Consolidated BRCA1/2 Variant Interpretation by MH BRCA Correlates with Predicted PARP Inhibitor Efficacy Association by MH Guide

 , , ,
, , ,
Abstract

1. Introduction
2. Results
2.1. Pre-Evaluation of MH BRCA
2.2. Impact of Japanese Genomic Cohort Data
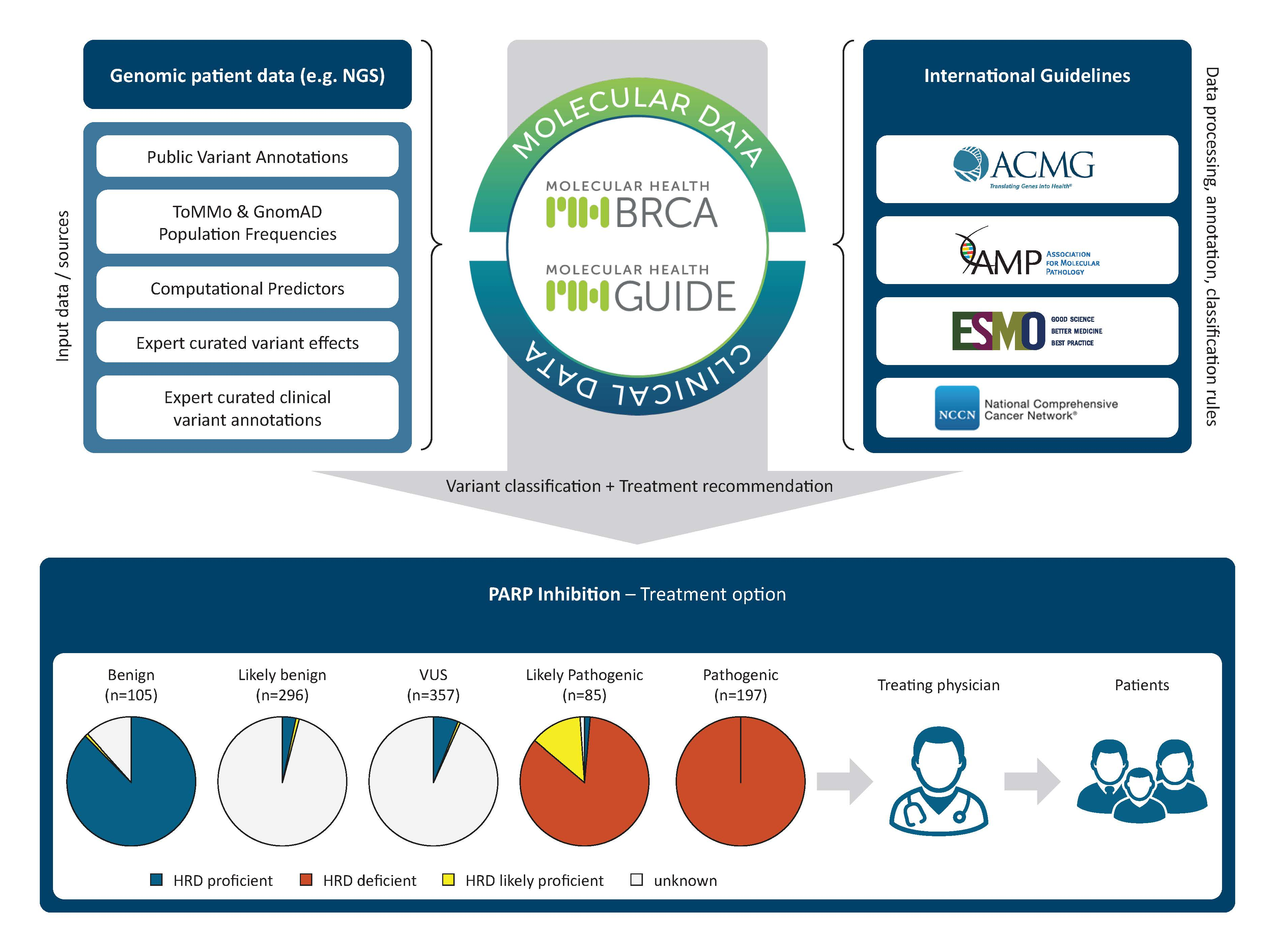
2.3. Performance Indication of Consolidated BRCA1/2 Variant Interpretation
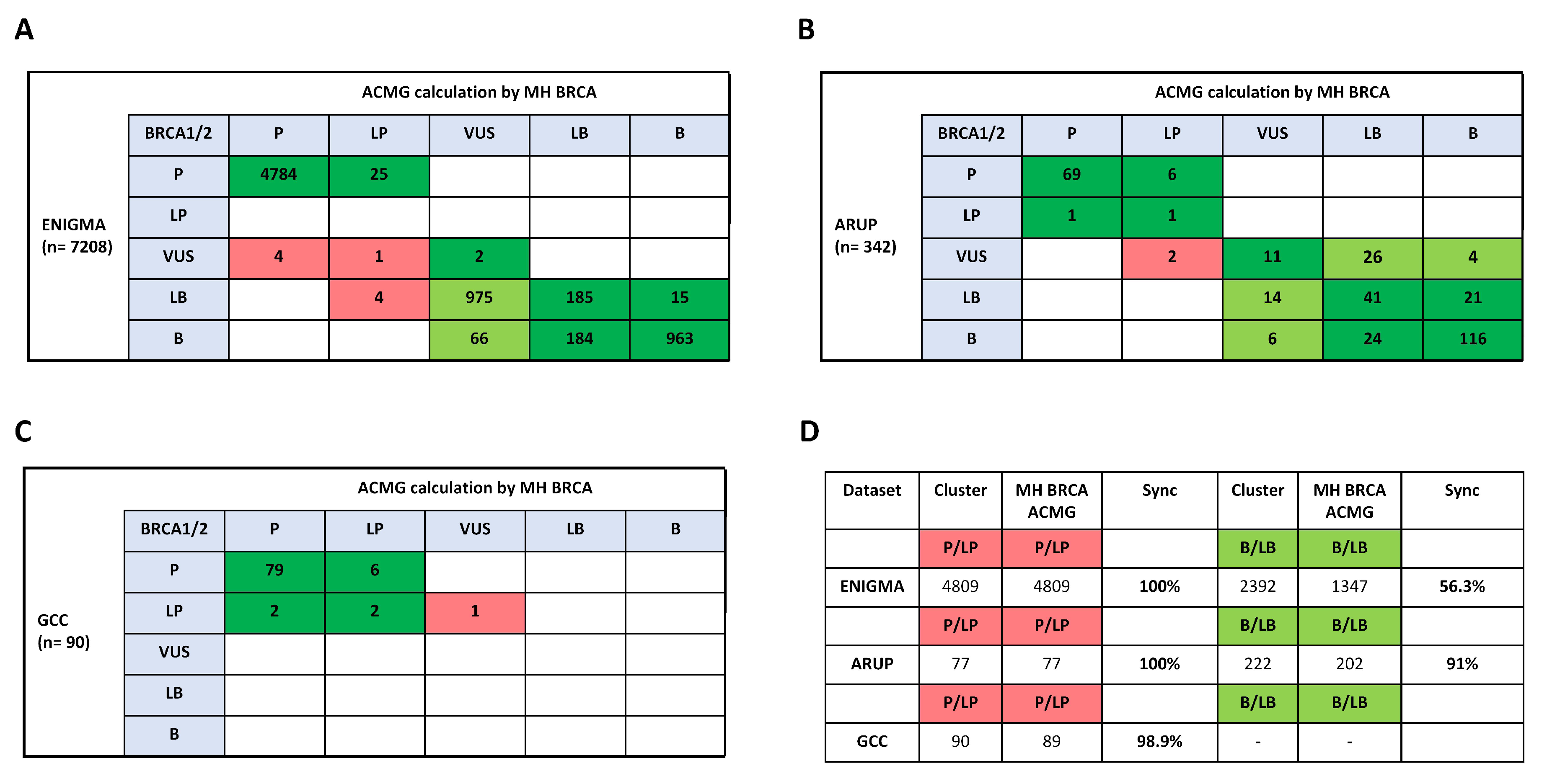
2.4. Comparison to ClinVar Submitter Content for BRCA1/2 Variant Classification
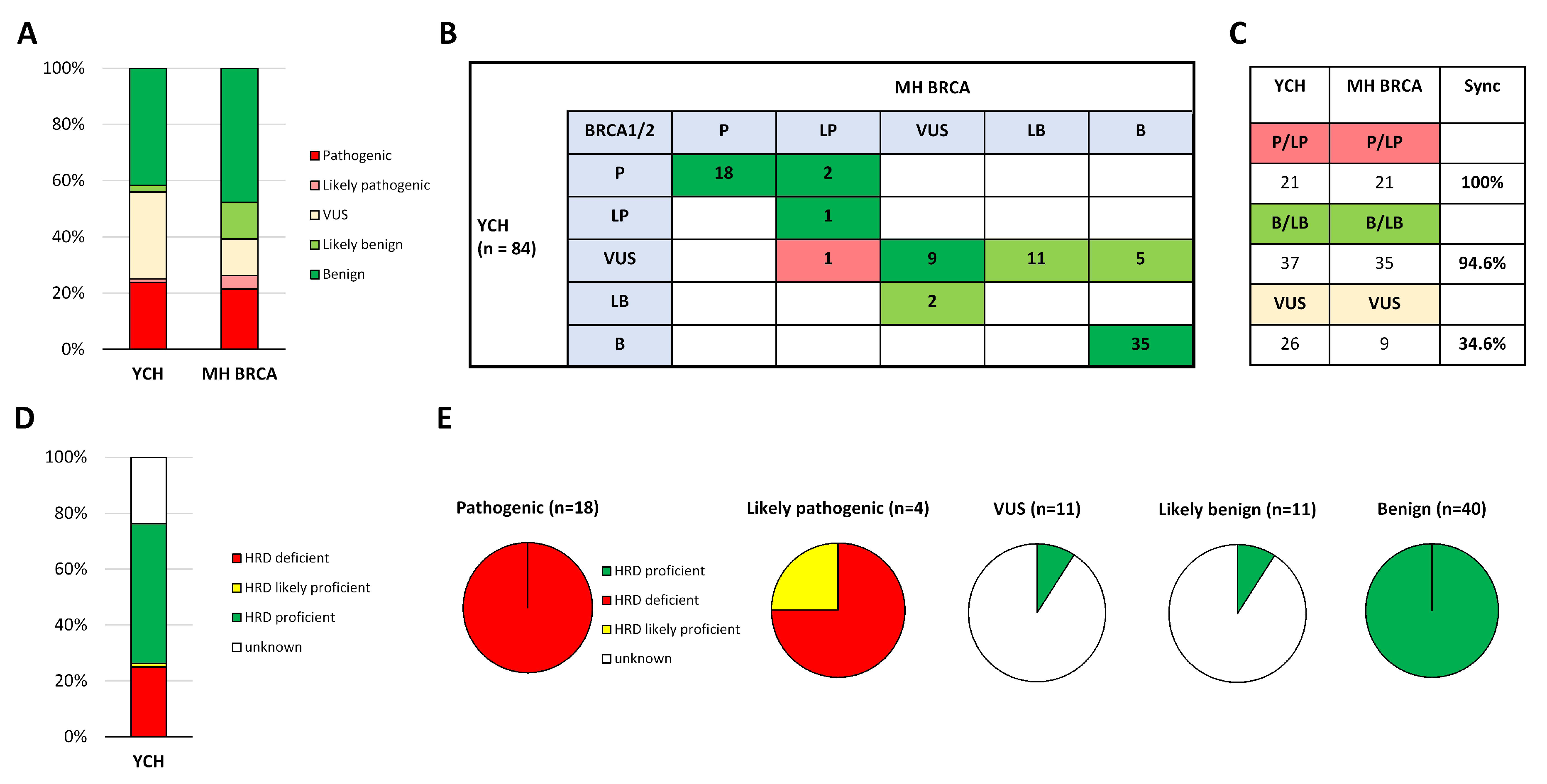
2.4.1. Clinical Evaluation of MH BRCA
2.4.2. Correlation of Predicted Efficacy Association to PARP Inhibition by MH Guide
3. Discussion
4. Material and Methods
4.1. Patients and Methods
4.2. Targeted Next-Generation Sequencing (NGS)
4.3. Data Analysis for Clinical Assessment of BRCA1/2 Interpretation
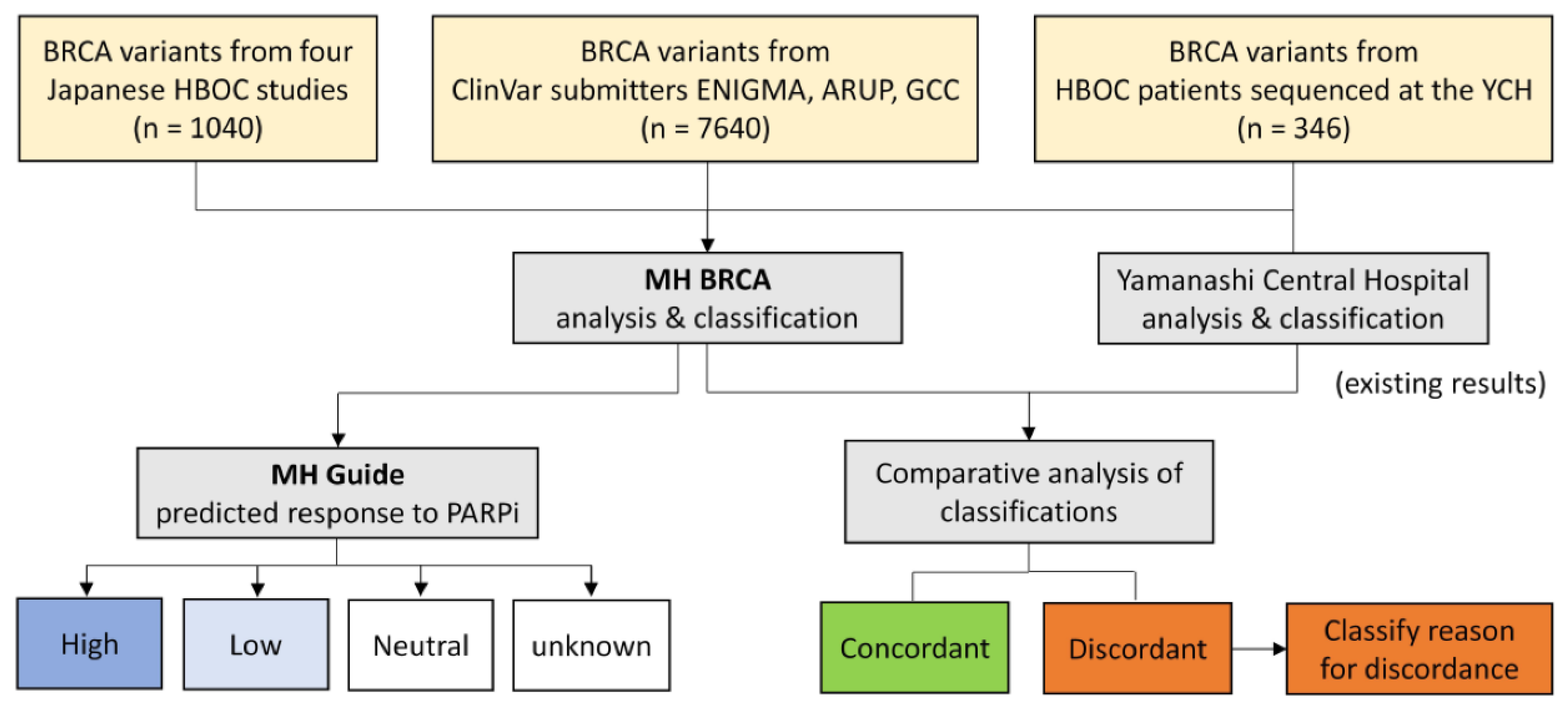
4.4. Study Design for Validation of MH BRCA
4.5. BRCA1/2 Interpretation by MH BRCA
4.6. BRCA1/2 Interpretation for Predicted Treatment Response to PARP Inhibition
4.7. Data Availability Statement
4.8. Code Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.; Puhalla, S. BRCA 1/2-Mutation Related and Sporadic Breast and Ovarian Cancers: More Alike than Different. Front. Oncol. 2014, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu. Rev. Pathol. 2009, 4, 461–487. [Google Scholar] [CrossRef]
