
- Valor, L.M.; Viosca, J.; Lopez-Atalaya, J.P.; Barco, A. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr. Pharm. Des. 2013, 19, 5051–5064. [Google Scholar] [CrossRef]
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V. Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut 2017, 66, 168–179. [Google Scholar] [CrossRef]
- Yonemori, K.; Kurahara, H.; Maemura, K.; Natsugoe, S. MicroRNA in pancreatic cancer. J. Hum. Genet. 2017, 62, 33–40. [Google Scholar] [CrossRef]
- Hwang, H.W.; Mendell, J.T. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer 2006, 94, 776–780. [Google Scholar] [CrossRef]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef]
- Hampton, T. MicroRNAs linked to pancreatic cancer. JAMA J. Am. Med Assoc. 2007, 297, 937. [Google Scholar]
- Szafranska, A.E.; Davison, T.S.; John, J.; Cannon, T.; Sipos, B.; Maghnouj, A.; Labourier, E.; Hahn, S.A. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene 2007, 26, 4442–4452. [Google Scholar] [CrossRef]
- Hong, T.H.; Park, I.Y. MicroRNA expression profiling of diagnostic needle aspirates from surgical pancreatic cancer specimens. Ann. Surg. Treat. Res. 2014, 87, 290–297. [Google Scholar] [CrossRef]
- Daoud, A.Z.; Mulholland, E.J.; Cole, G.; McCarthy, H.O. MicroRNAs in Pancreatic Cancer: Biomarkers, prognostic, and therapeutic modulators. BMC Cancer 2019, 19, 1130. [Google Scholar] [CrossRef]
- Yonemori, K.; Seki, N.; Idichi, T.; Kurahara, H.; Osako, Y.; Koshizuka, K.; Arai, T.; Okato, A.; Kita, Y.; Arigami, T.; et al. The microRNA expression signature of pancreatic ductal adenocarcinoma by RNA sequencing: Anti-tumour functions of the microRNA-216 cluster. Oncotarget 2017, 8, 70097–70115. [Google Scholar] [CrossRef]
- Wang, H.; Peng, R.; Wang, J.; Qin, Z.; Xue, L. Circulating microRNAs as potential cancer biomarkers: The advantage and disadvantage. Clin. Epigenetics 2018, 10, 59. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Komatsu, S.; Ichikawa, D.; Morimura, R.; Tsujiura, M.; Konishi, H.; Takeshita, H.; Nagata, H.; Arita, T.; Hirajima, S.; et al. Clinical impact of circulating miR-221 in plasma of patients with pancreatic cancer. Br. J. Cancer 2013, 108, 361–369. [Google Scholar] [CrossRef]
- Diaz-Riascos, Z.V.; Ginesta, M.M.; Fabregat, J.; Serrano, T.; Busquets, J.; Buscail, L.; Cordelier, P.; Capellá, G. Expression and Role of MicroRNAs from the miR-200 Family in the Tumor Formation and Metastatic Propensity of Pancreatic Cancer. Mol. Ther. Nucleic Acids 2019, 17, 491–503. [Google Scholar] [CrossRef]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef]
- Khoshchehreh, R.; Totonchi, M.; Carlos Ramirez, J.; Torres, R.; Baharvand, H.; Aicher, A.; Ebrahimi, M.; Heeschen, C. Epigenetic reprogramming of primary pancreatic cancer cells counteracts their in vivo tumourigenicity. Oncogene 2019, 38, 6226–6239. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Neureiter, D.; Jäger, T.; Ocker, M.; Kiesslich, T. Epigenetics and pancreatic cancer: Pathophysiology and novel treatment aspects. World J. Gastroenterol. 2014, 20, 7830–7848. [Google Scholar] [CrossRef]
- Zagorac, S.; Garcia-Bermejo, L.; Sainz, B. The Epigenetic Landscape of Pancreatic Cancer Stem Cells. Epigenomes 2018, 2, 10. [Google Scholar]
