Extracellular Matrix Analysis of Human Renal Arteries in Both Quiescent and Active Vascular State

 , ,
, ,  , , and
, , and
Abstract
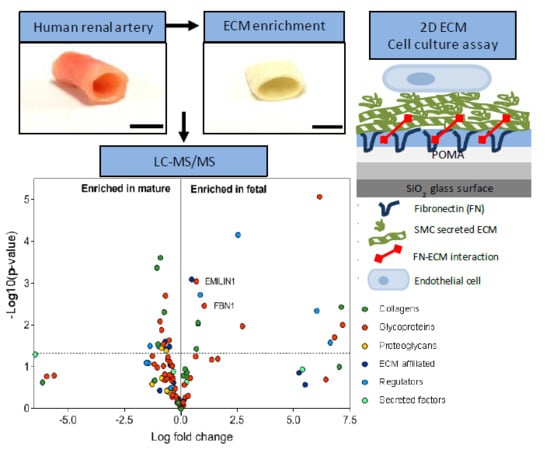
1. Introduction
2. Results
2.1. Enrichment of ECM Proteins in Vascular Tissue Prior to LC-MS/MS
2.2. Matrisome Protein Expression Differs between Human Fetal and Mature Renal Arteries
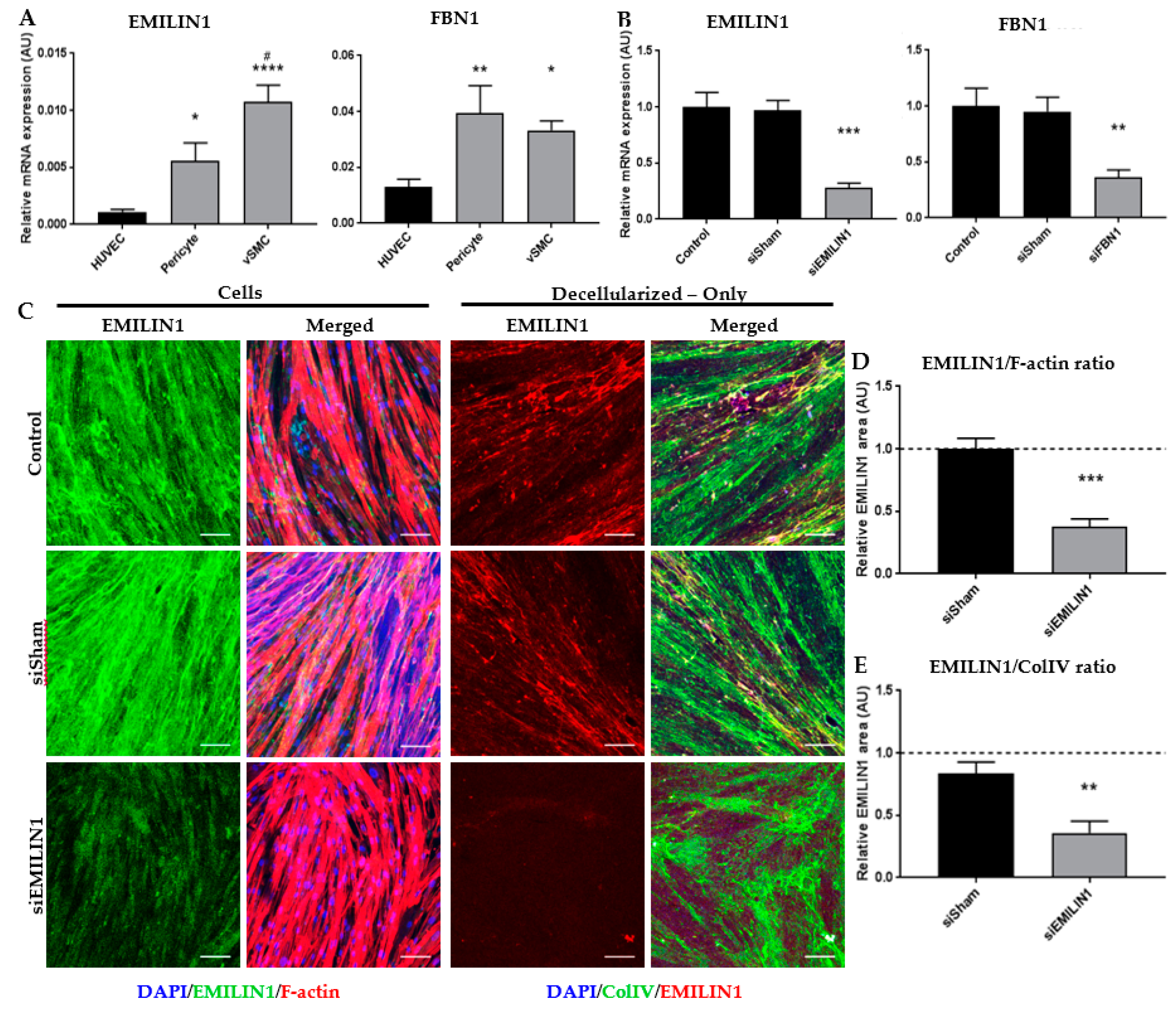
2.3. Glycoproteins EMILIN1 and FBN1 are Enriched in Fetal Renal Arteries and are Produced by Cells of the Mesenchymal Lineage
2.4. ECM Secreted by SMCs Can Be Altered by Depleting Specific ECM Components Using siRNA
2.5. Loss of EMILIN1 or FBN1 in the ECM Alters Transcriptome of ECs that Interacted with the Depleted ECM
3. Discussion
4. Materials and Methods
4.1. Human Tissue
4.2. Sample Preparation
4.3. LC-MS/MS Analysis
4.4. MS Data Analysis
4.5. Immunohistochemistry
4.6. Cell Culture
4.7. POMA Slides for Tight Anchoring of ECM
4.8. Immunocytochemistry
4.9. Quantitative PCR
4.10. Endothelial Cell Assays
4.10.1. RNA Sequencing
4.10.2. Proliferation Assay
4.10.3. PicoGreen Assay
4.10.4. PrestoBlue Assay
4.10.5. Adhesion Assay
4.10.6. Migration Assay
4.10.7. RhoA GTPase Activity Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APTES | (3-Aminopropyl)triethoxysilane |
| αSMA | Alpha-smooth muscle actin |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| EGM-2 | Endothelial growth medium-2 |
| EMILIN1 | Elastin microfibril interfacer 1 |
| FBN1 | Fibrillin-1 |
| FN | Fibronectin |
| HUVEC | Human umbilical vein endothelial cell |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| POMA | poly(maleic anhydride-alt-1-octadecene) |
| POMA-FN | poly(maleic anhydride-alt-1-octadecene) with fibronectin |
| SMC | Smooth muscle cell |
| TGF-β | Transforming growth factor beta |
References
- Stratman, A.N.; Malotte, K.M.; Mahan, R.D.; Davis, M.J.; Davis, G.E. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 2009, 114, 5091–5101. [Google Scholar] [CrossRef] [PubMed]
- Stratman, A.N.; Davis, G.E. Endothelial cell-pericyte interactions stimulate basement membrane matrix assembly, influence on vascular tube remodeling, maturation, and stabilization. Microsc. Microanal. 2012, 18, 68-80002E. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome, in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteom 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix, biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef]
- Liu, Y.; Senger, D.R. Matrix-specific activation of Src and Rho initiates capillary morphogenesis of endothelial cells. FASEB J. 2004, 18, 457–468. [Google Scholar] [CrossRef]