- Drew, Y.; Ledermann, J.; Hall, G.; Rea, D.; Glasspool, R.; Highley, M.; Jayson, G.; Sludden, J.; Murray, J.; Jamieson, D.; et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br. J. Cancer 2016, 114, 723–730. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Nagasaki, M.; Yasuda, J.; Katsuoka, F.; Nariai, N.; Kojima, K.; Kawai, Y.; Yamaguchi-Kabata, Y.; Yokozawa, J.; Danjoh, I.; Saito, S.; et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun. 2015, 6, 8018. [Google Scholar] [CrossRef] [PubMed]
- Higasa, K.; Miyake, N.; Yoshimura, J.; Okamura, K.; Niihori, T.; Saitsu, H.; Doi, K.; Shimizu, M.; Nakabayashi, K.; Aoki, Y.; et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J. Hum. Genet. 2016, 61, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Momozawa, Y.; Iwasaki, Y.; Parsons, M.T.; Kamatani, Y.; Takahashi, A.; Tamura, C.; Katagiri, T.; Yoshida, T.; Nakamura, S.; Sugano, K.; et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat. Commun. 2018, 9, 4083. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Yokoyama, S.; Watanabe, C.; Yoshida, R.; Kita, M.; Okawa, M.; Sakurai, A.; Sekine, M.; Yotsumoto, J.; Nomura, H.; et al. Genetic and clinical characteristics in Japanese hereditary breast and ovarian cancer: First report after establishment of HBOC registration system in Japan. J. Hum. Genet. 2018, 63, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Nakagomi, H.; Sakamoto, I.; Amemiya, K.; Mochizuki, H.; Omata, M. Detection of BRCA1 and BRCA2 germline mutations in Japanese population using next-generation sequencing. Mol. Genet. Genom. Med. 2015, 3, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Nakagomi, H.; Mochizuki, H.; Inoue, M.; Hirotsu, Y.; Amemiya, K.; Sakamoto, I.; Nakagomi, S.; Kubota, T.; Omata, M. Combined annotation-dependent depletion score for BRCA1/2 variants in patients with breast and/or ovarian cancer. Cancer Sci. 2018, 109, 453–461. [Google Scholar] [CrossRef]
