1. Introduction
Cyclic nucleotide phosphodiesterases (PDEs) are cellular enzymes responsible for the hydrolysis of phosphodiester bonds in two second messengers‘—adenosine 3′,5′-cyclic monophosphate (cAMP) or guanosine 3′,5′-cyclic monophosphate (cGMP). PDEs are grouped into 11 subfamilies (PDE1–PDE11) characterized by different substrate specificities: cAMP-specific (PDE4, PDE7, and PDE8), cGMP-specific (PDE5, PDE6, and PDE9) and both, cAMP and cGMP-specific (PDE1, PDE2, PDE3, PDE10, and PDE11) [
1]. Therefore, the range of available inhibitors and their therapeutic potential is enormous [
1]. Their potential therapeutic application covers disorders from heart failure and fertility, through Parkinson’s and Alzheimer’s disease, as well as depression and schizophrenia, to a wide range of inflammatory diseases [
1]. Anti-inflammatory and anti-fibrotic properties of cAMP were demonstrated in different cellular models [
2]. The cAMP was reported to be involved in regulating the function of both inflammatory cells as well as lung and bronchi structural cells in respiratory diseases, which inspired a growing interest in the potential therapeutic applications of PDE inhibitors [
3,
4]. A substantial body of evidence indicates that elevated cAMP levels promote airway smooth muscle cell (ASMC) relaxation and decrease ASMC/lung fibroblast proliferation, migration, and the ability to synthesize extracellular matrix (ECM) proteins, as well as reduce lung fibroblast to myofibroblast transition (FMT) [
2,
3,
5]. Also, cGMP inhibitors are currently receiving increasingly more interest [
1,
4,
6], owing to the facts that (1) cigarette smoking was associated with a decreased level of guanylyl cyclase [
7] and (2) that stimulation of this cGMP-producing enzyme can reduce oxidative stress in chronic obstructive pulmonary disease (COPD) [
8]. Additionally, it is known that smoking can induce PDE3 and PDE4 expression in the lungs [
9], which not only indicates PDEs involvement in COPD but is also good further evidence confirming the validity of using PDE inhibitors in the treatment of lung diseases.
The first non-selective PDE inhibitor used in asthma therapy was theophylline (1,3-dimethylpurine-2,6-dione), a compound that belongs to the group of methylxanthines [
10]. However, a number of side effects often limit its therapeutic application. Recently, this and other naturally occurring methylxanthines were proposed as a potential treatments for pediatric respiratory tract diseases [
11]. Since PDE1, PDE3, PDE4, PDE5, and PDE7 are most closely associated with the pathogenesis of asthma or COPD [
1,
3], there has been a need for more selective inhibitors suitable for targeting isoforms of these PDE families. The aim of this approach is not only to achieve a more precise blockade of these enzymes but also to reduce the side effects of non-selective PDE inhibitors. Several inhibitors have undergone clinical trials, and some of them, such as roflumilast (a PDE4 inhibitor), are currently used in the clinic [
12]. Another example is the inhaled PDE4 inhibitor, CHF6001 currently in phase II clinical trials, which also exhibits preferential anti-inflammatory properties in COPD [
13]. However, since the efficacy of PDE inhibitors has not been markedly improved by the increased selectivity toward a specific PDE, the focus of the research was moved towards dual or pan-PDE inhibitors. Pan-PDE inhibitors represent compounds that are able to inhibit various isoforms within different PDE classes. Another reason why such inhibitors could be desirable is the cell-specific expression and compartmentalized intracellular localization of individual PDEs in the cell, which could account for the synergistic effects of targeting multiple subtypes simultaneously [