- Hallmann, R.; Horn, N.; Selg, M.; Wendler, O.; Pausch, F.; Sorokin, L.M. Expression and function of laminins in the embryonic and mature vasculature. Physiol. Rev. 2005, 85, 979–1000. [Google Scholar] [CrossRef]
- Thottappillil, N.; Nair, P.D. Scaffolds in vascular regeneration, current status. Vasc. Health Risk Manag. 2015, 11, 79–91. [Google Scholar]
- Bouten, C.V.C.; Smits, A.; Baaijens, F.P.T. Can We Grow Valves Inside the Heart? Perspective on Material-based In Situ Heart Valve Tissue Engineering. Front. Cardiovasc. Med. 2018, 5, 54. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix, Tools and insights for the “omics” era. Matrix. Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Barallobre-Barreiro, J.; Oklu, R.; Lynch, M.; Fava, M.; Baig, F.; Yin, X.; Barwari, T.; Potier, D.N.; Albadawi, H.; Jahangiri, M.; et al. Extracellular matrix remodelling in response to venous hypertension, proteomics of human varicose veins. Cardiovasc. Res. 2016, 110, 419–430. [Google Scholar] [CrossRef]
- Mustafa, D.A.; Dekker, L.J.; Stingl, C.; Kremer, A.; Stoop, M.; Sillevis Smitt, P.A.; Kros, J.M.; Luider, T.M. A proteome comparison between physiological angiogenesis and angiogenesis in glioblastoma. Mol. Cell Proteom 2012, 11, M111.008466. [Google Scholar] [CrossRef]
- Bressan, G.M.; Daga-Gordini, D.; Colombatti, A.; Castellani, I.; Marigo, V.; Volpin, D. Emilin, a component of elastic fibers preferentially located at the elastin-microfibrils interface. J. Cell Biol. 1993, 121, 201–212. [Google Scholar] [CrossRef]
- Kielty, C.M.; Sherratt, M.J.; Shuttleworth, C.A. Elastic fibres. J. Cell Sci. 2002, 115, 2817–2828. [Google Scholar]
- Colombatti, A.; Spessotto, P.; Doliana, R.; Mongiat, M.; Bressan, G.M.; Esposito, G. The EMILIN/Multimerin family. Front. Immunol. 2011, 2, 93. [Google Scholar] [CrossRef]
- Verdone, G.; Corazza, A.; Colebrooke, S.A.; Cicero, D.; Eliseo, T.; Boyd, J.; Doliana, R.; Fogolari, F.; Viglino, P.; Colombatti, A.; et al. NMR-based homology model for the solution structure of the C-terminal globular domain of EMILIN1. J. Biomol. NMR 2009, 43, 79–96. [Google Scholar] [CrossRef]
- Verdone, G.; Doliana, R.; Corazza, A.; Colebrooke, S.A.; Spessotto, P.; Bot, S.; Bucciotti, F.; Capuano, A.; Silvestri, A.; Viglino, P.; et al. The solution structure of EMILIN1 globular C1q domain reveals a disordered insertion necessary for interaction with the alpha4beta1 integrin. J. Biol. Chem. 2008, 283, 18947–18956. [Google Scholar] [CrossRef]