- Genomes Project, C.; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Easton, D.F.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wenstrup, R.J.; Allen-Brady, K.; Tavtigian, S.V.; Monteiro, A.N.; Iversen, E.S.; Couch, F.J.; et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am. J. Hum. Genet. 2007, 81, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Mesman, R.L.S.; Calleja, F.; Hendriks, G.; Morolli, B.; Misovic, B.; Devilee, P.; van Asperen, C.J.; Vrieling, H.; Vreeswijk, M.P.G. The functional impact of variants of uncertain significance in BRCA2. Genet. Med. 2019, 21, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Yang, S.; Nykamp, K.; Garcia, J.; Lincoln, S.E.; Topper, S.E. Pathogenic variant burden in the ExAC database: An empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Lee, E.J.; Lee, Y.J.; Kim, J.; Kim, Y.; Kim, K.; Lee, S.W.; Chang, S.; Lee, Y.J.; Lee, J.W.; et al. Impact of proactive high-throughput functional assay data on BRCA1 variant interpretation in 3684 patients with breast or ovarian cancer. J. Hum. Genet. 2020, 65, 209–220. [Google Scholar] [CrossRef]
- Lee, M.S.; Green, R.; Marsillac, S.M.; Coquelle, N.; Williams, R.S.; Yeung, T.; Foo, D.; Hau, D.D.; Hui, B.; Monteiro, A.N.; et al. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 2010, 70, 4880–4890. [Google Scholar] [CrossRef]
- Nakamura, S.; Takahashi, M.; Tozaki, M.; Nakayama, T.; Nomizu, T.; Miki, Y.; Murakami, Y.; Aoki, D.; Iwase, T.; Nishimura, S.; et al. Prevalence and differentiation of hereditary breast and ovarian cancers in Japan. Breast Cancer 2015, 22, 462–468. [Google Scholar] [CrossRef]
