The Emerging Role of Innate Immunity in Chronic Kidney Diseases

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Inflammatory and Fibrotic Pathways in CKD
2.1. Inflammatory Pathway
2.1.1. NF-κB Signaling
2.1.2. Toll-like Receptors
2.2. Fibrotic Pathway
2.2.1. TGF-β1/Smads Signaling
2.2.2. Wnt/β-catenin Signaling
2.2.3. MAP Kinases Signaling
2.2.4. JAK/STAT Signaling
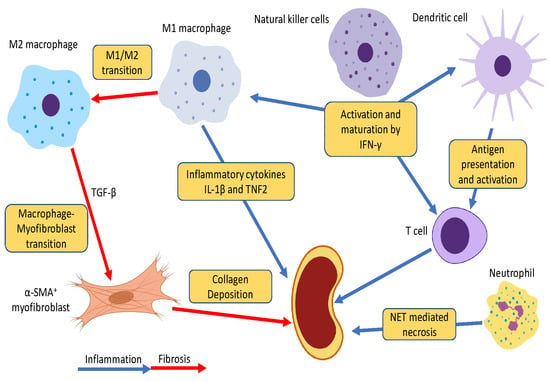
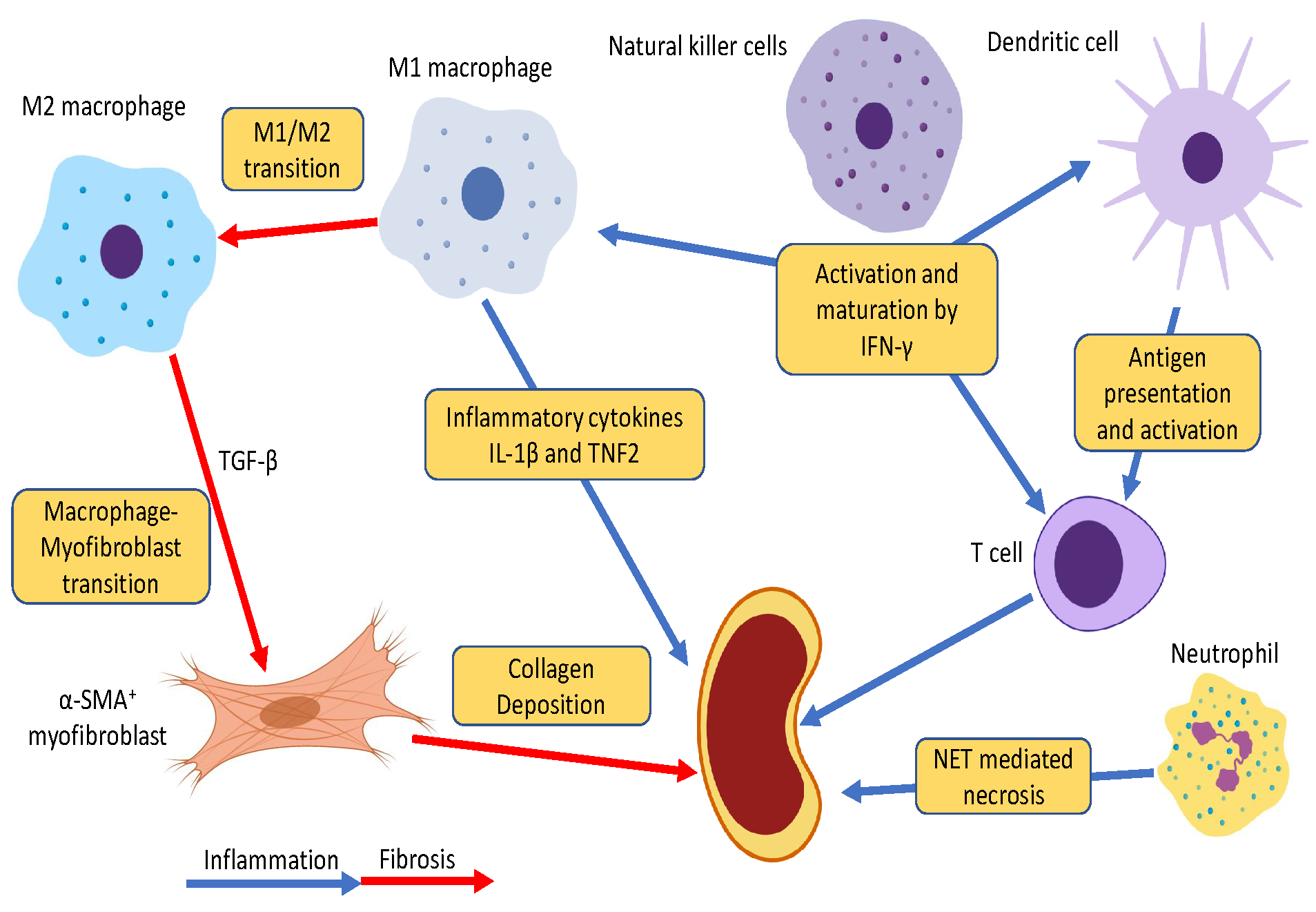
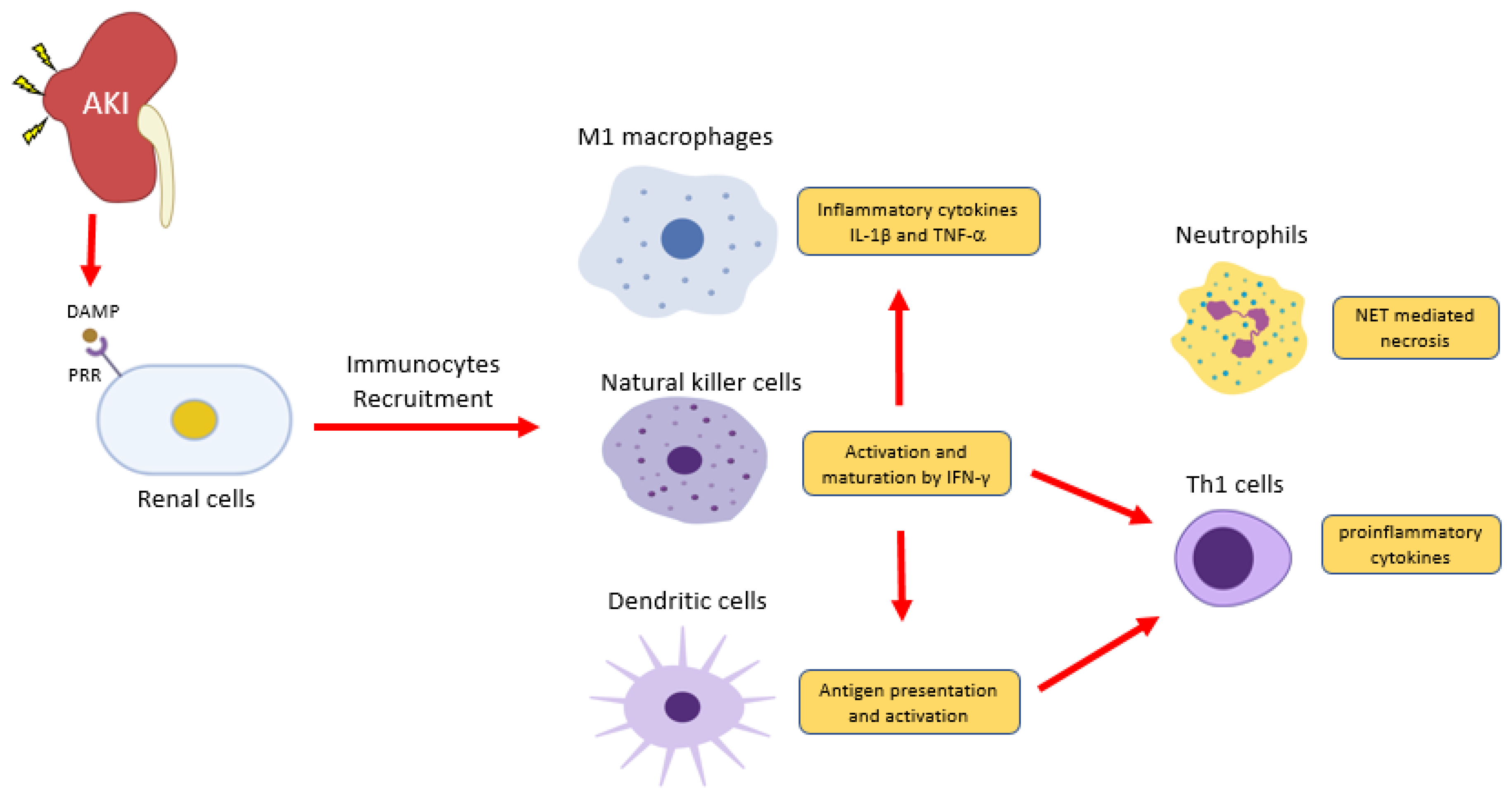
3. Roles of Innate Immunity in CKD
3.1. Neutrophils
3.2. Dendritic Cells
3.3. Natural Killer Cells
3.4. Macrophages
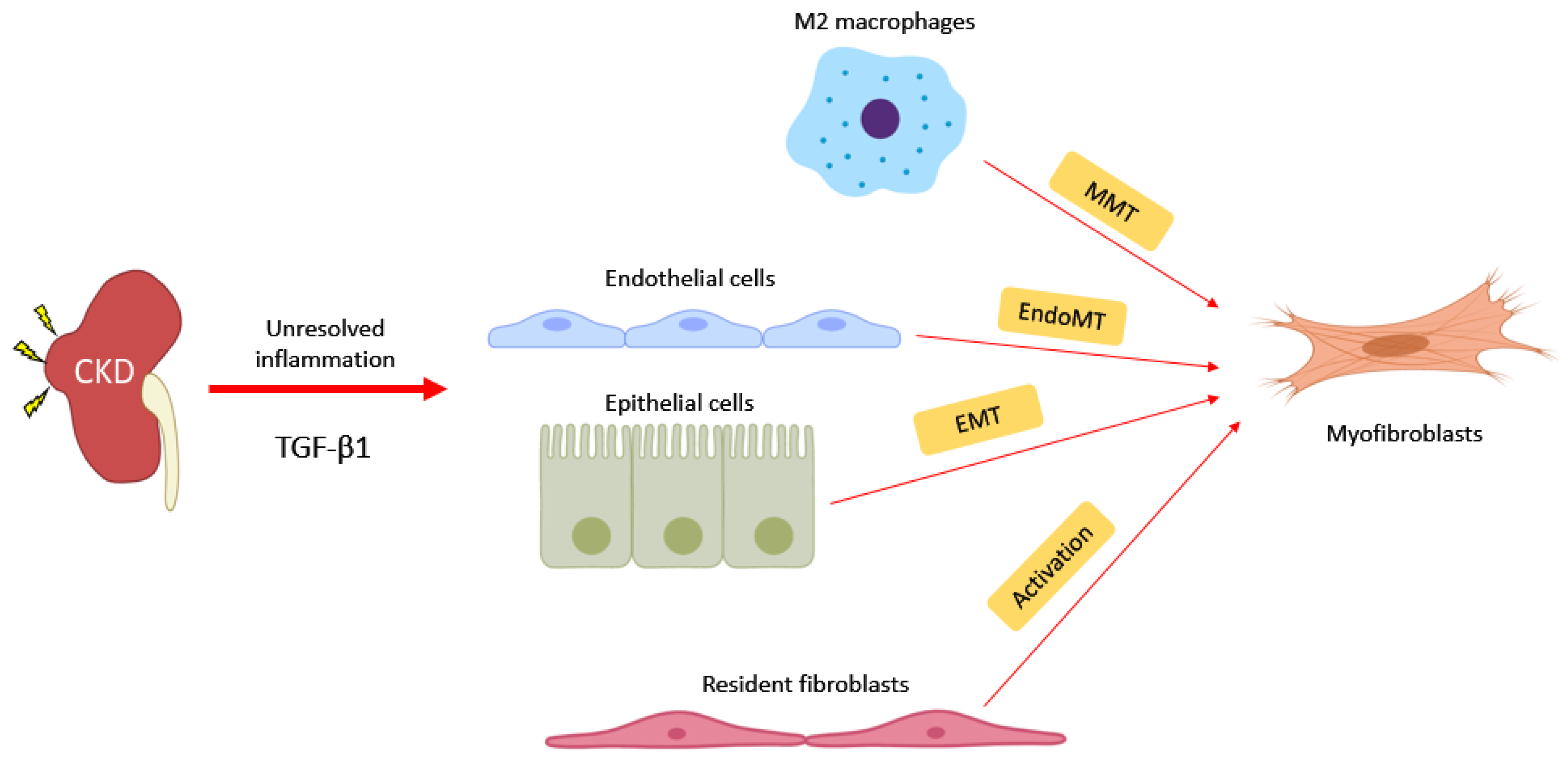
4. Novel Pathogenic Mechanism: Macrophage-Myofibroblast Transition
5. The Perspective of Immunosuppressive for CKD
5.1. Clinical Ready Immunotherapy for CKD
5.1.1. T cell-targeted Therapy
5.1.2. B cell-targeted Therapy
5.1.3. Mesenchymal Stem Cells (MSCs) Therapy
5.1.4. Chimeric Antigen Receptor T (CAR T) Cells Therapy
5.1.5. Inflammatory Reflex Targeted Therapy
5.2. Effect of Immunotherapy in Experimental CKD Models
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ActA | Activin A |
| α7nAChR | α7 Nicotinic Acetylcholine Receptor |
| AKI | Acute Kidney Injury |
| AP-1 | Activator Protein-1 |
| APC | Antigen-Presenting Cell |
| α-SMA | Alpha-Smooth Muscle Actin |
| BAFF | B-Cell Activating Factor |
| CAR-T | Chimeric Antigen Receptor T Cells |
| CD- | Cluster of Differentiation- |
| ChAT+ | Choline Acetyltransferase Positive T Cells |
| CKD | Chronic Kidney Disease |
| CNIs | Calcineurin Inhibitors |
| CTLA4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| DAMPs | Damage-Associated Molecular Patterns |
| DC | Dendritic Cell |
| DKK1 | Dickkopf One |
| EMT | Epithelial-Mesenchymal Transition |
| EndoMT | Endothelial-Mesenchymal Transition |
| ERK | Extracellular-Signal-Regulated Kinase |
| ESRD | End-Stage Renal Disease |
| FGF-23 | Fibroblast Growth Factor 23 |
| FSGS | Focal Segmental Glomerulosclerosis |
| gGT1-Cre | Gamma-Glutamyltransferase 1, Promoter. Cre |
| GVHD | Graft Versus Host Disease |
| IFN- | Interferon- |
| IKK | Iκb Kinase |
| IL- | Interleukin- |
| IMN | Idiopathic Membranous Nephropathy |
| IRI | Ischemia/Reperfusion Injury |
| JAK | Janus Kinase |
| JNK | C-Jun N-Terminal Kinase |
| Ksp-Cre | Cadherin 16 Promoter. Cre |
| LAP | Latency-Associated Peptide |
| LPS | Lipopolysaccharides |
| LTBP | Latent TGF-β Binding Proteins |
| MAPK | Mitogen-Activated Protein Kinases |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| MHC | Major Histocompatibility Complex |
| MMP- | Matrix Metallopeptidase- |
| MMT | Macrophage-To-Myofibroblast Transition |
| MSCs | Mesenchymal Stem Cells |
| MYD88 | Myeloid Differentiation Factor 88 |
| NETs | Neutrophil Extracellular Traps |
| NFAT | Nuclear Factor of Activated T-Cells |
| NF-kB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| NK | Natural Killer Cell |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PDGF | Platelet-Derived Growth Factor |
| ROS | Reactive Oxygen Species |
| SMADs | Mothers Against Decapentaplegic Homologs |
| SRC | Proto-Oncogene Tyrosine-Protein Kinase Src |
| STAT | Signal Transducer and Activator Of Transcription |
| TCR | T-Cell Receptor |
| TGF-β | Transforming Growth Factor-Βeta |
| TGFBR2 | Transforming Growth Factor Beta Receptor 2 |
| Th1/2 | T Helper Type 1/2 |
| TLRs | Toll-Like Receptors |
| TNF- | Tumor Necrosis Factor |
| TSP-1 | Thrombospondin-1 |
| TΒRI | TGF-β Receptor Type I |
| UUO | Unilateral Ureter Obstruction |
| WNT | Wingless-Type MMTV Integration Site Family |
References
- Lv, J.C.; Zhang, L.X. Prevalence and Disease Burden of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [PubMed]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M.; Martinez-Salgado, C.; Rodriguez-Pena, A.B.; Lopez-Hernandez, F.J. Common pathophysiological mechanisms of chronic kidney disease: Therapeutic perspectives. Pharmacol. Ther. 2010, 128, 61–81. [Google Scholar] [CrossRef]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef]
- El Nahas, A.M. Glomerulosclerosis: Intrinsic and extrinsic pathways. Nephrol. Dial. Transplant. 1996, 11, 773–777. [Google Scholar] [CrossRef]
- Tang, P.M.; Zhang, Y.Y.; Lan, H.Y. LncRNAs in TGF-β-Driven Tissue Fibrosis. Noncoding RNA 2018, 4, 26. [Google Scholar] [CrossRef]
- Lan, H.Y. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr. Opin. Nephrol. Hypertens. 2003, 12, 25–29. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Tang, P.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef]
- Qi, R.; Yang, C. Renal tubular epithelial cells: The neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018, 9, 1126. [Google Scholar] [CrossRef]