14]. The reason for testing PDE inhibitors with a broader spectrum of activity also lies in the fact that the expression of individual PDEs may change in response to irritants to which patients are exposed, or those that cause asthma or COPD, e.g., tobacco smoke [
9]. Pan-PDE inhibitors may provide a more effective therapeutic approach than selectively acting agents, mainly through their improved spectrum of pharmacological activity [
1,
4]. Enhanced therapeutic potential of pan-PDE inhibitors is expected to result from their effect on a number of signaling pathways involved in the development of asthma and other lung or bronchial diseases [
1,
3].
Besides the above-mentioned PDEs, transient receptor potential ankyrin 1 (TRPA1) channels have recently been proposed to contribute to the pathogenesis of asthma and COPD [
15]. These non-selective calcium-permeable channels have been implicated in allergic reactions, including the late allergic response, airway hyperresponsiveness and bronchoconstriction, neurogenic inflammation, and cough; these receptors were also suggested to function as toxicant sensors [
16,
17]. TRPA1 expression was described in both immune and lung structural cells, including epithelial cells, smooth muscle cells, and fibroblasts, as well as sensory neurons. They can be activated by many different irritants, e.g., allyl isothiocyanate, allicin, or acrolein as well as pro-inflammatory mediators, including histamine, prostaglandins, or bradykinin [
15,
16,
17,
18]. Therefore, it is believed that TRPA1 antagonists may serve as a promising therapeutic alternative for lung diseases [
17,
18].
Our earlier studies revealed that, compared to selective PDE4 inhibitors (roflumilast or cilomilast), pan-PDE inhibitors might provide better inhibition of the transforming growth factor type β
1 (TGF-β
1)-induced ASMC remodeling [
19]. We have also reported that some of the recently synthesized 7,8-disubstituted purine-2,6-dione derivatives, in addition to being pan-selective PDE inhibitors, can interact with TRPA1 ion channels [
20,
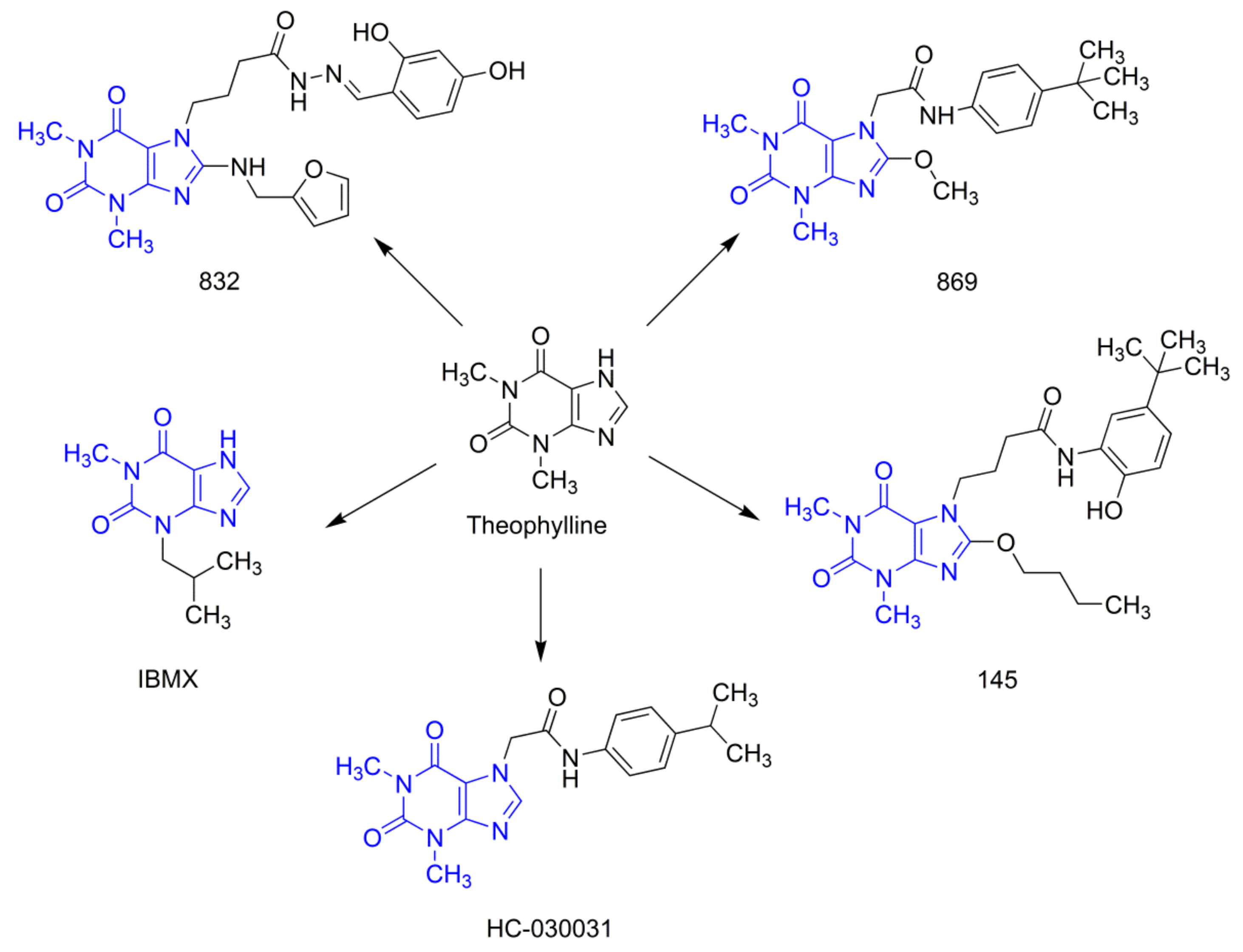
21]. In this study, we selected three 7,8-disubstituted purine-2,6-dione derivatives (
Figure 1): 832 (a pan-PDE inhibitor), 869 (a TRPA1 modulator), and 145 (a pan-PDE inhibitor and a TRPA1 modulator) and evaluated their ability to limit profibrotic responses of lung fibroblasts. For this purpose, we used TGF-β
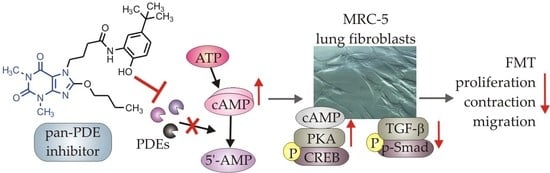
1 or fetal bovine serum (FBS)-activated MRC-5 cells and investigated proliferation, migration, contraction, expression of profibrotic genes, and phenotype transition into myofibroblasts under the influence of these 7,8-disubstituted purine-2,6-dione derivatives. Our goal was also to verify the TRPA1 contribution in the potential anti-fibrotic effects of 7,8-disubstituted purine-2,6-dione derivatives.
3. Discussion
Tissue remodeling is a widespread pathological process in which a number of structural changes occur in a tissue or organ that impairs its normal physiological functions [
24,
25,
26]. In the lungs or bronchi, remodeling in asthma, COPD and lung fibrosis is often associated with the imbalance in ECM protein turnover, disturbances that occur in the structure and functions of the epithelial layer, smooth muscle cell hyperplasia and hypertrophy, and increased levels of tissue myofibroblasts, predominantly as a result of fibroblast to myofibroblast transition [
27,
28]. Therefore, in the search for new anti-fibrotic drugs, particular attention should be paid to pleiotropic compounds that affect various cellular processes associated with remodeling. We have recently shown novel PDE inhibitors from the group of 7,8-disubstituted purine-2,6-dione derivatives as potent modulators of ASMC remodeling [