- Hart, S.N.; Hoskin, T.; Shimelis, H.; Moore, R.M.; Feng, B.; Thomas, A.; Lindor, N.M.; Polley, E.C.; Goldgar, D.E.; Iversen, E.; et al. Comprehensive annotation of BRCA1 and BRCA2 missense variants by functionally validated sequence-based computational prediction models. Genet. Med. 2019, 21, 71–80. [Google Scholar] [CrossRef]
- Lee, J.S.; Oh, S.; Park, S.K.; Lee, M.H.; Lee, J.W.; Kim, S.W.; Son, B.H.; Noh, D.Y.; Lee, J.E.; Park, H.L.; et al. Reclassification of BRCA1 and BRCA2 variants of uncertain significance: A multifactorial analysis of multicentre prospective cohort. J. Med. Genet. 2018, 55, 794–802. [Google Scholar] [CrossRef]
- So, M.K.; Jeong, T.D.; Lim, W.; Moon, B.I.; Paik, N.S.; Kim, S.C.; Huh, J. Reinterpretation of BRCA1 and BRCA2 variants of uncertain significance in patients with hereditary breast/ovarian cancer using the ACMG/AMP 2015 guidelines. Breast Cancer 2019. [Google Scholar] [CrossRef]
- Mondal, G.; Rowley, M.; Guidugli, L.; Wu, J.; Pankratz, V.S.; Couch, F.J. BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Dev. Cell 2012, 23, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.; Neuhausen, S.L.; Hansen, T.V.; Couch, F.J.; Vreeswijk, M.P.; et al. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum. Mutat. 