- Danussi, C.; Petrucco, A.; Wassermann, B.; Pivetta, E.; Modica, T.M.; Del Bel Belluz, L.; Colombatti, A.; Spessotto, P. EMILIN1-alpha4/alpha9 integrin interaction inhibits dermal fibroblast and keratinocyte proliferation. J. Cell Biol. 2011, 195, 131–145. [Google Scholar] [CrossRef]
- Danussi, C.; Del Bel Belluz, L.; Pivetta, E.; Modica, T.M.; Muro, A.; Wassermann, B.; Doliana, R.; Sabatelli, P.; Colombatti, A.; Spessotto, P. EMILIN1/alpha9beta1 integrin interaction is crucial in lymphatic valve formation and maintenance. Mol. Cell Biol. 2013, 33, 4381–4394. [Google Scholar] [CrossRef]
- Spessotto, P.; Cervi, M.; Mucignat, M.T.; Mungiguerra, G.; Sartoretto, I.; Doliana, R.; Colombatti, A. beta 1 Integrin-dependent cell adhesion to EMILIN-1 is mediated by the gC1q domain. J. Biol. Chem. 2003, 278, 6160–6167. [Google Scholar] [CrossRef]
- Angel, P.M.; Narmoneva, D.A.; Sewell-Loftin, M.K.; Munjal, C.; Dupuis, L.; Landis, B.J.; Jegga, A.; Kern, C.B.; Merryman, W.D.; Baldwin, H.S.; et al. Proteomic Alterations Associated with Biomechanical Dysfunction are Early Processes in the Emilin1 Deficient Mouse Model of Aortic Valve Disease. Ann. Biomed. Eng. 2017, 45, 2548–2562. [Google Scholar] [CrossRef]
- Mariko, B.; Pezet, M.; Escoubet, B.; Bouillot, S.; Andrieu, J.P.; Starcher, B.; Quaglino, D.; Jacob, M.P.; Huber, P.; Ramirez, F.; et al. Fibrillin-1 genetic deficiency leads to pathological ageing of arteries in mice. J. Pathol. 2011, 224, 33–44. [Google Scholar] [CrossRef]
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1, The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 591, 279–291. [Google Scholar] [CrossRef]
- Louzao-Martinez, L.; van Dijk, C.G.M.; Xu, Y.J.; Korn, A.; Bekker, N.J.; Brouwhuis, R.; Nicese, M.N.; Demmers, J.A.A.; Goumans, M.-J.T.H.; Masereeuw, R.; et al. A proteome comparison between human fetal and mature renal extracellular matrix identifies EMILIN1 as a regulator of renal epithelial cell adhesion. Matrix Biol. Plus 2019, 4, 100011. [Google Scholar] [CrossRef]
- Goumans, M.J.; Liu, Z.; ten Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Keely, P.J. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J. Cell Sci. 2011, 124, 1195–1205. [Google Scholar] [CrossRef]
- Hu, Y.L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci. Rep. 2014, 4, 6024. [Google Scholar] [CrossRef]
- Carta, L.; Pereira, L.; Arteaga-Solis, E.; Lee-Arteaga, S.Y.; Lenart, B.; Starcher, B.; Merkel, C.A.; Sukoyan, M.; Kerkis, A.; Hazeki, N.; et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J. Biol. Chem. 2006, 281, 8016–8023. [Google Scholar] [CrossRef]