19]. Here we demonstrated for the first time that a strong pan-PDE inhibitor from this group, compound 145 can also exert anti-fibrotic effects in TGF-β
1-induced human lung fibroblasts via limiting FMT.
A number of studies have earlier demonstrated FMT-limiting properties of cAMP elevating agents, such as PDE inhibitors or activators of soluble adenylyl cyclase [
29,
30,
31]. The potential benefits of using theophylline as an anti-inflammatory drug in the treatment of asthma have long been known, but it was only a few years ago when Yano et al. reported the anti-FMT effects of this substance in the lung [
32]. In turn, we demonstrated that other methylxanthines, i.e., pentoxifylline or lisofylline, can also reduce FMT in bronchial fibroblasts isolated from asthmatics [
33]. Other studies revealed that selective inhibitors, particularly those inhibiting PDE4, such as rolipram or roflumilast, also show anti-FMT effects [
31,
34,
35].
Our recently designed and synthesized 7,8-disubstituted purine-2,6-dione derivatives fill very well the gap between inhibitors highly selective for a single isoenzyme, and non-selective and weak non-specific inhibitors. Both our previous [
19] and current research show that 7,8-disubstituted purine-2,6-dione derivatives represent a group of pan-PDE inhibitors with high inhibitory potency against individual PDE isoenzymes. They can effectively and strongly inhibit many different PDEs, including those relevant to the pathogenesis of respiratory diseases [
1,
3]. Recently, we confirmed 145 favorable binding modes in the active site of PDE4B and PDE7A as well as to the TRPA1 model [
20]. Moreover, molecular docking studies of close analogs of compounds 832 and 869, which recognized crucial interactions in PDE4B/PDE7A and TRPA1 sites, respectively, were previously described [
20,
21]. Here, it was demonstrated for the first time that these potent, pan-PDE inhibitors exert a strong limiting effect on TGF-β
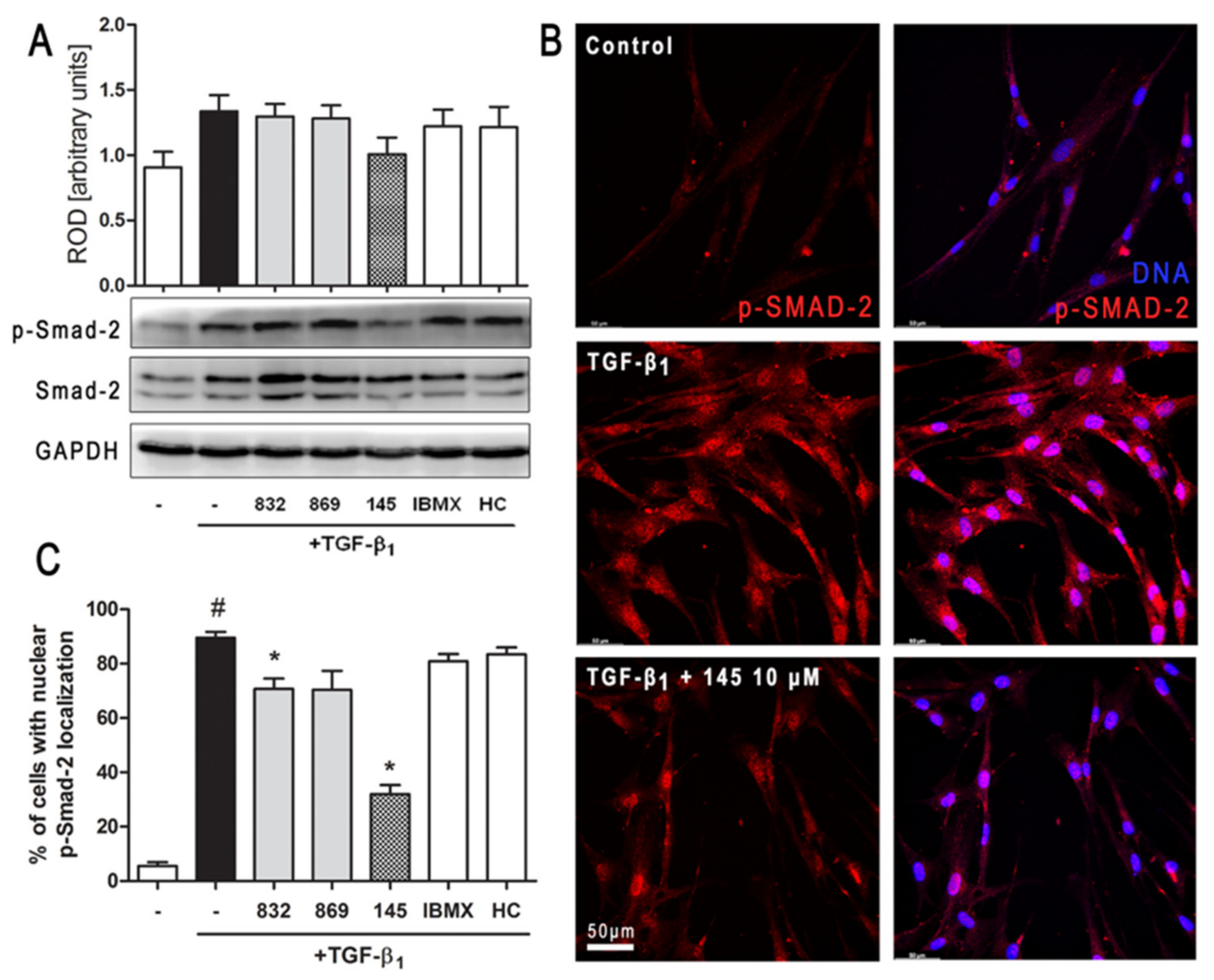
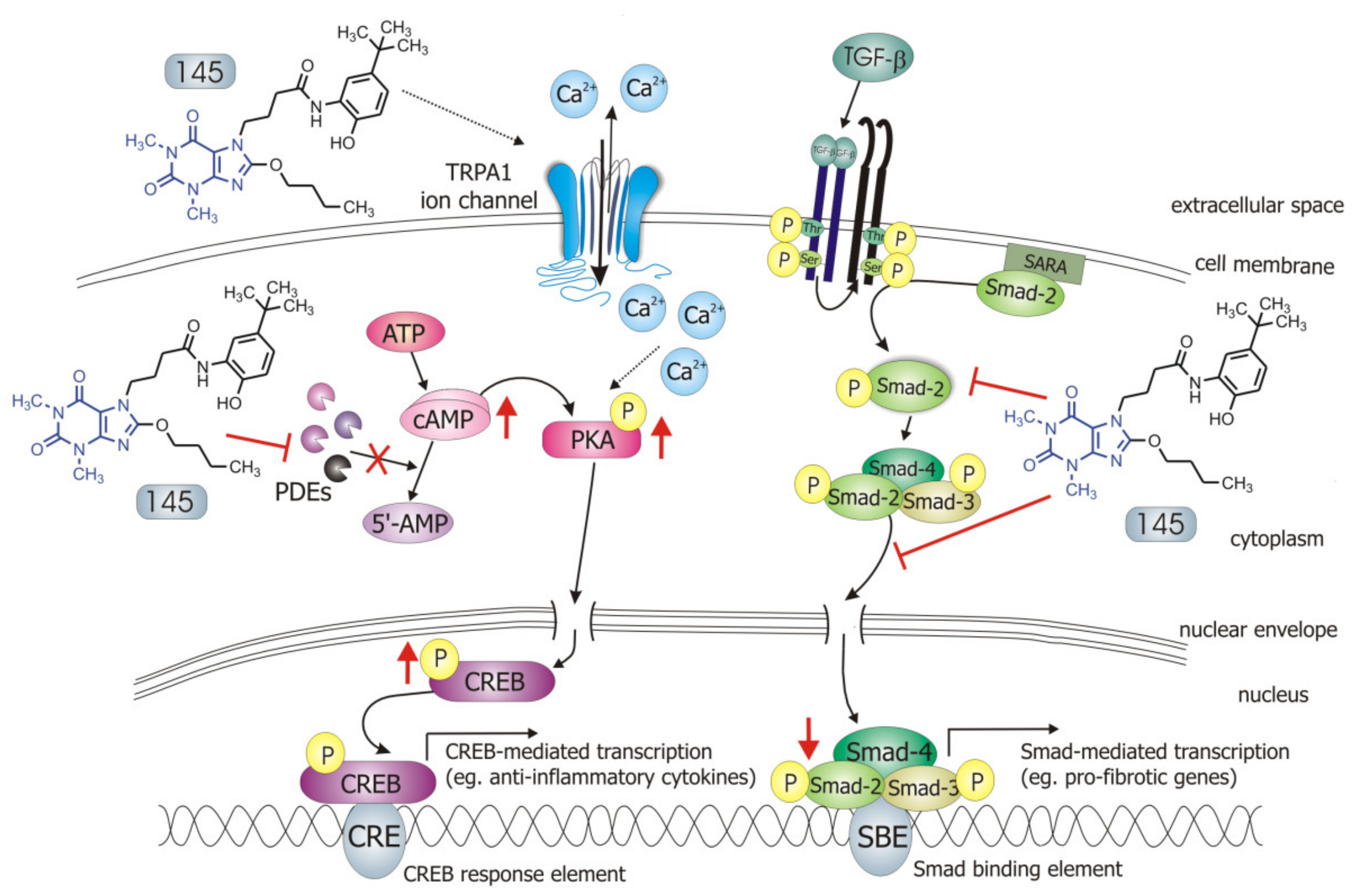
1-induced FMT in lung fibroblasts. Selected 7,8-disubstituted purine-2,6-dione derivatives, especially 145 inhibited the expression of genes and proteins characteristic for both myofibroblasts and extracellular matrix proteins secreted by these cells. We showed that some compounds belonging to 7,8-disubstituted purine-2,6-dione derivatives can also reduce proliferation, migration, and contraction of pulmonary fibroblasts. We also displayed that the observed anti-fibrotic effects of pan-PDE inhibitor, 145, are the result of its impact on the canonical TGF-β signaling pathway. This compound, decreased Smad-2 phosphorylation and subsequent translocation to the nucleus, thereby reducing TGF-β
1-induced and Smad-mediated transcription of profibrotic genes in lung fibroblasts (
Figure 8).
The Smad-dependent TGF-β signaling pathway has already been identified as a potential therapeutic target for preventing fibrosis in the lung and other organs [
36,
37]. This has been confirmed by our current and previous data, in which we have also shown that in asthmatic fibroblasts, pentoxifylline and lisofylline can limit TGF-β signaling [
33]. Further evidence is provided by Fehrholz et al. who demonstrated in lung epithelial cells that caffeine and rolipram might also affect Smad-dependent TGF-β signaling [
38]. Similar effects have been described for the new drugs used in idiopathic pulmonary fibrosis (IPF) therapy, pirfenidone, and nintedanib [
39]. Although the exact mechanism of action of these anti-inflammatory, antioxidant and anti-remodeling drugs is yet unknown, it is has been found that they can affect, among others, TGF-β/Smad-dependent signaling [
39,
40].