- Qian, Y.; Feldman, E.; Pennathur, S.; Kretzler, M.; Brosius, F.C., III. From fibrosis to sclerosis: Mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes 2008, 57, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Hickey, F.B.; Martin, F. Role of the Immune System in Diabetic Kidney Disease. Curr. Diab. Rep. 2018, 18, 20. [Google Scholar] [CrossRef]
- Tecklenborg, J.; Clayton, D.; Siebert, S.; Coley, S.M. The role of the immune system in kidney disease. Clin. Exp. Immunol. 2018, 192, 142–150. [Google Scholar] [CrossRef]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef]
- Wang, Y.H.; Zhang, Y.G. Kidney and innate immunity. Immunol. Lett. 2017, 183, 73–78. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Camara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef] [PubMed]
- Rangan, G.; Wang, Y.; Harris, D. NF-kappaB signalling in chronic kidney disease. Front Biosci. (Landmark Ed.) 2009, 14, 3496–3522. [Google Scholar] [CrossRef]
- Mezzano, S.; Aros, C.; Droguett, A.; Burgos, M.E.; Ardiles, L.; Flores, C.; Schneider, H.; Ruiz-Ortega, M.; Egido, J. NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol. Dial. Transplant. 2004, 19, 2505–2512. [Google Scholar] [CrossRef]
- Sanz, A.B.; Sanchez-Nino, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-kappaB in renal inflammation. J. Am. Soc. Nephrol. 2010, 21, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, X.R.; Chen, H.Y.; Penninger, J.M.; Lan, H.Y. Loss of angiotensin-converting enzyme 2 enhances TGF-beta/Smad-mediated renal fibrosis and NF-kappaB-driven renal inflammation in a mouse model of obstructive nephropathy. Lab. Investig. 2012, 92, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Ravichandran, K.; Wang, Q.; Ljubanovic, D.; Edelstein, C.L. NF-kappaB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol. Lett. 2016, 240, 105–113. [Google Scholar] [CrossRef]
- Zhu, L.; Han, J.; Yuan, R.; Xue, L.; Pang, W. Berberine ameliorates diabetic nephropathy by inhibiting TLR4/NF-kappaB pathway. Biol. Res. 2018, 51, 9. [Google Scholar] [CrossRef]
- Czaya, B.; Faul, C. FGF23 and inflammation-a vicious coalition in CKD. Kidney Int. 2019, 96, 813–815. [Google Scholar] [CrossRef]
- Cianciolo, G.; Galassi, A.; Capelli, I.; Schillaci, R.; La Manna, G.; Cozzolino, M. Klotho-FGF23, Cardiovascular Disease, and Vascular Calcification: Black or White? Curr. Vasc. Pharmacol. 2018, 16, 143–156. [Google Scholar] [CrossRef]
- Mattinzoli, D.; Ikehata, M.; Tsugawa, K.; Alfieri, C.M.; Dongiovanni, P.; Trombetta, E.; Valenti, L.; Puliti, A.; Lazzari, L.; Messa, P. FGF23 and Fetuin-A Interaction in the Liver and in the Circulation. Int. J. Biol. Sci. 2018, 14, 586–598. [Google Scholar] [CrossRef]
- Mehta, R.; Cai, X.; Lee, J.; Xie, D.; Wang, X.; Scialla, J.; Anderson, A.H.; Taliercio, J.; Dobre, M.; Chen, J.; et al. Serial Fibroblast Growth Factor 23 Measurements and Risk of Requirement for Kidney Replacement Therapy: The CRIC (Chronic Renal Insufficiency Cohort) Study. Am. J. Kidney Dis. 2019, 75, 908–918. [Google Scholar] [CrossRef]
- Anders, H.J.; Banas, B.; Schlondorff, D. Signaling danger: Toll-like receptors and their potential roles in kidney disease. J. Am. Soc. Nephrol. 2004, 15, 854–867. [Google Scholar] [CrossRef]
- Gluba, A.; Banach, M.; Hannam, S.; Mikhailidis, D.P.; Sakowicz, A.; Rysz, J. The role of Toll-like receptors in renal diseases. Nat. Rev. Nephrol. 2010, 6, 224–235. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Anders, H.J.; Schlondorff, D. Toll-like receptors: Emerging concepts in kidney disease. Curr. Opin. Nephrol. Hypertens. 2007, 16, 177–183. [Google Scholar] [CrossRef]
- O’ullivan, K.M.; Ford, S.L.; Longano, A.; Kitching, A.R.; Holdsworth, S.R. Intrarenal Toll-like receptor 4 and Toll-like receptor 2 expression correlates with injury in antineutrophil cytoplasmic antibody-associated vasculitis. Am. J. Physiol. Renal. Physiol. 2018, 315, F1283–F1294. [Google Scholar] [CrossRef]
- Gao, G.; Zhang, B.; Ramesh, G.; Betterly, D.; Tadagavadi, R.K.; Wang, W.; Reeves, W.B. TNF-alpha mediates increased susceptibility to ischemic AKI in diabetes. Am. J. Physiol. Renal. Physiol. 2013, 304, F515–F521. [Google Scholar] [CrossRef]
- Lin, M.; Yiu, W.H.; Li, R.X.; Wu, H.J.; Wong, D.W.; Chan, L.Y.; Leung, J.C.; Lai, K.N.; Tang, S.C. The TLR4 antagonist CRX-526 protects against advanced diabetic nephropathy. Kidney Int. 2013, 83, 887–900. [Google Scholar] [CrossRef]
- Chen, J.Q.; Szodoray, P.; Zeher, M. Toll-Like Receptor Pathways in Autoimmune Diseases. Clin. Rev. Allergy. Immunol. 2016, 50, 1–17. [Google Scholar] [CrossRef]
- Hou, B.; Reizis, B.; DeFranco, A.L. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008, 29, 272–282. [Google Scholar] [CrossRef]
- Zettel, K.; Korff, S.; Zamora, R.; Morelli, A.E.; Darwiche, S.; Loughran, P.A.; Elson, G.; Shang, L.; Salgado-Pires, S.; Scott, M.J.; et al. Toll-Like Receptor 4 on both Myeloid Cells and Dendritic Cells Is Required for Systemic Inflammation and Organ Damage after Hemorrhagic Shock with Tissue Trauma in Mice. Front. Immunol. 2017, 8, 1672. [Google Scholar] [CrossRef]