- Barbier, M.; Gross, M.S.; Aubart, M.; Hanna, N.; Kessler, K.; Guo, D.C.; Tosolini, L.; Ho-Tin-Noe, B.; Regalado, E.; Varret, M.; et al. MFAP5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am. J. Hum. Genet. 2014, 95, 736–743. [Google Scholar] [CrossRef]
- Combs, M.D.; Knutsen, R.H.; Broekelmann, T.J.; Toennies, H.M.; Brett, T.J.; Miller, C.A.; Kober, D.L.; Craft, C.S.; Atkinson, J.J.; Shipley, J.M.; et al. Microfibril-associated glycoprotein 2 (MAGP2) loss of function has pleiotropic effects in vivo. J. Biol. Chem. 2013, 288, 28869–28880. [Google Scholar] [CrossRef]
- Nakajima, Y.; Miyazono, K.; Kato, M.; Takase, M.; Yamagishi, T.; Nakamura, H. Extracellular fibrillar structure of latent TGF beta binding protein-1, role in TGF beta-dependent endothelial-mesenchymal transformation during endocardial cushion tissue formation in mouse embryonic heart. J. Cell Biol. 1997, 136, 193–204. [Google Scholar] [CrossRef]
- Rifkin, D.B.; Rifkin, W.J.; Zilberberg, L. LTBPs in biology and medicine, LTBP diseases. Matrix Biol. 2018, 71, 90–99. [Google Scholar] [CrossRef]
- Litteri, G.; Carnevale, D.; D’Urso, A.; Cifelli, G.; Braghetta, P.; Damato, A.; Bizzotto, D.; Landolfi, A.; Ros, F.D.; Sabatelli, P.; et al. Vascular smooth muscle Emilin-1 is a regulator of arteriolar myogenic response and blood pressure. Arter. Thromb. Vasc. Biol. 2012, 32, 2178–2184. [Google Scholar] [CrossRef]
- Braghetta, P.; Ferrari, A.; de Gemmis, P.; Zanetti, M.; Volpin, D.; Bonaldo, P.; Bressan, G.M. Expression of the EMILIN-1 gene during mouse development. Matrix Biol. 2002, 21, 603–609. [Google Scholar] [CrossRef]
- Zanetti, M.; Braghetta, P.; Sabatelli, P.; Mura, I.; Doliana, R.; Colombatti, A.; Volpin, D.; Bonaldo, P.; Bressan, G.M. EMILIN-1 deficiency induces elastogenesis and vascular cell defects. Mol. Cell Biol. 2004, 24, 638–650. [Google Scholar] [CrossRef]
- Zacchigna, L.; Vecchione, C.; Notte, A.; Cordenonsi, M.; Dupont, S.; Maretto, S.; Cifelli, G.; Ferrari, A.; Maffei, A.; Fabbro, C.; et al. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell 2006, 124, 929–942. [Google Scholar] [CrossRef]
- Raman, M.; Cobb, M.H. TGF-beta regulation by Emilin1, new links in the etiology of hypertension. Cell 2006, 124, 893–895. [Google Scholar] [CrossRef]
- Carnevale, D.; Facchinello, N.; Iodice, D.; Bizzotto, D.; Perrotta, M.; De Stefani, D.; Pallante, F.; Carnevale, L.; Ricciardi, F.; Cifelli, G.; et al. Loss of EMILIN-1 Enhances Arteriolar Myogenic Tone Through TGF-beta (Transforming Growth Factor-beta)-Dependent Transactivation of EGFR (Epidermal Growth Factor Receptor) and Is Relevant for Hypertension in Mice and Humans. Arter. Thromb. Vasc. Biol. 2018, 38, 2484–2497. [Google Scholar] [CrossRef]