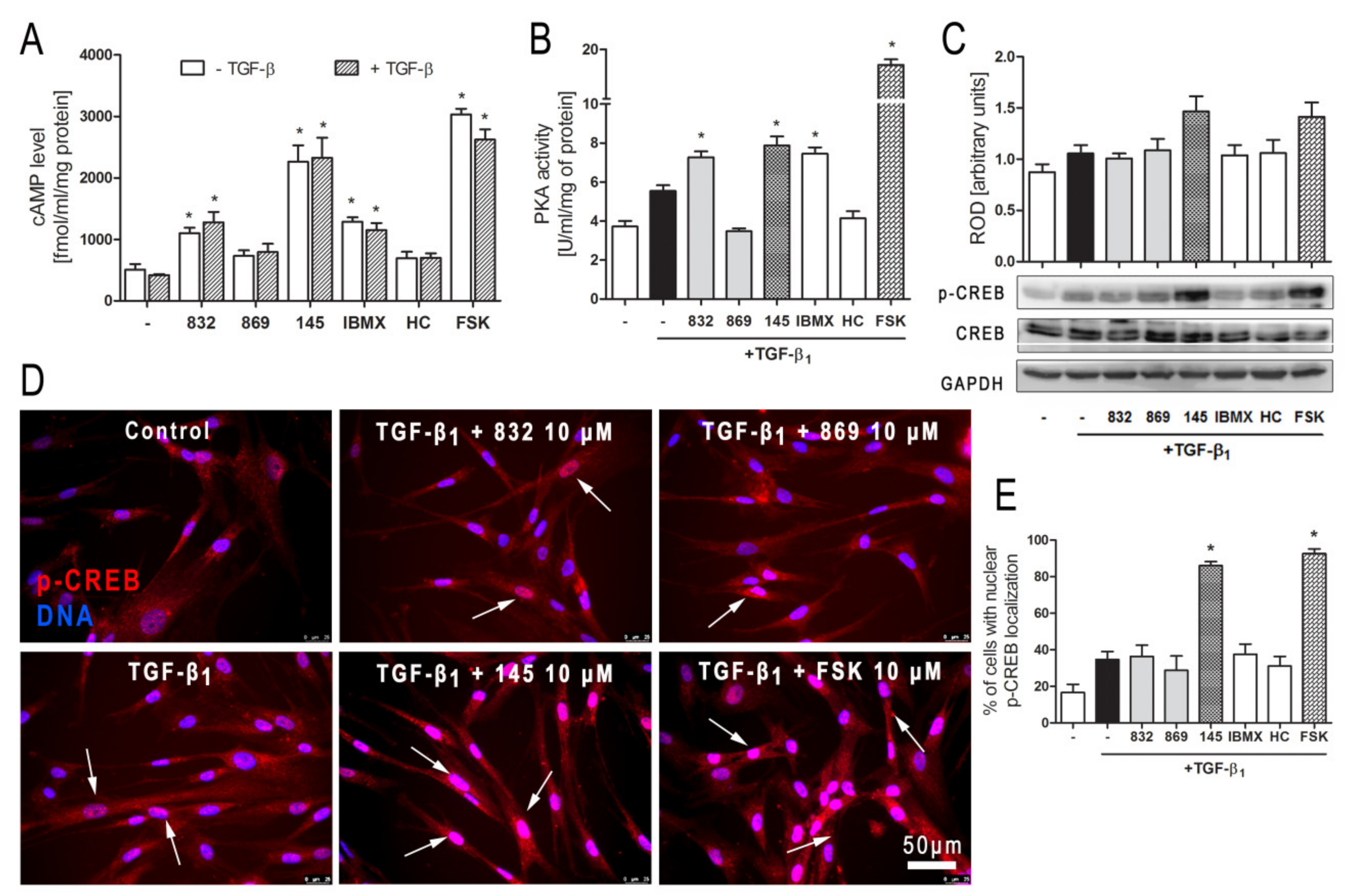
Some studies revealed that increased intracellular cAMP levels could directly affect the canonical TGF-β and Smad-dependent signaling pathway [
41,
42,
43,
44,
45]. After an increased CREB phosphorylation (e.g., in response to the elevated intracellular cAMP levels), a CREB-binding protein (CBP) effectively binds p-CREB resulting in enhancement of its transcriptional activity [
46]. On the other hand, it is known that CBP can directly interact with the Smad protein complex and operate as a Smad transcriptional co-activator [
47]. As such, competition between cAMP-dependent CREB/CBP binding and TGF-β-dependent Smad/CBP binding for the common co-activator CBP [
43,
45] is proposed as the mechanistic explanation for the functional inhibition of Smad-dependent signaling. Compound 145, as a strong PDE inhibitor, elevated cAMP levels in lung fibroblasts, activated PKA and, as a consequence, led to increased CREB phosphorylation, which in turn inhibited Smad signaling (
Figure 8). The presence of 145 caused the most prominent activation of the cAMP/PKA/CREB pathway in TGF-β
1-induced MRC-5 cells. At the same time, this compound had the best inhibitory effect on TGF-β
1/Smad-2 signaling. While a strong activation of cAMP-dependent CREB phosphorylation is not surprising given that 145 elevates intracellular cAMP, its inhibitory effect on Smad-2 phosphorylation indicates new and desirable properties of this compound (
Figure 8). Since other Smad proteins can also regulate TGF-β signaling, the effect of 145 on Smad-3/4 or Smad-1 should also be tested in future studies. The increased efficacy of compound 145 may be the result of the interaction with two independent signaling pathways, both of which promote anti-fibrotic effects. It is known that the reduction of TGF-β-induced and Smad-mediated transcription of profibrotic genes is one of the main goals of anti-fibrotic therapy [
36,
37] and out of the studied compounds, 145 best meets this requirement. Of note, is that there are also other reports that associate CREB phosphorylation and activation of CREB-mediated transcription with anti-inflammatory properties, e.g., increased IL-10 synthesis, inhibition of NF-κB signaling, Th17 cell differentiation, and survival of T
reg cells [
48,
49]. Therefore, it is likely that the high CREB phosphorylation induced by 145, compared to other tested compounds, may explain the prominent anti-fibrotic effects of this compound in TGF-β
1-induced MRC-5.
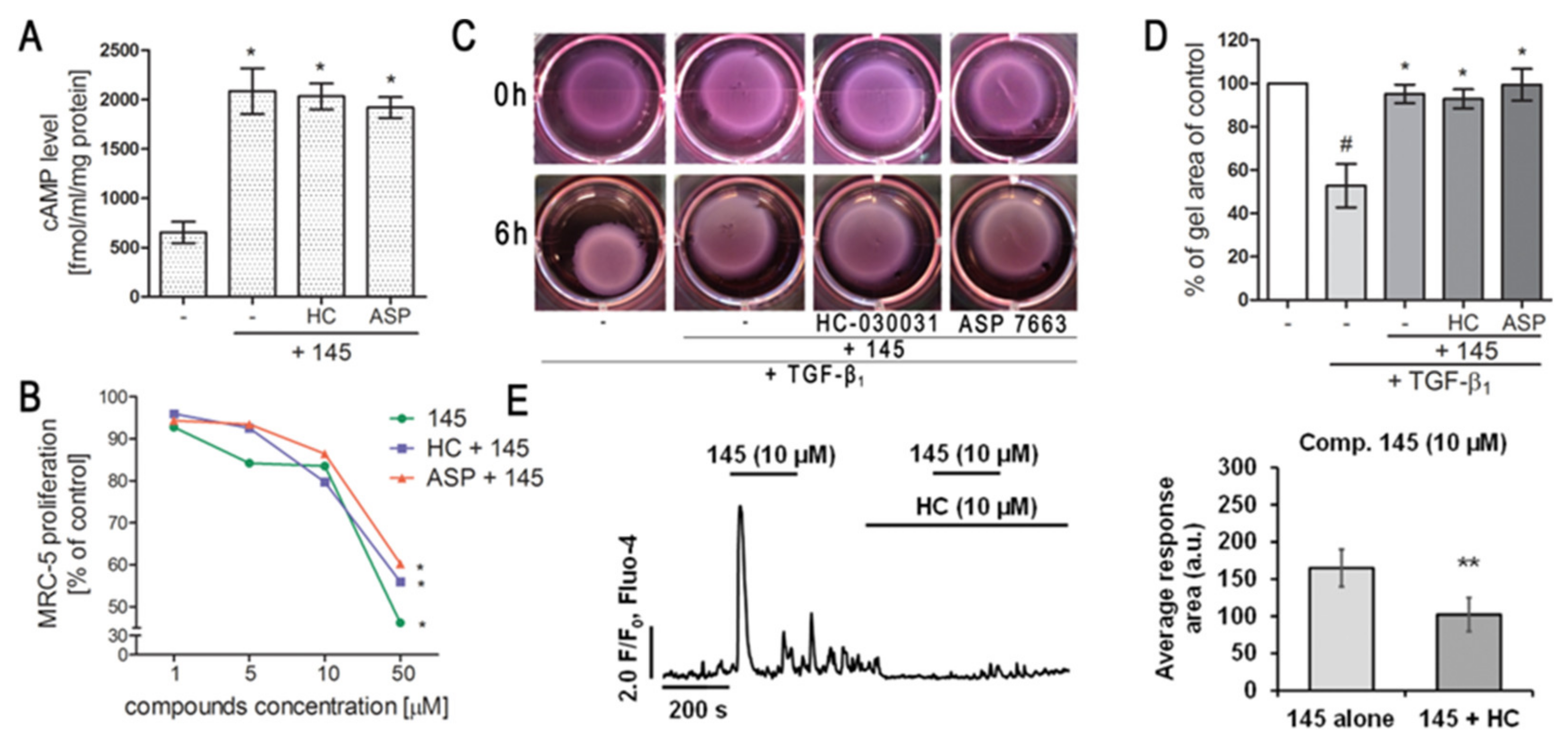
We have characterized compounds 832 and 145 as strong, pan-PDE inhibitors. However, despite the similar affinity of both compounds to, e.g., PDE1, PDE3, or PDE4, only 145 was able to inhibit, e.g., PDE4D, PDE5A, or PDE8A. Compound 145 ability to inhibit PDE8A is an important feature because even the currently known non-selective inhibitors, such as IBMX (also used in this study), are unable to inhibit PDE8, probably due to lack of the interaction in the binding site, i.e., hydrogen bond with Tyr748 [
50]. The literature data showed that PDE8 significantly differs from other PDE isoenzymes [
50]. Recent reports indicate that this isoform may be of particular importance for the development of effective PDE inhibitors [
4,
51]. Both the elevated cAMP levels in MRC-5 treated with 145, as well as the increased anti-fibrotic properties of this compound, may result from its interaction with PDE8. Another reason for enhanced 145 activity may be its ability to inhibit cGMP-specific, PDE5A. This also suggests a possible involvement of cGMP in 145 anti-fibrotic effects in MRC-5 cells. In turn, 145 PDE4D and PDE3 inhibition may result in inducing nausea and ionotropic effects, respectively. On the other hand, the potential side-effects caused by pan-PDE inhibitors can be mitigated by reducing their potential therapeutic dose, while maintaining a similar impact to selective PDE inhibitors.