- Lan, H.Y.; Chung, A.C. TGF-beta/Smad signaling in kidney disease. Semin. Nephrol. 2012, 32, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.; Zhang, Y.Y.; Mak, T.S.; Tang, P.C.; Huang, X.R.; Lan, H.Y. Transforming growth factor-beta signalling in renal fibrosis: From Smads to non-coding RNAs. J. Physiol. 2018, 596, 3493–3503. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.J.; Yu, X.; Huang, X.R.; Yu, J.W.; Lan, H.Y. Opposing roles for Smad2 and Smad3 in peritoneal fibrosis in vivo and in vitro. Am. J. Pathol. 2014, 184, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Huang, X.R.; Chung, A.C.; Qin, W.; Shao, X.; Igarashi, P.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1477–1487. [Google Scholar] [CrossRef]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chung, A.C.; Qin, W.; Chen, H.Y.; Lan, H.Y. Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012, 81, 266–279. [Google Scholar] [CrossRef]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, X.M.; Huang, X.R.; Wang, X.J.; Yang, L.; Lan, H.Y. Transforming growth factor-beta1 mediates psoriasis-like lesions via a Smad3-dependent mechanism in mice. Clin. Exp. Pharmacol. Physiol. 2014, 41, 921–932. [Google Scholar] [CrossRef]
- Feng, M.; Tang, P.M.; Huang, X.R.; Sun, S.F.; You, Y.K.; Xiao, J.; Lv, L.L.; Xu, A.P.; Lan, H.Y. TGF-beta Mediates Renal Fibrosis via the Smad3-Erbb4-IR Long Noncoding RNA Axis. Mol. Ther. 2018, 26, 148–161. [Google Scholar] [CrossRef]
- Ji, X.; Wang, H.; Wu, Z.; Zhong, X.; Zhu, M.; Zhang, Y.; Tan, R.; Liu, Y.; Li, J.; Wang, L. Specific Inhibitor of Smad3 (SIS3) Attenuates Fibrosis, Apoptosis, and Inflammation in Unilateral Ureteral Obstruction Kidneys by Inhibition of Transforming Growth Factor beta (TGF-beta)/Smad3 Signaling. Med. Sci. Monit. 2018, 24, 1633–1641. [Google Scholar] [CrossRef]
- Yang, Q.; Ren, G.L.; Wei, B.; Jin, J.; Huang, X.R.; Shao, W.; Li, J.; Meng, X.M.; Lan, H.Y. Conditional knockout of TGF-betaRII /Smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics 2019, 9, 8277–8293. [Google Scholar] [CrossRef]
- Gewin, L.; Vadivelu, S.; Neelisetty, S.; Srichai, M.B.; Paueksakon, P.; Pozzi, A.; Harris, R.C.; Zent, R. Deleting the TGF-beta receptor attenuates acute proximal tubule injury. J. Am. Soc. Nephrol. 2012, 23, 2001–2011. [Google Scholar] [CrossRef]
- Hollander, M.C.; Latour, L.L.; Yang, D.; Ishii, H.; Xiao, Z.; Min, Y.; Ray-Choudhury, A.; Munasinghe, J.; Merchant, A.S.; Lin, P.C.; et al. Attenuation of Myeloid-Specific TGFbeta Signaling Induces Inflammatory Cerebrovascular Disease and Stroke. Circ. Res. 2017, 121, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Wada, W.; Kuwano, H.; Hasegawa, Y.; Kojima, I. The dependence of transforming growth factor-beta-induced collagen production on autocrine factor activin A in hepatic stellate cells. Endocrinology 2004, 145, 2753–2759. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Maeshima, A.; Kojima, I.; Nojima, Y. Activin A is a potent activator of renal interstitial fibroblasts. J. Am. Soc. Nephrol. 2004, 15, 91–101. [Google Scholar] [CrossRef]
- Mehta, N.; Krepinsky, J.C. The emerging role of activins in renal disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 136–144. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, C.J.; Liu, Y. Wnt Signaling in Kidney Development and Disease. Prog. Mol. Biol. Transl. Sci. 2018, 153, 181–207. [Google Scholar]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, L.; Wang, Y.; Miao, J.; Hong, X.; Hou, F.F.; Liu, Y. (Pro)renin Receptor Is an Amplifier of Wnt/beta-Catenin Signaling in Kidney Injury and Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 2393–2408. [Google Scholar] [CrossRef]
- Cruciat, C.M.; Ohkawara, B.; Acebron, S.P.; Karaulanov, E.; Reinhard, C.; Ingelfinger, D.; Boutros, M.; Niehrs, C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 2010, 327, 459–463. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Y. Wnt/beta-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nat. Rev. Nephrol. 2015, 11, 535–545. [Google Scholar] [CrossRef]
- Naves, M.A.; Requiao-Moura, L.R.; Soares, M.F.; Silva-Junior, J.A.; Mastroianni-Kirsztajn, G.; Teixeira, V.P. Podocyte Wnt/ss-catenin pathway is activated by integrin-linked kinase in clinical and experimental focal segmental glomerulosclerosis. J. Nephrol. 2012, 25, 401–409. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; He, W.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Liu, Y. Mutual antagonism of Wilms’ tumor 1 and beta-catenin dictates podocyte health and disease. J. Am. Soc. Nephrol. 2015, 26, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.A.; Gonzalez, A.A.; Inestrosa, N.C.; Vio, C.P.; Prieto, M.C. Angiotensin II increases fibronectin and collagen I through the beta-catenin-dependent signaling in mouse collecting duct cells. Am. J. Physiol. Renal. Physiol. 2015, 308, F358–F365. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.J.; Zhou, D.; Zhou, L.; Liu, Y. Wnt/beta-catenin signaling and kidney fibrosis. Kidney Int. 2014, 4 (Suppl. 2011), 84–90. [Google Scholar] [CrossRef]