2014, 35, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Balia, C.; Galli, A.; Caligo, M.A. Effect of the overexpression of BRCA2 unclassified missense variants on spontaneous homologous recombination in human cells. Breast Cancer Res. Treat 2011, 129, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Islam, M.M.; Banerjee, T.; Adamovich, A.I.; Gullingsrud, J.; Fields, S.; Shendure, J.; Parvin, J.D. A Multiplex Homology-Directed DNA Repair Assay Reveals the Impact of More Than 1,000 BRCA1 Missense Substitution Variants on Protein Function. Am. J. Hum. Genet. 2018, 103, 498–508. [Google Scholar] [CrossRef]
- Brnich, S.E.; Rivera-Muñoz, E.A.; Berg, J.S. Quantifying the potential of functional evidence to reclassify variants of uncertain significance in the categorical and Bayesian interpretation frameworks. Hum. Mutat. 2018, 39, 1531–1541. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Ooka, Y.; Sakamoto, I.; Nakagomi, H.; Omata, M. Simultaneous detection of genetic and copy number alterations in BRCA1/2 genes. Oncotarget 2017, 8, 114463–114473. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case-ID | Gene | HGVS g. | HGVS c. | HGVS p. | SNP-ID Number | YCH | MH BRCA |
|---|---|---|---|---|---|---|---|
| BRCA006 | BRCA1 | 41222974C > A | 4957G > T | V1653L | rs80357261 | VUS | LP |
| BRCA2 | 32914623G > T | 6131G > T | G2044V | rs56191579 | B | B | |
| BRCA008 | BRCA1 | 41222974C > A | 4957G > T | V1653L | rs80357261 | VUS | LP |
| BRCA2 | 32914623G > T | 6131G > T | G2044V | rs56191579 | B | B | |
| BRCA1 | 41243841T > C | 3707A > G | N1236S | rs863224760 | VUS | VUS | |
| BRCA2 | 32910842A > G | 2350A > G | M784V | rs11571653 | B | B | |