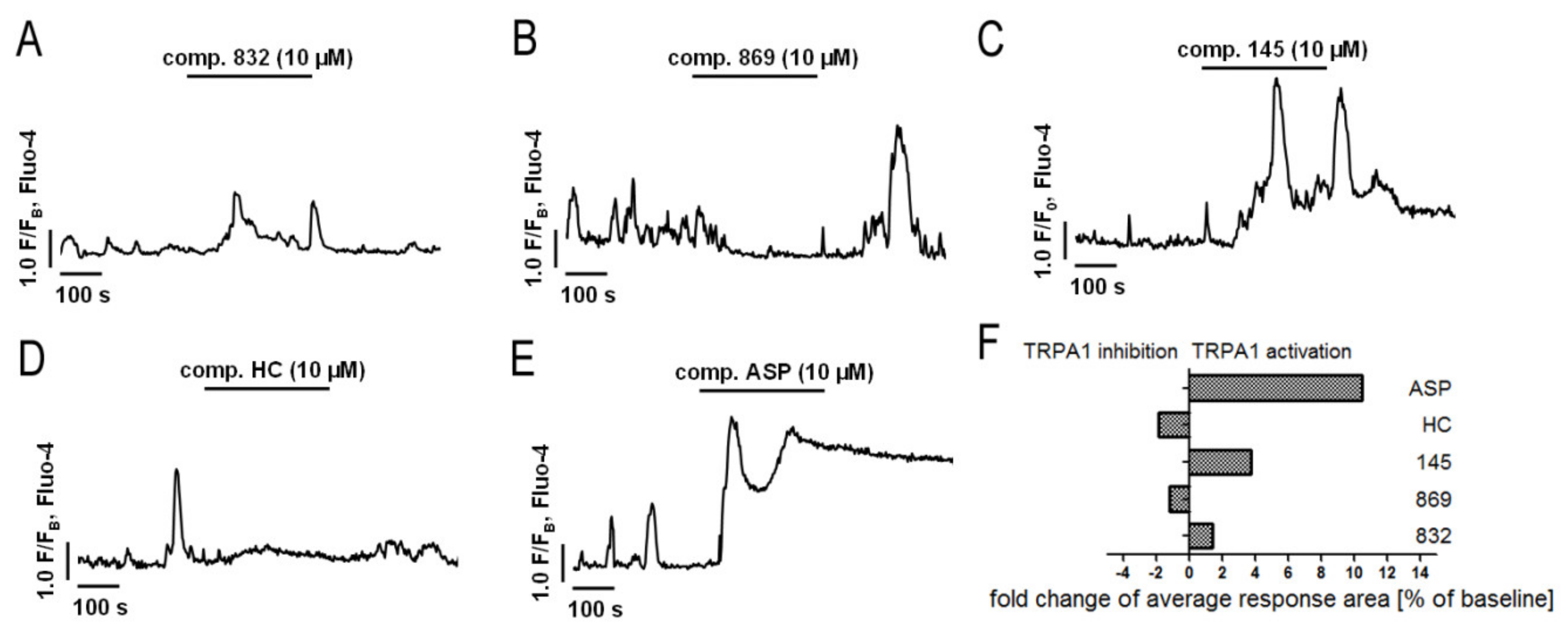
Previous studies [
20] made us aware that 145 may also interact with TRPA1 channels. In light of the reports indicating that TRPA1 channels may be a potential molecular target for lung diseases [
18,
52,
53], and since caffeine can suppress TRPA1 activity [
54], we examined whether 832, 869, and 145 affect the function of this channel. Our results do not support a very strong agonistic/antagonistic activity of 145 towards TRPA1, but functional analysis revealed that this compound can modify calcium influx in pulmonary fibroblasts in a way a weak agonist would. To assess the involvement of TRPA1 in the observed activity of compound 145, we performed a series of experiments in which we pharmacologically blocked or activated this ion channel and then compared the effects in the presence and absence of 145. The presence of 145 neither affected MRC-5 proliferation or cAMP levels, which indicates that the TRPA1 component may not play an important role in 145-mediated anti-fibrotic effects. We cannot exclude that 145 exerts such a strong inhibitory effect on PDE that the maximum anti-fibrotic potential is already achieved, and thus, any further modulation of TRPA1 activity may be negligible for the final outcome. The slight but noticeable effect of the TRPA1 antagonists—HC-030031 and 869, on some tested TGF-β
1-induced MRC-5 myofibroblast features associated with remodeling may support this notion. This is also in line with several clinical trials in which TRPA1 antagonists are tested to treat respiratory diseases [
53]. On the other hand, Kurahara et al. demonstrated that pirfenidone might exert anti-inflammatory and anti-fibrotic effects upon TRPA1 activation [
55]. We have not seen similar effects in our study; in fact, TRPA1 activation via ASP 7663 did not result in significant changes in the phenotype of TGF-β
1-induced MRC-5 cells. Considering the above reports, and since large and sustained intracellular Ca
2+ elevations are often noxious for cells and may trigger cell death [
56], the fact that compound 145 does not excessively activate Ca
2+ influx via TRPA1 might be desirable from the therapeutic perspective. Collectively, it could be argued that anti-fibrotic properties of compound 145 are accomplished by a strong PDE inhibitor activity, while the TRPA1 channel modulation is minor and may only slightly contribute to 145 activity.
One of the further mechanisms of methylxanthine interaction with its cellular targets is that these compounds may function as adenosine receptor antagonists. Our earlier studies carried out on airway smooth muscle cells did not support the concept of adenosine A
1 or A
2A receptor antagonism as a potent modulator of TGF-β-induced airway remodeling [
19]. In turn, Roberts et al. showed that selected agonists of the Gs-coupled receptors, including, e.g., adenosine A
2B or prostacyclin receptors, can inhibit lung fibroblast differentiation into myofibroblasts [
57]. The effect of these compounds was comparable to that achieved after administration of the adenylate cyclase activator, forskolin, but it was not consistently associated with the level of cAMP [
57]. Here, we demonstrate a link between PDE inhibition, increased cAMP levels, activation of the PKA/CREB pathway, and anti-fibrotic properties for both, compound 145 and forskolin. Considering the results of SAR studies indicating which chemical substituents predispose a compound to possess adenosine receptor antagonist activity [
58], and our earlier data indicating a lack of effect of adenosine antagonists on TGF-β
1-induced remodeling in ASMC, we believe that adenosine antagonistic effects do not contribute to the observed effects of the 7,8-disubstituted purine-2,6-dione derivatives in our study. Calcium signals have also been implicated in the mechanism of action of methylxanthine compounds [
59,
60]. Disturbances in calcium homeostasis may also lead to the synthesis of the extracellular matrix and other profibrotic proteins in pulmonary fibroblasts [
56]. 7,8-disubstituted purine-2,6-dione derivatives turned out to have poor activity towards calcium-dependent TRPA1 channels.