- DiRocco, D.P.; Kobayashi, A.; Taketo, M.M.; McMahon, A.P.; Humphreys, B.D. Wnt4/beta-catenin signaling in medullary kidney myofibroblasts. J. Am. Soc. Nephrol. 2013, 24, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Li, B.; Rao, S.; Yeo, E.J.; Hudson, T.E.; Nowlin, B.T.; Pei, H.; Chen, L.; Zheng, J.J.; Carroll, T.J.; et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 4194–4199. [Google Scholar] [CrossRef]
- Zhou, D.; Li, Y.; Lin, L.; Zhou, L.; Igarashi, P.; Liu, Y. Tubule-specific ablation of endogenous beta-catenin aggravates acute kidney injury in mice. Kidney Int. 2012, 82, 537–547. [Google Scholar] [CrossRef]
- Nishikawa, K.; Osawa, Y.; Kimura, K. Wnt/beta-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. Int. J. Mol. Sci. 2018, 19, 3103. [Google Scholar] [CrossRef]
- Okazaki, H.; Sato, S.; Koyama, K.; Morizumi, S.; Abe, S.; Azuma, M.; Chen, Y.; Goto, H.; Aono, Y.; Ogawa, H.; et al. The novel inhibitor PRI-724 for Wnt/beta-catenin/CBP signaling ameliorates bleomycin-induced pulmonary fibrosis in mice. Exp. Lung. Res. 2019, 45, 188–199. [Google Scholar] [CrossRef]
- Ma, F.Y.; Sachchithananthan, M.; Flanc, R.S.; Nikolic-Paterson, D.J. Mitogen activated protein kinases in renal fibrosis. Front. Biosci. (Schol. Ed.) 2009, 1, 171–187. [Google Scholar] [CrossRef]
- Sekine, S.; Nitta, K.; Uchida, K.; Yumura, W.; Nihei, H. Possible involvement of mitogen-activated protein kinase in the angiotensin II-induced fibronectin synthesis in renal interstitial fibroblasts. Arch. Biochem. Biophys. 2003, 415, 63–68. [Google Scholar] [CrossRef]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK Signaling Pathway in Renal Fibrosis. Front. Physiol. 2017, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Inoki, K.; Isono, M.; Mori, H.; Kanasaki, K.; Sugimoto, T.; Akiba, S.; Sato, T.; Yang, B.; Kikkawa, R.; et al. MAPK/AP-1-dependent regulation of PAI-1 gene expression by TGF-beta in rat mesangial cells. Kidney Int. 2005, 68, 972–984. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Deng, D.; Zhang, Q.; Liu, W. Renalase Protects against Renal Fibrosis by Inhibiting the Activation of the ERK Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 855. [Google Scholar] [CrossRef]
- Ma, F.Y.; Flanc, R.S.; Tesch, G.H.; Han, Y.; Atkins, R.C.; Bennett, B.L.; Friedman, G.C.; Fan, J.H.; Nikolic-Paterson, D.J. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J. Am. Soc. Nephrol. 2007, 18, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Masaki, T.; Nikolic-Paterson, D.J.; Tanji, C.; Yorioka, N.; Kohno, N. Angiotensin II induces thrombospondin-1 production in human mesangial cells via p38 MAPK and JNK: A mechanism for activation of latent TGF-beta1. Am. J. Physiol. Renal. Physiol. 2004, 286, F278–F287. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, X.; Wang, B.; Nie, Y.; Wen, J.; Wang, Q.; Gu, C. Pirfenidone suppresses MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology (Carlton.) 2017, 22, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Matsui, F.; Meldrum, K.K. The role of the Janus kinase family/signal transducer and activator of transcription signaling pathway in fibrotic renal disease. J. Surg. Res. 2012, 178, 339–345. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Bienaime, F.; Muorah, M.; Yammine, L.; Burtin, M.; Nguyen, C.; Baron, W.; Garbay, S.; Viau, A.; Broueilh, M.; Blanc, T.; et al. Stat3 Controls Tubulointerstitial Communication during CKD. J. Am. Soc. Nephrol. 2016, 27, 3690–3705. [Google Scholar] [CrossRef] [PubMed]
- Berthier, C.C.; Zhang, H.; Schin, M.; Henger, A.; Nelson, R.G.; Yee, B.; Boucherot, A.; Neusser, M.A.; Cohen, C.D.; Carter-Su, C.; et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 2009, 58, 469–477. [Google Scholar] [CrossRef]
- Yokota, T.; Omachi, K.; Suico, M.A.; Kamura, M.; Kojima, H.; Fukuda, R.; Motomura, K.; Teramoto, K.; Kaseda, S.; Kuwazuru, J.; et al. STAT3 inhibition attenuates the progressive phenotypes of Alport syndrome mouse model. Nephrol. Dial. Transplant. 2018, 33, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Fu, P.; Huang, X.R.; Liu, F.; Lai, K.N.; Lan, H.Y. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J. Am. Soc. Nephrol. 2010, 21, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Bae, E.; Kwon, S.H.; Yu, M.Y.; Cha, R.H.; Lee, H.; Kim, D.K.; Lee, J.P.; Ye, S.K.; Yoo, J.Y.; et al. Transcriptional modulation of the T helper 17/interleukin 17 axis ameliorates renal ischemia-reperfusion injury. Nephrol. Dial. Transplant. 2019, 34, 1481–1498. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.V.; Wu, Y.Y.; Liu, Q.; Wang, D.; Nguyen, S.; Loh, R.; Pang, J.; Friedman, K.; Orlofsky, A.; Augenlicht, L.; et al. STAT3 in epithelial cells regulates inflammation and tumor progression to malignant state in colon. Neoplasia 2013, 15, 998–1008. [Google Scholar] [CrossRef]