- Spessotto, P.; Bulla, R.; Danussi, C.; Radillo, O.; Cervi, M.; Monami, G.; Bossi, F.; Tedesco, F.; Doliana, R.; Colombatti, A. EMILIN1 represents a major stromal element determining human trophoblast invasion of the uterine wall. J. Cell Sci. 2006, 119, 4574–4584. [Google Scholar] [CrossRef]
- Mariko, B.; Ghandour, Z.; Raveaud, S.; Quentin, M.; Usson, Y.; Verdetti, J.; Huber, P.; Kielty, C.; Faury, G. Microfibrils and fibrillin-1 induce integrin-mediated signaling, proliferation and migration in human endothelial cells. Am. J. Physiol. Cell Physiol. 2010, 299, C977–C987. [Google Scholar] [CrossRef]
- Cheng, A.; Cain, S.A.; Tian, P.; Baldwin, A.K.; Uppanan, P.; Kielty, C.M.; Kimber, S.J. Recombinant Extracellular Matrix Protein Fragments Support Human Embryonic Stem Cell Chondrogenesis. Tissue Eng. Part A 2018, 24, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Hajian, H.; Wise, S.G.; Bax, D.V.; Kondyurin, A.; Waterhouse, A.; Dunn, L.L.; Kielty, C.M.; Yu, Y.; Weiss, A.S.; Bilek, M.M.; et al. Immobilisation of a fibrillin-1 fragment enhances the biocompatibility of PTFE. Colloids Surf. B Biointerfaces 2014, 116, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Sacks, T.; Moldow, C.F.; Craddock, P.R.; Bowers, T.K.; Jacob, H.S. Oxygen radicals mediate endothelial cell damage by complement-stimulated granulocytes. An in vitro model of immune vascular damage. J. Clin. Invest. 1978, 61, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Wilm, M.; Shevchenko, A.; Houthaeve, T.; Breit, S.; Schweigerer, L.; Fotsis, T.; Mann, M. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 1996, 379, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Sap, K.A.; Bezstarosti, K.; Dekkers, D.H.W.; Voets, O.; Demmers, J.A.A. Quantitative Proteomics Reveals Extensive Changes in the Ubiquitinome after Perturbation of the Proteasome by Targeted dsRNA-Mediated Subunit Knockdown in Drosophila. J. Proteome Res. 2017, 16, 2848–2862. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Labit, H.; Goldar, A.; Guilbaud, G.; Douarche, C.; Hyrien, O.; Marheineke, K. A simple and optimized method of producing silanized surfaces for FISH and replication mapping on combed DNA fibers. Biotechniques 2008, 45, 649–658. [Google Scholar] [CrossRef]