Our previous research demonstrated that compounds from the group of 7,8-disubstituted purine-2,6-dione derivatives are able to limit ASMC remodeling response, and here we confirm 145 preferential activity in reducing lung fibroblast features associated with remodeling. Although both the previous and current studies provide solid evidence of the anti-fibrotic effect of compound 145, the studies so far have been performed in vitro in the presence of one growth factor. Therefore, the activity of 145, a new pan-PDE inhibitor, should be verified in vivo in a model of chronic lung diseases and compared with known anti-inflammatory drugs or PDE inhibitors already used clinically. In the in vivo model, it would be possible to know the 145 pleiotropic effects. Given the complexity of bronchial and lung remodeling, the search for compounds with a wide spectrum of activity is extremely valuable. Considering the data obtained with ASMCs and presented here lung fibroblasts, it can be concluded that the group of 7,8-disubstituted purine-2,6-dione derivatives offers promising drug candidates targeting chronic lung disease therapy.
4. Materials and Methods
4.1. Tested Compounds
N′-(2,4-dihydroxybenzylidene)-4-(8-((furan-2-ylmethyl)amino)-1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7
H-purin-7-yl)butanehydrazide (working name 832, described in [
21]),
N-(4-(
tert-butyl) phenyl)-2-(8-methoxy-1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7
H-purin-7-yl)acetamide (working name 869, described in [
20]), and 4-(8-butoxy-1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro -7
H-purin-7-yl)-
N-(5-(
tert-butyl)-2-hydroxyphenyl)butanamide (working name 145, described in [
20]) were used in this study. These compounds were synthesized according to a multistep procedure, as described previously [
20,
21]. Briefly, to obtain 832, 8-bromo-1,3-dimethyl-3,7-dihydro-1
H-purine-2,6-dione was treated with furan-2-ylmethanamine in refluxing 2-methoxyethan-1-ol. Next, the 8-[(furan-2-yl)methyl]amine derivative was alkylated at position 7 using ethyl 4-bromobutanoate in the presence of K
2CO
3 and a catalytic amount of
N-benzyl-
N,
N-diethylethanaminium chloride (TEBA) in refluxing acetone. Treatment of the obtained ester with hydrazine hydrate in anhydrous ethanol gave the corresponding hydrazide. In the final step, the hydrazide was condensed with 2,4-dihydroxybenzaldehyde in methanol in the presence of a catalytic amount of HCl. To obtain 869, 8-bromo-1,3-dimethyl-3,7-dihydro-1
H-purine-2,6-dione was alkylated at position 7 with ethyl 2-chloroacetate in the presence of K
2CO
3 and a catalytic amount of TEBA in refluxing acetone. The synthesized ethyl 2-(8-bromo-1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7
H-purin-7-yl)acetate was treated with sodium methanolate in methanol, then hydrolyzed using potassium hydroxide in a water-acetone mixture and acidified with concentrated HCl to obtain the corresponding acid. In the final step, the obtained acid was condensed with 4-
tert-butylaniline in
N,N-dimethylformamide in the presence of di(1
H-imidazol-1-yl)methanone. 145 was obtained in a similar manner, but ethyl 4-bromobutanoate was used for the alkylation, the transesterification reaction was carried out using sodium butanolate in butanol, and in the final stage, the resulting acid was condensed with 2-amino-4-(
tert-butyl)phenol [
20]. Reference substances used in the studies were: 3-isobutyl-1-methyl-3,7-dihydro-1
H-purine-2,6-dione (IBMX, Sigma Aldrich, St. Louis, MO, USA), 2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7
H-purin-7-yl)-
N-(4-isopropylphenyl)acetamide (HC-030031, Sigma Aldrich, St. Louis, MO, USA) and (
E)-2-(7-fluoro-1-isobutyl-2-oxoindolin-3-ylidene)acetic acid (ASP 7663, Sigma Aldrich, St. Louis, MO, USA). In some experiments, the adenylyl cyclase activator, forskolin (Sigma Aldrich, St. Louis, MO, USA) was used. All evaluated compounds were dissolved in dimethylsulfoxide (DMSO, PAN-Biotech, Germany) and diluted in culture medium (the final DMSO concentration did not exceed 0.5% and was not harmful to the cells).
4.2. PDE Subtype Selectivity
Inhibitory activities of the investigated compounds against various subtypes of human recombinant (hr)PDEs, namely PDE1A, PDE1B, PDE1C, PDE3A, PDE3B, PDE4A, PDE4B, PDE4D, PDE7A, and PDE8A were assessed using a PDE-GloTM Assay Kit (Promega, Madison, WI, USA) according to the protocol provided by the manufacturer. Briefly, appropriate amounts of each enzyme (SignalChem, Richmond, B.C., Canada), diluted with PDE-Glo reaction buffer, were distributed on a 384-well plate (Thermo Scientific, Waltham, MA, USA). The investigated compounds were dissolved in DMSO, diluted with the same vehicle, and mixed with the reaction buffer at a v/v ratio of 1:5. Thereafter, 1 µL of these mixtures and 2.5 µL of the cAMP solution as a substrate were added. The final cAMP concentration was 0.05 µM. The samples were incubated for 10 min at 30 °C. Then, 2.5 µL of termination buffer and 2.5 µL of PDE-Glo detection solution were added to each well and incubated for 20 min. Finally, 10 µL of PDE-Glo kinase reagent was added, and luminescence was measured using the POLARstar Omega plate reader (BMG LABTECH, Ortenberg, Germany). Each sample was prepared in quadruplicate. The relative activities (A, %) of all samples were calculated from the following equation: A = [(LUsample − LUblank)/(LUcontrol − LUblank)]·100%; where LUcontrol is the luminescence of a control sample in the absence of a PDE inhibitor, LUsample is a luminescence of an investigated sample in the presence of an inhibitor, and LUblank is a luminescence of a sample in the absence of enzyme (activity = 0). IC50 values were estimated using non-linear regression.
4.3. TRPA1 Assay
The TRPA1 agonist and antagonist fluorescent imaging plate reader (FLIPR) Assay (functional, HEK293 cell-based) was performed at Eurofins Panlabs, Inc. (St. Charles, MO, USA). The compounds were prepared in assay buffer to the final dilutions 10 and 50 µM. In each experiment, the respective reference compounds were tested: allyl isothiocyanate (the agonist assay) and ruthenium red (the antagonist assay). The electrophysiological assays were conducted on a FLIPRTETRA instrument where the test compounds, vehicle controls, and reference agonists or antagonists were added to the assay plate after a fluorescence baseline was established. The agonist or antagonist incubation during the assay was for a total of 180 s and was used to assess each compound’s ability to activate or inactivate TRPA1. All plates were subjected to appropriate baseline corrections. Once baseline corrections were processed, maximum fluorescence values were exported, and data was manipulated to calculate percentage activation or percentage inhibition. Results showing activity greater than 25% were considered to represent significant effects of test compounds. Experiments were run three times, and at least 20 individual cells were recorded in each experiment.