- Reindl, W.; Weiss, S.; Lehr, H.A.; Forster, I. Essential crosstalk between myeloid and lymphoid cells for development of chronic colitis in myeloid-specific signal transducer and activator of transcription 3-deficient mice. Immunology 2007, 120, 19–27. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Brosius, F.C., III; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transplant. 2018, 33, 1950–1959. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, H.C.; Fogo, A.B. A perspective on chronic kidney disease progression. Am. J. Physiol. Renal. Physiol. 2017, 312, F375–F384. [Google Scholar] [CrossRef]
- Bonavia, A.; Singbartl, K. A review of the role of immune cells in acute kidney injury. Pediatr. Nephrol. 2018, 33, 1629–1639. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive Oxygen Species and Neutrophil Function. Annu. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef] [PubMed]
- Rabadi, M.; Kim, M.; D’Agati, V.; Lee, H.T. Peptidyl arginine deiminase-4-deficient mice are protected against kidney and liver injury after renal ischemia and reperfusion. Am. J. Physiol. Renal. Physiol. 2016, 311, F437–F449. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Marschner, J.A.; Platen, L.; Anders, H.J. Extracellular traps in kidney disease. Kidney Int. 2018, 94, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Liang, D.; Gajjalaiahvari, U.R.; Kabbaj, M.H.; Paik, J.; Gunjan, A. Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle 2010, 9, 4236–4244. [Google Scholar] [CrossRef]
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.W.; et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J. Am. Soc. Nephrol. 2017, 28, 1753–1768. [Google Scholar] [CrossRef]
- Liu, Y.J. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell 2001, 106, 259–262. [Google Scholar] [CrossRef]
- Qian, C.; Cao, X. Dendritic cells in the regulation of immunity and inflammation. Semin. Immunol. 2018, 35, 3–11. [Google Scholar] [CrossRef]
- Segura, E.; Amigorena, S. Inflammatory dendritic cells in mice and humans. Trends Immunol. 2013, 34, 440–445. [Google Scholar] [CrossRef]
- Wang, R.; Chen, T.; Wang, C.; Zhang, Z.; Wang, X.M.; Li, Q.; Lee, V.W.S.; Wang, Y.M.; Zheng, G.; Alexander, S.I.; et al. Flt3 inhibition alleviates chronic kidney disease by suppressing CD103+ dendritic cell-mediated T cell activation. Nephrol. Dial. Transplant. 2019, 34, 1853–1863. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Vivier, E.; Nunes, J.A.; Vely, F. Natural killer cell signaling pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Bajenoff, M.; Breart, B.; Huang, A.Y.; Qi, H.; Cazareth, J.; Braud, V.M.; Germain, R.N.; Glaichenhaus, N. Natural killer cell behavior in lymph nodes revealed by static and real-time imaging. J. Exp. Med. 2006, 203, 619–631. [Google Scholar] [CrossRef]
- Moretta, L.; Ferlazzo, G.; Bottino, C.; Vitale, M.; Pende, D.; Mingari, M.C.; Moretta, A. Effector and regulatory events during natural killer-dendritic cell interactions. Immunol. Rev. 2006, 214, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Ikezumi, Y.; Hurst, L.; Atkins, R.C.; Nikolic-Paterson, D.J. Macrophage-mediated renal injury is dependent on signaling via the JNK pathway. J. Am. Soc. Nephrol. 2004, 15, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Sweet, M.J.; Hume, D.A. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology 2006, 211, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lee, J.; Raz, E.; Corr, M.; Carson, D.A. Necessity of oligonucleotide aggregation for toll-like receptor 9 activation. J. Biol. Chem. 2004, 279, 33071–33078. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.; Kulkarni, O.P.; Radomska, E.; Miosge, N.; Gross, O.; Anders, H.J. Bacterial CpG-DNA accelerates Alport glomerulosclerosis by inducing an M1 macrophage phenotype and tumor necrosis factor-alpha-mediated podocyte loss. Kidney Int. 2011, 79, 189–198. [Google Scholar] [CrossRef]
- Tomosugi, N.I.; Cashman, S.J.; Hay, H.; Pusey, C.D.; Evans, D.J.; Shaw, A.; Rees, A.J. Modulation of antibody-mediated glomerular injury in vivo by bacterial lipopolysaccharide, tumor necrosis factor, and IL-1. J. Immunol. 1989, 142, 3083–3090. [Google Scholar]
- Timoshanko, J.R.; Sedgwick, J.D.; Holdsworth, S.R.; Tipping, P.G. Intrinsic renal cells are the major source of tumor necrosis factor contributing to renal injury in murine crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2003, 14, 1785–1793. [Google Scholar] [CrossRef]