- Brandt, M.M.; Meddens, C.A.; Louzao-Martinez, L.; van den Dungen, N.A.M.; Lansu, N.R.; Nieuwenhuis, E.E.S.; Duncker, D.J.; Verhaar, M.C.; Joles, J.A.; Mokry, M.; et al. Chromatin Conformation Links Distal Target Genes to CKD Loci. J. Am. Soc. Nephrol. 2018, 29, 462–476. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins Detected | Fetal Renal Artery | Mature Renal Artery |
|---|---|---|
| Total number of proteins detected | 206 | 246 |
| Matrisome proteins | 79 (38.3% of total) | 87 (35.4% of total) |
| Non-matrisome proteins | 127 (61.7% of total) | 159 (64.6% of total) |
| Matrisome core proteins | 58 (73.4% of matrisome) | 63 (73.3% of matrisome) |
| Glycoproteins | 38 (65.6% of core) | 39 (61.9% of core) |
| Collagens | 14 (24.1% of core) | 15 (23.8% of core) |
| Proteoglycans | 6 (10.3% of core) | 9 (14.3% of core) |
| Matrisome-associated proteins | 21 (26.6% of matrisome) | 23 (26.7% of matrisome) |
| ECM-affiliated | 9 (42.9% of associated) | 9 (39.1% of associated) |
| ECM regulators | 7 (33.3% of associated) | 10 (43.5% of associated) |
| Secreted factors | 5 (23.8% of associated) | 4 (17.4% of associated) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Dijk, C.G.M.; Louzao-Martinez, L.; van Mulligen, E.; Boermans, B.; Demmers, J.A.A.; van den Bosch, T.P.P.; Goumans, M.-J.; Duncker, D.J.; Verhaar, M.C.; Cheng, C. Extracellular Matrix Analysis of Human Renal Arteries in Both Quiescent and Active Vascular State. Int. J. Mol. Sci. 2020, 21, 3905. https://doi.org/10.3390/ijms21113905
van Dijk CGM, Louzao-Martinez L, van Mulligen E, Boermans B, Demmers JAA, van den Bosch TPP, Goumans M-J, Duncker DJ, Verhaar MC, Cheng C. Extracellular Matrix Analysis of Human Renal Arteries in Both Quiescent and Active Vascular State. International Journal of Molecular Sciences. 2020; 21(11):3905. https://doi.org/10.3390/ijms21113905
Chicago/Turabian Stylevan Dijk, Christian G.M., Laura Louzao-Martinez, Elise van Mulligen, Bart Boermans, Jeroen A.A. Demmers, Thierry P.P. van den Bosch, Marie-José Goumans, Dirk J. Duncker, Marianne C. Verhaar, and Caroline Cheng. 2020. "Extracellular Matrix Analysis of Human Renal Arteries in Both Quiescent and Active Vascular State" International Journal of Molecular Sciences 21, no. 11: 3905. https://doi.org/10.3390/ijms21113905
APA Stylevan Dijk, C. G. M., Louzao-Martinez, L., van Mulligen, E., Boermans, B., Demmers, J. A. A., van den Bosch, T. P. P., Goumans, M.-J., Duncker, D. J., Verhaar, M. C., & Cheng, C. (2020). Extracellular Matrix Analysis of Human Renal Arteries in Both Quiescent and Active Vascular State. International Journal of Molecular Sciences, 21(11), 3905. https://doi.org/10.3390/ijms21113905