- Zhang, Y.; Wei, J.; Wang, H.; Xue, X.; An, Y.; Tang, D.; Yuan, Z.; Wang, F.; Wu, J.; Zhang, J. Epithelial mesenchymal transition correlates with CD24+ CD44+ and CD133+ cells in pancreatic cancer. Oncol. Rep. 2012, 27, 1599–1605. [Google Scholar]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Klymkowsky, M.W.; Savagner, P. Epithelial-mesenchymal transition: A cancer researcher’s conceptual friend and foe. Am. J. Pathol. 2009, 174, 1588–1593. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Voulgari, A.; Pintzas, A. Epithelial–mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta (BBA) Rev. Cancer 2009, 1796, 75–90. [Google Scholar] [CrossRef]
- Vetter, G.; Saumet, A.; Moes, M.; Vallar, L.; Le Béchec, A.; Laurini, C.; Sabbah, M.; Arar, K.; Theillet, C.; Lecellier, C.H.; et al. miR-661 expression in SNAI1-induced epithelial to mesenchymal transition contributes to breast cancer cell invasion by targeting Nectin-1 and StarD10 messengers. Oncogene 2010, 29, 4436–4448. [Google Scholar] [CrossRef]
- Kotsafti, A.; Farinati, F.; Cardin, R.; Cillo, U.; Nitti, D.; Bortolami, M. Autophagy and apoptosis-related genes in chronic liver disease and hepatocellular carcinoma. BMC Gastroenterol. 2012, 12, 118. [Google Scholar]
- Liu, J.; Hu, G.; Chen, D.; Gong, A.; Soori, G.; Dobleman, T.; Chen, X.-M. Suppression of SCARA5 by Snail1 is essential for EMT-associated cell migration of A549 cells. Oncogenesis 2013, 2, e73. [Google Scholar]
- Yang, M.-H.; Wu, M.-Z.; Chiou, S.-H.; Chen, P.-M.; Chang, S.-Y.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar]
- Christofori, G.; Semb, H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci. 1999, 24, 73–76. [Google Scholar]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar]
- Von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361–371.e3715. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Paik, W.H.; Song, B.J.; Kim, H.W.; Kim, H.R.; Hwang, J.H. MicroRNA-200c as a Prognostic Biomarker for Pancreatic Cancer. Korean J. Gastroenterol. 2015, 66, 215–220. [Google Scholar] [CrossRef]
- Song, Y.; Washington, M.K.; Crawford, H.C. Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res. 2010, 70, 2115–2125. [Google Scholar] [CrossRef]
- Moletta, L.; Serafini, S.; Valmasoni, M.; Pierobon, E.S.; Ponzoni, A.; Sperti, C. Surgery for Recurrent Pancreatic Cancer: Is It Effective? Cancers 2019, 11, 991. [Google Scholar] [CrossRef]
- Buscail, E.; Maulat, C.; Muscari, F.; Chiche, L.; Cordelier, P.; Dabernat, S.; Alix-Panabières, C.; Buscail, L. Liquid Biopsy Approach for Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 852. [Google Scholar] [CrossRef]
- Zhang, L.; Sanagapalli, S.; Stoita, A. Challenges in diagnosis of pancreatic cancer. World J. Gastroenterol. 2018, 24, 2047. [Google Scholar]
- Ankeny, J.; Court, C.; Hou, S.; Li, Q.; Song, M.; Wu, D.; Chen, J.; Lee, T.; Lin, M.; Sho, S. Circulating tumour cells as a biomarker for diagnosis and staging in pancreatic cancer. Br. J. Cancer 2016, 114, 1367–1375. [Google Scholar]
- Leung, F.; Kulasingam, V.; Diamandis, E.P.; Hoon, D.S.B.; Kinzler, K.; Pantel, K.; Alix-Panabières, C. Circulating Tumor DNA as a Cancer Biomarker: Fact or Fiction? Clin. Chem. 2016, 62, 1054–1060. [Google Scholar] [CrossRef]
- Sho, S.; Court, C.M.; Kim, S.; Braxton, D.R.; Hou, S.; Muthusamy, V.R.; Watson, R.R.; Sedarat, A.; Tseng, H.-R.; Tomlinson, J.S. Digital PCR improves mutation analysis in pancreas fine needle aspiration biopsy specimens. PLoS ONE 2017, 12, e0170897. [Google Scholar]
- Imamura, T.; Komatsu, S.; Ichikawa, D.; Kawaguchi, T.; Miyamae, M.; Okajima, W.; Ohashi, T.; Arita, T.; Konishi, H.; Shiozaki, A. Liquid biopsy in patients with pancreatic cancer: Circulating tumor cells and cell-free nucleic acids. World J. Gastroenterol. 2016, 22, 5627. [Google Scholar]