- Tesch, G.H.; Yang, N.; Yu, H.; Lan, H.Y.; Foti, R.; Chadban, S.J.; Atkins, R.C.; Nikolic-Paterson, D.J. Intrinsic renal cells are the major source of interleukin-1 beta synthesis in normal and diseased rat kidney. Nephrol. Dial. Transplant. 1997, 12, 1109–1115. [Google Scholar] [CrossRef]
- Han, Y.; Ma, F.Y.; Tesch, G.H.; Manthey, C.L.; Nikolic-Paterson, D.J. c-fms blockade reverses glomerular macrophage infiltration and halts development of crescentic anti-GBM glomerulonephritis in the rat. Lab. Investig. 2011, 91, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.G.; Kim, S.C.; Ko, Y.S.; Lee, H.Y.; Jo, S.K.; Cho, W. The Role of M2 Macrophages in the Progression of Chronic Kidney Disease following Acute Kidney Injury. PLoS ONE 2015, 10, e0143961. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Liu, X.; Fan, Y.; Qiu, J. Macrophages regulate renal fibrosis through modulating TGFbeta superfamily signaling. Inflammation 2014, 37, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Huen, S.C.; Cantley, L.G. Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr. Nephrol. 2015, 30, 199–209. [Google Scholar] [CrossRef]
- Lin, L.; Hu, K. Tissue-type plasminogen activator modulates macrophage M2 to M1 phenotypic change through annexin A2-mediated NF-kappaB pathway. Oncotarget 2017, 8, 88094–88103. [Google Scholar] [CrossRef]
- Han, Y.; Ma, F.Y.; Tesch, G.H.; Manthey, C.L.; Nikolic-Paterson, D.J. Role of macrophages in the fibrotic phase of rat crescentic glomerulonephritis. Am. J. Physiol. Renal. Physiol. 2013, 304, F1043–F1053. [Google Scholar] [CrossRef]
- Belliere, J.; Casemayou, A.; Ducasse, L.; Zakaroff-Girard, A.; Martins, F.; Iacovoni, J.S.; Guilbeau-Frugier, C.; Buffin-Meyer, B.; Pipy, B.; Chauveau, D.; et al. Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J. Am. Soc. Nephrol. 2015, 26, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; DHT, I.J. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transplant. 2017, 32, 1322–1329. [Google Scholar] [CrossRef]
- Ikezumi, Y.; Suzuki, T.; Yamada, T.; Hasegawa, H.; Kaneko, U.; Hara, M.; Yanagihara, T.; Nikolic-Paterson, D.J.; Saitoh, A. Alternatively activated macrophages in the pathogenesis of chronic kidney allograft injury. Pediatr. Nephrol. 2015, 30, 1007–1017. [Google Scholar] [CrossRef]
- Du, X.; Shimizu, A.; Masuda, Y.; Kuwahara, N.; Arai, T.; Kataoka, M.; Uchiyama, M.; Kaneko, T.; Akimoto, T.; Iino, Y.; et al. Involvement of matrix metalloproteinase-2 in the development of renal interstitial fibrosis in mouse obstructive nephropathy. Lab. Investig. 2012, 92, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Kipari, T.; Haslett, C.; Iredale, J.P.; Liu, F.T.; Hughes, J.; Sethi, T. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 2008, 172, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y.; et al. TGF-beta/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar] [CrossRef]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Jiang, H.; Pan, J.; Huang, X.R.; Wang, Y.C.; Huang, H.F.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y.; Chen, J.H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’onnell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Fujimoto, M.; Maezawa, Y.; Yokote, K.; Joh, K.; Kobayashi, K.; Kawamura, H.; Nishimura, M.; Roberts, A.B.; Saito, Y.; Mori, S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem. Biophys. Res. Commun. 2003, 305, 1002–1007. [Google Scholar] [CrossRef]
- Moon, J.A.; Kim, H.T.; Cho, I.S.; Sheen, Y.Y.; Kim, D.K. IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006, 70, 1234–1243. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, S. Src family kinases in chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2017, 313, F721–F728. [Google Scholar] [CrossRef]
- Tang, P.M.; Zhou, S.; Li, C.J.; Liao, J.; Xiao, J.; Wang, Q.M.; Lian, G.Y.; Li, J.; Huang, X.R.; To, K.F.; et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018, 93, 173–187. [Google Scholar] [CrossRef]
- Wiseman, A.C. Immunosuppressive Medications. Clin. J. Am. Soc. Nephrol. 2016, 11, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Praga, M.; Barrio, V.; Juarez, G.F.; Luno, J.; Grupo Espanol de Estudio de la Nefropatia Membranosa. Tacrolimus monotherapy in membranous nephropathy: A randomized controlled trial. Kidney Int. 2007, 71, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Donnelly, M.; Merscher-Gomez, S.; Chang, Y.H.; Franz, S.; Delfgaauw, J.; Chang, J.M.; Choi, H.Y.; Campbell, K.N.; Kim, K.; et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 2008, 14, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Braun, N.; Schmutzler, F.; Lange, C.; Perna, A.; Remuzzi, G.; Risler, T.; Willis, N.S. Immunosuppressive treatment for focal segmental glomerulosclerosis in adults. Cochrane Database Syst. Rev. 2008, CD003233. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.P.; Pearson, T.C.; Adams, A.B.; Tso, P.; Shirasugi, N.; Strobert, E.; Anderson, D.; Cowan, S.; Price, K.; Naemura, J.; et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am. J. Transplant. 2005, 5, 443–453. [Google Scholar] [CrossRef]