4.4. In Vitro Lung Fibroblast Culture
The normal human lung fibroblast cell line—MRC-5 (ATCC® CCL-171™, Manassas, VA, USA)—was used in the study. Cells were cultured in Eagle’s Minimum Essential Medium (EMEM, ATCC® 30-2003™, Manassas, VA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and antibiotics mixture (penicillin, streptomycin, amphotericin B; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) (standard culture medium) in standard culture conditions (5% CO2, 37 °C, 95% humidity). Unless otherwise specified, for the experiments, cells were seeded at a final density (5 × 103/cm2) in standard culture medium for 24 h and then cultured in EMEM at reduced serum content (0.5%; v/v) for additional 24 h. TGF-β1 (BD Biosciences, San Jose, CA, USA) was administered at 5 ng/mL concentration in all experiments.
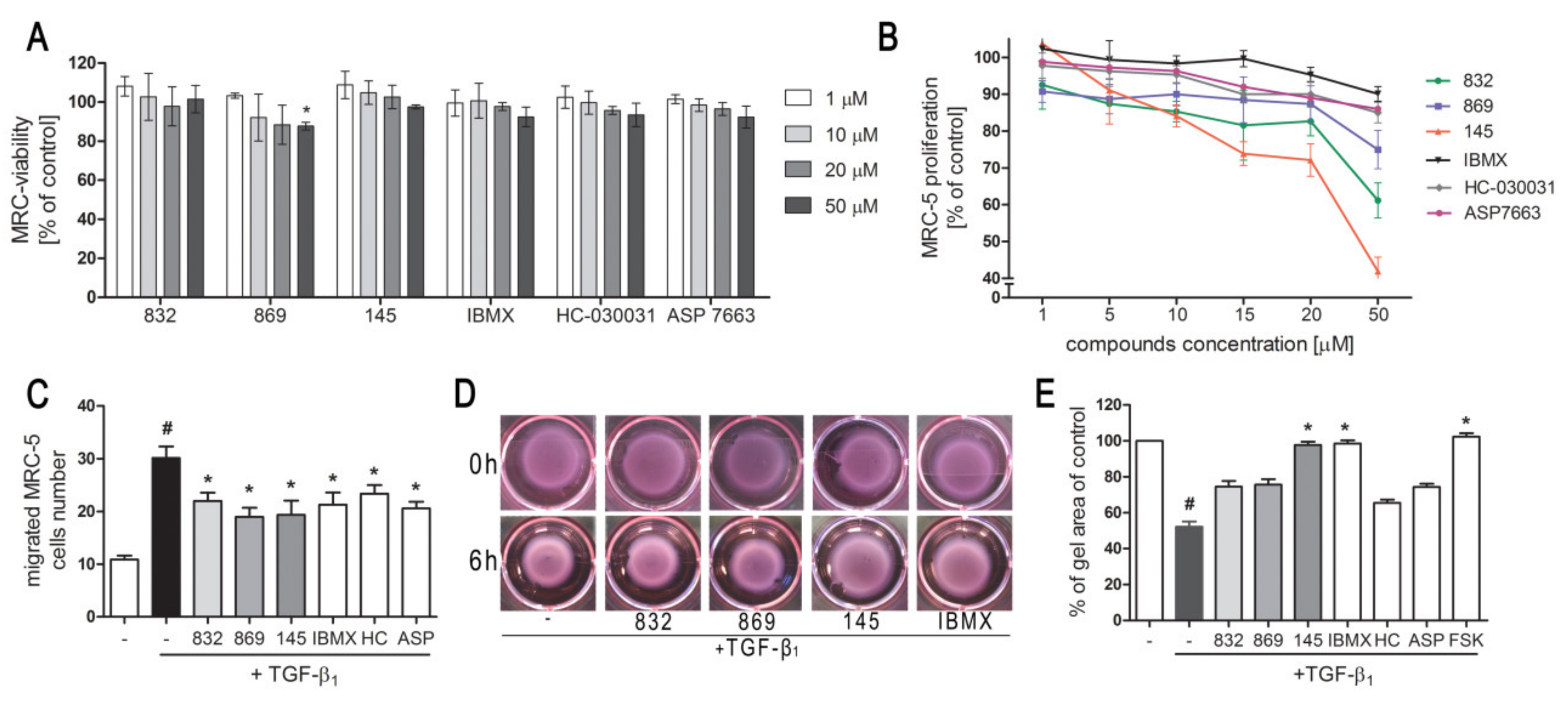
4.5. Viability and Proliferation Assays
MRC-5 cell viability was evaluated using CytoTox-ONE™ Homogeneous Membrane Integrity Assay (Promega Corporation, Madison, WI, USA) according to the manufacturer’s protocol. The average fluorescence intensity with an excitation wavelength of 560 nm and an emission wavelength of 590 nm was measured using a microplate reader (SpectraMax® iD3, Molecular Devices, San Jose, CA, USA). Experiments were run three times in duplicates. MRC-5 proliferation was determined using the alamarBlue® assay. Cells were seeded for 24 h in standard culture medium, then serum-starved for an additional three days. Evaluated compounds (1-50 µM) were added 1 h prior FBS (10%, v/v) for an additional 48 h. After incubation, the alamarBlue® reagent (Invitrogen, Life Technologies, Carlsbad, CA, USA), dissolved in Hank’s Balanced Salt Solution (10% v/v; Gibco, Thermo Fisher Scientific, Waltham, MA, USA), was added for 30 min. After the observed color change, the supernatant was transferred to 96-well plates, and the absorbance was measured at 570 and 600 nm. The percent difference in reduction between treated and control cells was calculated according to the manufacturer’s protocol. The experiments were run three times in duplicates.
4.6. Transwell Migration Assay
MRC-5 migration was evaluated using 6.5 mm Transwell culture plates with an 8.0 µm pore polycarbonate membrane (Corning Incorporated, NY, USA). Cells were serum-deprived for 24 h, trypsinized and seeded in the upper chamber in serum-free culture medium supplemented with or without study compounds (10 µM). The lower compartment was filled with the same culture medium, and after 1 h TGF-β1 was added to the wells. After 24 h of incubation cells were fixed with 4% formaldehyde solution (Sigma Aldrich, St. Louis, MO, USA) and stained with 0.5% crystal violet solution (Sigma Aldrich, St. Louis, MO, USA). The non-migrated cells were removed from the upper face of the membranes using a cotton swab. The number of migrated cells was counted under an inverted microscope (Nikon Eclipse TS 100, 10× objective) using 10 randomly selected fields of view. Experiments were run twice, in a blind-folded manner.
4.7. Cell Contraction Assay
A collagen cell contraction assay was performed according to manufactures’ instructions (Cell Biolabs, INC., San Diego, CA, USA). Briefly, MRC-5 was harvested and mixed with a collagen solution. After polymerization, collagen gel lattices were cultured for two days, during which mechanical tension develops. Before releasing the stressed matrix, MRC-5 cells were pre-treated with study compounds (10 µM) for 1 h. Then collagen gels were gently released from the sides of the culture dishes and contraction in the presence of study compounds, and TGF-β1 was initiated. The collagen gel size was monitored at various time points and quantified with the Fiji ImageJ Software (National Institutes of Health, Bethesda, MD, USA, version 1.52q). The experiments were run three times.
4.8. Intracellular Calcium Measurements
MRC-5 cells were briefly washed with NaHEPES buffer (NaHEPES buffer was prepared as follows: NaCl 140 mM, KCl 4.7 mM, HEPES 10 mM, MgCl2 1 mM, glucose 10 mM; pH 7.3) and then loaded with 1 µM Fluo-4 AM (Thermo Fisher Scientific, Waltham, MA, USA) for 1 h, 37 °C in NaHEPES. After the incubation, the cells were washed again with fresh NaHEPES and used for experiments in a flow chamber perfused with NaHEPES-based extracellular solution supplemented with 1 mM CaCl2. All experiments were carried out at room temperature using Zeiss LSM 880 confocal microscope (40× oil objective), excitation was set to 488 nm, and the emission was between 500 and 600 nm. Each series of images was recorded at 256 × 256-pixel resolution, two consecutive frames were averaged, and the interval between the averaged images was set at 2 s. Fluorescence signals were plotted as F/F0, where F0 was baseline fluorescence defined as the averaged signal from ten consecutive baseline images.