| BRCA052 | BRCA1 | 41222974C > A | 4957G > T | V1653L | rs80357261 | VUS | LP |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirotsu, Y.; Schmidt-Edelkraut, U.; Nakagomi, H.; Sakamoto, I.; Hartenfeller, M.; Narang, R.; Soldatos, T.G.; Kaduthanam, S.; Wang, X.; Hettich, S.; et al. Consolidated BRCA1/2 Variant Interpretation by MH BRCA Correlates with Predicted PARP Inhibitor Efficacy Association by MH Guide. Int. J. Mol. Sci. 2020, 21, 3895. https://doi.org/10.3390/ijms21113895
Hirotsu Y, Schmidt-Edelkraut U, Nakagomi H, Sakamoto I, Hartenfeller M, Narang R, Soldatos TG, Kaduthanam S, Wang X, Hettich S, et al. Consolidated BRCA1/2 Variant Interpretation by MH BRCA Correlates with Predicted PARP Inhibitor Efficacy Association by MH Guide. International Journal of Molecular Sciences. 2020; 21(11):3895. https://doi.org/10.3390/ijms21113895
Chicago/Turabian StyleHirotsu, Yosuke, Udo Schmidt-Edelkraut, Hiroshi Nakagomi, Ikuko Sakamoto, Markus Hartenfeller, Ram Narang, Theodoros G. Soldatos, Sajo Kaduthanam, Xiaoyue Wang, Stephan Hettich, and et al. 2020. "Consolidated BRCA1/2 Variant Interpretation by MH BRCA Correlates with Predicted PARP Inhibitor Efficacy Association by MH Guide" International Journal of Molecular Sciences 21, no. 11: 3895. https://doi.org/10.3390/ijms21113895
APA StyleHirotsu, Y., Schmidt-Edelkraut, U., Nakagomi, H., Sakamoto, I., Hartenfeller, M., Narang, R., Soldatos, T. G., Kaduthanam, S., Wang, X., Hettich, S., Brock, S., Jackson, D. B., & Omata, M. (2020). Consolidated BRCA1/2 Variant Interpretation by MH BRCA Correlates with Predicted PARP Inhibitor Efficacy Association by MH Guide. International Journal of Molecular Sciences, 21(11), 3895. https://doi.org/10.3390/ijms21113895