- Maxwell, L.; Singh, J.A. Abatacept for rheumatoid arthritis. Cochrane Database Syst. Rev. 2009, CD007277. [Google Scholar] [CrossRef]
- Salama, A.D.; Pusey, C.D. Drug insight: Rituximab in renal disease and transplantation. Nat. Clin. Pract. Nephrol. 2006, 2, 221–230. [Google Scholar] [CrossRef]
- Manzi, S.; Sanchez-Guerrero, J.; Merrill, J.T.; Furie, R.; Gladman, D.; Navarra, S.V.; Ginzler, E.M.; D’Cruz, D.P.; Doria, A.; Cooper, S.; et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: Combined results from two phase III trials. Ann. Rheum. Dis. 2012, 71, 1833–1838. [Google Scholar] [CrossRef]
- Gaballa, M.R.; Laubach, J.P.; Schlossman, R.L.; Redman, K.; Noonan, K.; Mitsiades, C.S.; Ghobrial, I.M.; Munshi, N.; Anderson, K.C.; Richardson, P.G. Management of myeloma-associated renal dysfunction in the era of novel therapies. Expert. Rev. Hematol. 2012, 5, 51–66, quiz 67–68. [Google Scholar] [CrossRef]
- Zhuang, Q.; Ma, R.; Yin, Y.; Lan, T.; Yu, M.; Ming, Y. Mesenchymal Stem Cells in Renal Fibrosis: The Flame of Cytotherapy. Stem. Cells Int. 2019, 2019, 8387350. [Google Scholar] [CrossRef]
- Kuppe, C.; Kramann, R. Role of mesenchymal stem cells in kidney injury and fibrosis. Curr. Opin. Nephrol. Hypertens. 2016, 25, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Rota, C.; Morigi, M.; Cerullo, D.; Introna, M.; Colpani, O.; Corna, D.; Capelli, C.; Rabelink, T.J.; Leuning, D.G.; Rottoli, D.; et al. Therapeutic potential of stromal cells of non-renal or renal origin in experimental chronic kidney disease. Stem. Cell Res. Ther. 2018, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Togel, F.; Cohen, A.; Zhang, P.; Yang, Y.; Hu, Z.; Westenfelder, C. Autologous and allogeneic marrow stromal cells are safe and effective for the treatment of acute kidney injury. Stem. Cells Dev. 2009, 18, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Huuskes, B.M.; Wise, A.F.; Cox, A.J.; Lim, E.X.; Payne, N.L.; Kelly, D.J.; Samuel, C.S.; Ricardo, S.D. Combination therapy of mesenchymal stem cells and serelaxin effectively attenuates renal fibrosis in obstructive nephropathy. FASEB J. 2015, 29, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Alatab, S.; Shekarchian, S.; Najafi, I.; Moghadasali, R.; Ahmadbeigi, N.; Pourmand, M.R.; Bolurieh, T.; Jaroughi, N.; Pourmand, G.; Aghdami, N. Systemic Infusion of Autologous Adipose Tissue-Derived Mesenchymal Stem Cells in Peritoneal Dialysis Patients: Feasibility and Safety. Cell J. 2019, 20, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018, 15, 31–46. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Gupta, S.; Seethapathy, H.; Strohbehn, I.A.; Frigault, M.J.; O’Donnell, E.K.; Jacobson, C.A.; Motwani, S.S.; Parikh, S.M.; Curhan, G.C.; Reynolds, K.L.; et al. Acute Kidney Injury and Electrolyte Abnormalities After Chimeric Antigen Receptor T-Cell (CAR-T) Therapy for Diffuse Large B-Cell Lymphoma. Am. J. Kidney Dis. 2020. [Google Scholar] [CrossRef]
- Kitching, A.R.; Jaw, J. Chimeric antigen receptor T (CAR T) cells: Another cancer therapy with potential applications in kidney disease and transplantation? Kidney Int. 2018, 94, 4–6. [Google Scholar] [CrossRef]
- Hosoi, T.; Okuma, Y.; Matsuda, T.; Nomura, Y. Novel pathway for LPS-induced afferent vagus nerve activation: Possible role of nodose ganglion. Auton. Neurosci. 2005, 120, 104–107. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Ochani, M.; Parrish, W.R.; Ochani, K.; Harris, Y.T.; Huston, J.M.; Chavan, S.; Tracey, K.J. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. USA 2008, 105, 11008–11013. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Gigliotti, J.C.; Huang, L.; Bajwa, A.; Ye, H.; Mace, E.H.; Hossack, J.A.; Kalantari, K.; Inoue, T.; Rosin, D.L.; Okusa, M.D. Ultrasound Modulates the Splenic Neuroimmune Axis in Attenuating AKI. J. Am. Soc. Nephrol. 2015, 26, 2470–2481. [Google Scholar] [CrossRef] [PubMed]
- Gigliotti, J.C.; Huang, L.; Ye, H.; Bajwa, A.; Chattrabhuti, K.; Lee, S.; Klibanov, A.L.; Kalantari, K.; Rosin, D.L.; Okusa, M.D. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J. Am. Soc. Nephrol. 2013, 24, 1451–1460. [Google Scholar] [CrossRef]
- Ribes, D.; Casemayou, A.; El Hachem, H.; Laurent, C.; Guilbeau-Frugier, C.; Vergez, F.; Tavitian, S.; Schanstra, J.P.; Chauveau, D.; Bascands, J.L.; et al. Asymptomatic circulating T-cell clone cause renal polymorphic inflammatory fibrosis. Clin. Exp. Nephrol. 2017, 21, 781–786. [Google Scholar] [CrossRef]
- Wen, Y.; Rudemiller, N.P.; Zhang, J.; Jeffs, A.D.; Griffiths, R.; Lu, X.; Ren, J.; Privratsky, J.; Crowley, S.D. Stimulating Type 1 Angiotensin Receptors on T Lymphocytes Attenuates Renal Fibrosis. Am. J. Pathol. 2019, 189, 981–988. [Google Scholar] [CrossRef]