- Lewis, A.R.; Valle, J.W.; McNamara, M.G. Pancreatic cancer: Are “liquid biopsies” ready for prime-time? World J. Gastroenterol. 2016, 22, 7175. [Google Scholar]
- Singh, N.; Rashid, S.; Dash, N.R.; Gupta, S.; Saraya, A. Clinical significance of promoter methylation status of tumor suppressor genes in circulating DNA of pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 2020, 146, 897–907. [Google Scholar] [CrossRef]
- Madhavan, B.; Yue, S.; Galli, U.; Rana, S.; Gross, W.; Müller, M.; Giese, N.A.; Kalthoff, H.; Becker, T.; Büchler, M.W. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int. J. Cancer 2015, 136, 2616–2627. [Google Scholar]
- Karasek, P.; Gablo, N.; Hlavsa, J.; Kiss, I.; Vychytilova-Faltejskova, P.; Hermanova, M.; Kala, Z.; Slaby, O.; Prochazka, V. Pre-operative plasma miR-21-5p is a sensitive biomarker and independent prognostic factor in patients with pancreatic ductal adenocarcinoma undergoing surgical resection. Cancer Genom. Proteom. 2018, 15, 321–327. [Google Scholar]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Juiz, N.A.; Iovanna, J.; Dusetti, N. Pancreatic Cancer Heterogeneity Can Be Explained Beyond the Genome. Front. Oncol. 2019, 9, 246. [Google Scholar] [CrossRef]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139–152. [Google Scholar]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]
- Arensman, M.D.; Telesca, D.; Lay, A.R.; Kershaw, K.M.; Wu, N.; Donahue, T.R.; Dawson, D.W. The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol. Cancer Ther. 2014, 13, 2303–2314. [Google Scholar] [CrossRef]
- Ono, H.; Basson, M.D.; Ito, H. P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget 2016, 7, 51301–51310. [Google Scholar] [CrossRef]
- Manegold, P.; Lai, K.K.Y.; Wu, Y.; Teo, J.L.; Lenz, H.J.; Genyk, Y.S.; Pandol, S.J.; Wu, K.; Lin, D.P.; Chen, Y.; et al. Differentiation Therapy Targeting the β-Catenin/CBP Interaction in Pancreatic Cancer. Cancers 2018, 10, 95. [Google Scholar] [CrossRef]
- Gerrard, D.L.; Boyd, J.R.; Stein, G.S.; Jin, V.X.; Frietze, S. Disruption of Broad Epigenetic Domains in PDAC Cells by HAT Inhibitors. Epigenomes 2019, 3, 11. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciernikova, S.; Earl, J.; García Bermejo, M.L.; Stevurkova, V.; Carrato, A.; Smolkova, B. Epigenetic Landscape in Pancreatic Ductal Adenocarcinoma: On the Way to Overcoming Drug Resistance? Int. J. Mol. Sci. 2020, 21, 4091. https://doi.org/10.3390/ijms21114091
Ciernikova S, Earl J, García Bermejo ML, Stevurkova V, Carrato A, Smolkova B. Epigenetic Landscape in Pancreatic Ductal Adenocarcinoma: On the Way to Overcoming Drug Resistance? International Journal of Molecular Sciences. 2020; 21(11):4091. https://doi.org/10.3390/ijms21114091
Chicago/Turabian StyleCiernikova, Sona, Julie Earl, María Laura García Bermejo, Viola Stevurkova, Alfredo Carrato, and Bozena Smolkova. 2020. "Epigenetic Landscape in Pancreatic Ductal Adenocarcinoma: On the Way to Overcoming Drug Resistance?" International Journal of Molecular Sciences 21, no. 11: 4091. https://doi.org/10.3390/ijms21114091
APA StyleCiernikova, S., Earl, J., García Bermejo, M. L., Stevurkova, V., Carrato, A., & Smolkova, B. (2020). Epigenetic Landscape in Pancreatic Ductal Adenocarcinoma: On the Way to Overcoming Drug Resistance? International Journal of Molecular Sciences, 21(11), 4091. https://doi.org/10.3390/ijms21114091