4.9. cAMP ELISA
MRC-5 cells were seeded in standard culture medium for 24 h and then serum-starved for 2 h in HBSS. The study compounds (10 µM) were added to the MRC-5 cultures and after 30 min of incubation, the intracellular cAMP level in cell lysates was measured using a colorimetric cAMP ELISA Kit according to the manufacturer’s protocol (Cell Biolabs, INC., San Diego, CA, USA). Obtained results were normalized to total protein content in each sample. The experiments were run two times in duplicates.
4.10. In-Cell ELISA
The relative α-SMA and collagen I level in MRC-5 cells were measured using an in-cell ELISA assay. Study compounds (10 µM) were added 1 h before TGF-β1 treatment, and the content of both proteins in fibroblasts was determined after 48 h of incubation. Briefly, MRC-5 were fixed in methanol, permeabilized and blocked in 1% bovine serum albumin (BSA) with 0.1% Tween® 20 and then immunostained with primary mouse, monoclonal anti-α-SMA or anti-collagen type I antibodies (#A2547 or #SAB4200678, respectively, Sigma Aldrich, St. Louis, MO, USA), followed by secondary, anti-mouse, peroxidase-conjugated antibody (#A9044, Sigma Aldrich, St. Louis, MO, USA). The colorimetric reaction resulting from the addition of peroxidase substrate was stopped, and the absorbance was measured at 450 nm. The relative protein amount was normalized to the total cell number in each sample (Janus Green staining). The experiments were run four times in duplicates.
4.11. Immunofluorescence Labelling
For immunocytochemical labeling, MRC-5 cells were seeded on glass coverslips. Study compounds (10 µM) were added 1 h before TGF-β1 treatment, and analysis was performed after 1 h (p-Smad-2 and p-CREB detection) or 48 h (α-SMA detection) of incubation. Cells were fixed in methanol or 4% formaldehyde solution, permeabilized in 0.2% or 0.1% Triton X-100 solution (for p-Smad-2 and p-CREB or α-SMA, respectively). After incubation in blocking solution the following primary antibodies: rabbit polyclonal, anti-phospho-Smad-2 (Ser465, Ser467) antibody (#44-244G, Thermo Fisher Scientific, Waltham, MA, USA); rabbit polyclonal, anti-phospho-CREB1 (Ser133) antibody (#orb213775, Biorbyt, Cambridge, United Kingdom); mouse monoclonal, anti-α-SMA antibody (#MA5-11547, Thermo Fisher Scientific, Waltham, MA, USA) and secondary antibodies: goat anti-rabbit Alexa Fluor 546 conjugated antibody and goat anti-mouse Alexa Fluor 488 conjugated antibody (#A-11010 and #A-11001, respectively, Invitrogen, Carlsbad, CA, USA) were used. Nuclei were counterstained with Hoechst 33342 dye (Thermo Fisher Scientific, Waltham, MA, USA). Slides were mounted in ProLong™ Glass Antifade Mountant (Invitrogen, Carlsbad, CA, USA) and analyzed using Leica DMiL LED Fluo microscope (for α-SMA and p-CREB visualization, 40× objective) or Leica DMi8 with Thunder Imaging System (for p-Smad-2 visualization, 40× objective), all equipped with LAS-X Software (Leica Microsystems GmbH, Wetzlar, Germany). Experiments were run four times using five randomly selected fields of view, at the same fluorescent time exposure and in a blind-folded manner.
4.12. Quantitative PCR
Quantitative polymerase chain reaction (qPCR) was performed to assess the expression level of selected human genes:
ACTA2, MYH11, SM22, COL1A1, TNC, FN1, and
VCAN. MRC-5 cells were preincubated with study compounds (10 µM) for 1 h, followed by 24 h incubation with TGF-β
1, and then total RNA was extracted using Total RNA Mini Kit (A&A Biotechnology, Gdynia, Poland) according to the manufacturer’s protocol. RNA concentration was measured with a BioSpectrometer
® basic (Eppendorf), and equal amounts of total RNA (about 1 µg) were reverse-transcribed using the iScript™ cDNA Synthesis Kit (BioRad, Hercules, CA, USA). qPCR assays were performed using the CFX96 Touch Real-Time PCR Detection System (BioRad, Hercules, CA, USA) and SsoAdvanced
TM Universal SYBR
® Green Supermix (BioRad, Hercules, CA, USA). qPCR cycling was performed with denaturation at 94 °C for 30 s, annealing at 59 °C for 30 s and an extension at 72 °C for 30 s. Specific primers used to quantify the transcripts are listed in
Table 3. The relative abundance of specific mRNA transcripts was estimated based on cycle threshold (Ct) value and recalculated against the endogenous reference 18S ribosomal RNA gene (
18S rRNA) using the ∆Ct method. The experiments were run three times in duplicates.
4.13. Western Blotting
Western Blot analysis was performed according to a standard protocol. Briefly, MRC-5 cells were pre-incubated with the study compounds (10 µM) for 1 h, followed by 48 h incubation with TGF-β1 and then lysed in RIPA buffer (RIPA Lysis and Extraction Buffer, Thermo Fisher Scientific, Waltham, MA, USA) and supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail (both from Sigma Aldrich, St. Louis, MO, USA). Equal amounts of protein (30 µg) were separated using 10% SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Scientific, Waltham, MA, USA). Then membranes were washed with Tris Buffered Saline with Tween 20 (TBST) buffer (150 mM NaCl, 50 mM Tris, 0.05% Tween 20), blocked with 5% skim milk in TBST and incubated overnight with the appropriate primary antibody: rabbit, polyclonal anti-SMAD2 antibody, dilution 1:250, #51-1300 and rabbit, polyclonal anti-phospho-SMAD2 (Ser465, Ser467) antibody, dilution 1:500, #44-244G (both from Thermo Fisher Scientific, Waltham, MA, USA); rabbit, polyclonal anti-CREB1 antibody, dilution 1:500, #orb213777 and rabbit, polyclonal anti-phospho-CREB1 (Ser133) antibody, dilution 1:500, #orb213775 (both from Biorbyt LLC, San Francisco, CA, USA); mouse monoclonal anti-GAPDH antibody, dilution 1:5000, #G8795 (Sigma Aldrich, St. Louis, MO, USA). The next day, after gentle rising, membranes were exposed to secondary, rabbit anti-mouse IgG (whole molecule) peroxidase antibody (#A9044, dilution 1:3000, Sigma Aldrich, St. Louis, MO, USA) and goat anti-rabbit IgG (H+L) peroxidase-conjugated antibody (#32460, dilution 1:3000, Thermo Fisher Scientific, Waltham, MA, USA). Blots were visualized using a chemiluminescence method (Clarity Max™ Western ECL Substrate—Peroxide Solution, BioRad, Hercules, CA, USA) with a C-DiGit® Blot Scanner (LI-COR Biosciences, Lincoln, NE, USA). ROD was measured according to the NIH guidelines with Fiji ImageJ Software. The experiments were run four times.
4.14. Protein Kinase A Activity
Protein Kinase A (PKA) activity in MRC-5 cells was determined using a PKA Colorimetric Activity Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. MRC-5 cells were seeded in standard culture medium, and after 48 h they were serum-starved for 2 h in HBSS, followed by the addition of study compounds (10 µM) and incubation for 30 min. Finally, TGF-β1 was administered, and the samples were incubated for an additional 30 min. Whole cell lysates were used in the assay. The amount of PKA was normalized to the total protein content in each sample. The experiments were run two times in duplicates.
4.15. Statistical Analysis
Statistical analysis was performed with GraphPad Prism (GraphPad Software, Inc., San Diego, CA, USA, version 5.01). The comparisons between experimental conditions were performed using a non-parametric Wilcoxon test for paired data and a non-parametric Mann–Whitney U test (for qPCR data). Values presented in the graphs correspond to the mean ± standard error of the mean (S.E.M.). P < 0.05 was considered statistically significant.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}