- Han, H.; Zhu, J.; Wang, Y.; Zhu, Z.; Chen, Y.; Lu, L.; Jin, W.; Yan, X.; Zhang, R. Renal recruitment of B lymphocytes exacerbates tubulointerstitial fibrosis by promoting monocyte mobilization and infiltration after unilateral ureteral obstruction. J. Pathol. 2017, 241, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.; Emal, D.; Teske, G.J.; Dessing, M.C.; Florquin, S.; Roelofs, J.J. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. 2017, 91, 352–364. [Google Scholar] [CrossRef]
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Muller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef] [PubMed]
- Westhorpe, C.L.; Bayard, J.E.; O’Sullivan, K.M.; Hall, P.; Cheng, Q.; Kitching, A.R.; Hickey, M.J. In Vivo Imaging of Inflamed Glomeruli Reveals Dynamics of Neutrophil Extracellular Trap Formation in Glomerular Capillaries. Am. J. Pathol. 2017, 187, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Lu, J.; Li, Q.; Wang, C.; Wang, X.M.; Lee, V.W.; Wang, C.; Nguyen, H.; Zheng, G.; Zhao, Y.; et al. CD103+ Dendritic Cells Elicit CD8+ T Cell Responses to Accelerate Kidney Injury in Adriamycin Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 1344–1360. [Google Scholar] [CrossRef] [PubMed]
- Spada, R.; Rojas, J.M.; Perez-Yague, S.; Mulens, V.; Cannata-Ortiz, P.; Bragado, R.; Barber, D.F. NKG2D ligand overexpression in lupus nephritis correlates with increased NK cell activity and differentiation in kidneys but not in the periphery. J. Leukoc. Biol. 2015, 97, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.X.; Wang, S.; Huang, X.; Min, W.P.; Sun, H.; Liu, W.; Garcia, B.; Jevnikar, A.M. NK cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J. Immunol. 2008, 181, 7489–7498. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Wang, S.; Huang, X.R.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016, 7, e2495. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Diseases | Models | Role of Inflammatory Cells | Ref. |
|---|---|---|---|
| Neutrophils | |||
| AKI | Renal I/R injury | Neutrophils release extracellular DNA (NET) to stimulate inflammation via toll-like receptor signaling and platelet activation. | [17] |
| AKI | Renal I/R injury | Neutrophils induce tubular necrosis via PAD-mediated NET formation | [18] |
| Glomerulo-nephritis | Anti-GBM Nephritis | Histones released by neutrophils induce glomerular vascular injury by direct killing of endothelial cells | [19,20] |
| Dendritic cells | |||
| Focal segmental glomerulo-sclerosis | Adriamycin nephropathy | CD103+ dendritic cells activate CD8+ T cells to induce apoptosis of tubular epithelial cells and inflammatory cytokines (TNF-α and IFN-γ) release. | [21,22] |
| Natural killer cells | |||
| Lupus nephritis | MRL/MpJ, MRL/lpr mice | Infiltrated NK cells secret IFN-γ to promote renal inflammation | [23] |
| AKI | Renal I/R injury | Activated NK cells induce kidney injury via attacking tubular epithelial cells | [24] |
| Macrophages | |||
| Crescentic glomerulonephritis | Anti-GBM Nephritis | Macrophages express pro-inflammatory molecules (tumor necrosis factor, MMP-12, CCL2, and IL-12) in crescentic injury. | [25,26] |
| Renal fibrosis | Unilateral Ureter Obstruction | Alternative activated macrophage produces pro-fibrotic molecules (MMPs and Galectin 3) for the development of renal fibrosis | [27,28] |
| Renal fibrosis | Unilateral Ureter Obstruction Kidney Trans-plantation | Alternative activated macrophage further transits into α-SMA+ collagen-producing myofibroblast for extensive extracellular matrix deposition | [29,30,31] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, P.C.-T.; Zhang, Y.-Y.; Chan, M.K.-K.; Lam, W.W.-Y.; Chung, J.Y.-F.; Kang, W.; To, K.-F.; Lan, H.-Y.; Tang, P.M.-K. The Emerging Role of Innate Immunity in Chronic Kidney Diseases. Int. J. Mol. Sci. 2020, 21, 4018. https://doi.org/10.3390/ijms21114018
Tang PC-T, Zhang Y-Y, Chan MK-K, Lam WW-Y, Chung JY-F, Kang W, To K-F, Lan H-Y, Tang PM-K. The Emerging Role of Innate Immunity in Chronic Kidney Diseases. International Journal of Molecular Sciences. 2020; 21(11):4018. https://doi.org/10.3390/ijms21114018
Chicago/Turabian StyleTang, Philip Chiu-Tsun, Ying-Ying Zhang, Max Kam-Kwan Chan, Winson Wing-Yin Lam, Jeff Yat-Fai Chung, Wei Kang, Ka-Fai To, Hui-Yao Lan, and Patrick Ming-Kuen Tang. 2020. "The Emerging Role of Innate Immunity in Chronic Kidney Diseases" International Journal of Molecular Sciences 21, no. 11: 4018. https://doi.org/10.3390/ijms21114018
APA StyleTang, P. C.-T., Zhang, Y.-Y., Chan, M. K.-K., Lam, W. W.-Y., Chung, J. Y.-F., Kang, W., To, K.-F., Lan, H.-Y., & Tang, P. M.-K. (2020). The Emerging Role of Innate Immunity in Chronic Kidney Diseases. International Journal of Molecular Sciences, 21(11), 4018. https://doi.org/10.3390/ijms21114018